





Abstract
Congenital muscular dystrophy type 1A (MDC1A) is caused by recessive variants in laminin α2 (LAMA2). Patients have been found to have white matter signal abnormalities on magnetic resonance imaging (MRI) but rarely structural brain abnormalities. We describe the autopsy neuropathology in a 17-year-old with white matter signal abnormalities on brain MRI. Dystrophic pathology was observed in skeletal muscle, and the sural nerve manifested a mild degree of segmental demyelination and remyelination. A diffuse, bilateral cobblestone appearance, and numerous points of fusion between adjacent gyri were apparent on gross examination of the cerebrum. Brain histopathology included focal disruptions of the glia limitans associated with abnormal cerebral cortical lamination or arrested cerebellar granule cell migration. Subcortical nodular heterotopia was present within the cerebellar hemispheres. Sampling of the centrum semiovale revealed no light microscopic evidence of leukoencephalopathy. Three additional MDC1A patients were diagnosed with cobblestone malformation on brain MRI. Unlike the autopsied patient whose brain had a symmetric distribution of cobblestone pathology, the latter patients had asymmetric involvement, most severe in the occipital lobes. These cases demonstrate that cobblestone malformation may be an important manifestation of the brain pathology in MDC1A and can be present even when patients have a structurally normal brain MRI.
INTRODUCTION
Congenital muscular dystrophy type 1A (MDC1A), also known as merosin-deficient congenital muscular dystrophy (CMD), is a rare autosomal recessive condition caused by deficient merosin expression (1–3). It is the most common cause of what was originally called classical, or occidental, CMD without structural involvement of the central nervous system (CNS) in distinction to CMDs with CNS involvement (Fukuyama CMD, muscle-eye-brain disease, and Walker-Warburg syndrome) (4).
MDC1A results from variants in the LAMA2 gene on 6p22-23 leading to partial or complete absence of laminin α2 chains (2). Merosin (laminin 211), a structural protein of the basement membrane, is a heterotrimer composed of laminin α2, γ1, and β1 chains. It is the predominant basement membrane laminin expressed in striated muscle and Schwann cells (5). In the brain, laminin α2 is expressed by astrocyte foot processes at the glia limitans and blood-brain barrier (6). The C-terminal G domain of merosin binds with high affinity to matriglycan, a multiple repeat polymer of xylose-glucuronic acid on α-dystroglycan, an extracellular component of the dystrophin-glycoprotein complex (DGC) (7–9). This helps influence adhesion, differentiation, growth, shape, and migration of cells. Through its linkage to the subsarcolemmal cytoskeleton via the DGC, it has a crucial role in maintaining the integrity of the cell membrane (10–12). With reduced (partial merosin deficiency) or absent (complete merosin deficiency) levels of merosin, the protein is not able to serve as an acceptor for matriglycan. This results in a loss of the stabilizing connection to the DGC, leading to a loss of structural integrity and function of various cells, including myocytes and Schwann cells (13).
MDC1A is most commonly caused by complete merosin deficiency resulting in severe muscle weakness with onset within the first 6 months of life (typically at or near birth), contractures, respiratory insufficiency, inability to achieve independent ambulation, and elevated creatinine kinase (CK) exceeding 1000 IU/L. Less frequent clinical features include seizures, intellectual disability, and neuropathy (14). Patients with less severe forms of the disease (partial merosin deficiency) present later in infancy or childhood with delayed gross motor milestones, hypotonia, and elevated CK. They have slowly progressive weakness that mimics limb-girdle muscular dystrophy. The phenotype depends, in part, on the amount of laminin α2 present (15, 16).
Bilateral subcortical T2 hyperintensities, sparing the corpus callosum and brainstem, are seen on brain magnetic resonance imaging (MRI) of most individuals with MDC1A. These signal abnormalities are typically first seen between 6 months and 1 year of age (16–18). More extensive brain abnormalities associated with MDC1A have been observed by brain imaging in a small minority of patients. These include cerebral atrophy with dilatation of ventricles, hypoplastic cerebellum and pons, focal cortical dysplasia, and polymicrogyria (perhaps more accurately termed cobblestone malformation) involving the occipital regions (10, 15, 19, 20). Pathologic correlation with brain imaging is limited to a single autopsy (21).
Cobblestone malformation (a.k.a. cobblestone lissencephaly or type II lissencephaly) can be differentiated from most other cortical malformations based on brain imaging appearance, especially on MRI (22). The extra-axial space overlying the cortex is often narrow or absent. The cerebral surface is undersulcated, usually with diffuse apparent agyria. The cerebral cortex is moderately thick, usually ∼7–10 mm unless thinned by hydrocephalus. The cortical-white matter border is jagged with frequent vertical (perpendicular to the cortical-white matter border) striations, which differs from classic lissencephaly and the chaotic striations seen in classic polymicrogyria. Streaks of discontinuous or beaded subcortical heterotopia are often seen just beneath the cortex. The white matter has very abnormal signal (bright on T2 and dark on T1 MRI sequences; dark on CT scan) and may have small cysts. The white matter volume may be normal or thinned by hydrocephalus. The third and lateral ventricles are at least mildly enlarged and may be hydrocephalic. The corpus callosum is present, although often thin.
In contrast to classic (thick 4-layer) lissencephaly, in which there is a retardation in neuronal migration, the pathogenesis of cobblestone malformation hinges on neuronal and glial precursor migration through disruptions in the glia limitans and into the subarachnoid space (23, 24). This developmental abnormality in cell migration leads to subarachnoid glioneuronal heterotopia and the characteristic ‘cobblestone’ brain surface appearance. These abnormalities may occur in isolation or within autosomal recessive inherited syndromes associated with CMDs, namely Walker-Warburg syndrome, Muscle-Eye-Brain disease, and Fukuyama CMD (23). Although MDC1A has been classified as CMD without brain involvement, cobblestone malformation diagnosed on MRI imaging studies has been reported to be a rare feature of MDC1A (23).
Here, we describe the clinical, pathologic, and radiographic features of 4 previously unreported individuals with genetically proven MDC1A and cobblestone malformation. Surprisingly, extensive, symmetric cobblestone pathology was observed at autopsy in the 1 patient of these 4 whose brain MRI showed only subtle structural abnormalities. This underscores the point that cobblestone malformation may be a more common pathological finding in patients with MDC1A than has previously been reported, even in patients with apparently normal brain MRIs.
MATERIALS AND METHODS
Patient Ascertainment
Patients were selected because they had a diagnosis of MDC1A and either MRI or autopsy evidence of cobblestone malformation. All patients were followed clinically by at least one of the coauthors. All brain MRIs were reviewed by author W.B.D. Diagnostic muscle biopsy evaluations (Patients 1, 2, and 4) and postmortem examination of brain, sural nerve, skeletal muscle, and heart (Patient 1) were completed by author S.A.M.
Institutional Review Board approval (200510769) was obtained for the use of clinical presentations, MRI, muscle biopsy, and autopsy data in the manuscript. All procedures performed in these studies involving human participants were in accordance with the ethical standards of the University of Iowa Institutional Review Board and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Pathology
Skeletal muscle from biopsies or autopsy were mounted for cross-sectional histology and frozen in isopentane precooled to −160°C. A portion of the autopsy heart was frozen in an identical manner. Skeletal muscle and heart were also fixed in formalin and processed routinely into paraffin blocks. Histopathology was evaluated in paraffin sections or cryosections of muscle and heart stained with hematoxylin and eosin (H&E). Immunofluorescence staining for muscular dystrophy associated proteins was carried out in cryosections with antibodies to dystrophin (rabbit polyclonal ab15277, Abcam, Cambridge, MA), α-dystroglycan (mouse monoclonal IIH6 and VIA4-1 from the Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA [DSHB]), β-dystroglycan (mouse monoclonal 7D11, DSHB, The University of Iowa, Iowa City, IA), laminin (rabbit polyclonal L9393, Sigma, St. Louis, MO), laminin α5 (mouse monoclonal 4B12, Millipore, Billerica, MA), and merosin (mouse monoclonal 5H2, Millipore; mouse monoclonal Mer3/22B2, Leica Microsystems Inc, Buffalo Grove, IL; rat monoclonal 4H8, Abcam, Cambridge, MA) following methods described previously (25). Species-specific secondary antibodies were used.
A segment of sural nerve was fixed in isosmolar, 2.5% glutaraldehyde then processed for epon embedment or teased fiber preparation. Ultrathin sections of epon-embedded nerve were used for electron microscopy. Teased fibers were evaluated by routine light microscopy.
The brain and spinal cord were removed at autopsy from Patient 1. A coronal slice through the frontal lobes and a parasagittal section of cerebellar hemisphere were frozen at −80°C. The remainder of the brain and spinal cord were fixed in 10% buffered formalin for 10 days prior to further sectioning. Selected regions were sampled for histologic evaluation as paraffin sections stained with H&E, reticulin, or luxol fast blue.
Immunoperoxidase staining of paraffin sections was performed according to the manufacturer’s instructions using the Dako Real Detection System, Alkaline Phosphatase/RED, Rabbit/Mouse (K5005, Dako, Glostrup, Denmark) in an automated immunostaining instrument (Dako Autostainer Plus); primary antibodies were to glial fibrillary acidic protein ([GFAP], mouse monoclonal and rabbit polyclonal, BioGenex, Fremont, CA), neuN (A60, Millipore), neurofilament (mouse monoclonal 2F11, Dako), and vimentin (mouse monoclonal V9, Dako). Using immunofluorescence methods similar to that used in muscle, frozen sections of cerebral and cerebellar cortex were stained for laminin (rabbit polyclonal L9393, Sigma), laminin α5 (mouse monoclonal 4B12, Millipore), and merosin (specific antibodies and vendors listed above).
RESULTS
Patient 1 (also see Table)
Patient 1 was born at term with a birth weight of 5 pounds 11 ounces, to nonconsanguineous parents. Pregnancy was monitored for intrauterine growth retardation. He was mildly hypotonic at birth but did not require respiratory support and did not have feeding problems. He was followed by his pediatrician for failure to thrive. At 5 months, he was referred for evaluation of hypotonia. He was an alert baby with full extraocular movements. He had reduced muscle mass, profound hypotonia and was weak in all 4 limbs. He had poor head control and was areflexic. His serum CK was elevated at 5951 IU/L. Electromyogram/nerve conduction velocity (EMG/NCV) study in infancy suggested a myopathic process with normal NCVs. His brain MRI was normal at 4 months of age (Fig. 1A–C). Repeat MRI at 2 years of age showed diffuse high signal in the white matter without structural abnormalities (Fig. 1D–F). On subsequent review, author W.B.D. identified focal, subtle cortical malformation (Fig. 1E). At 1 year of age, a muscle biopsy showed a necrotizing myopathy with absent immunostaining for merosin (Fig. 2A).
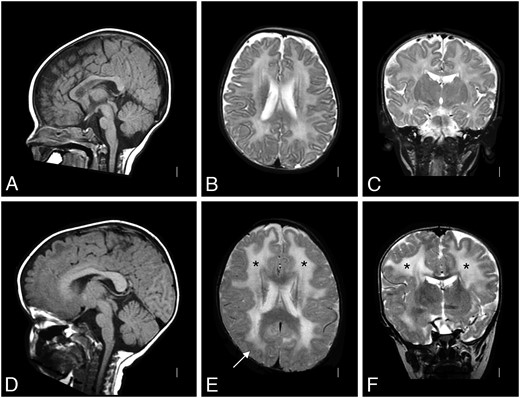
Brain MRI of Patient 1. A T2-weighted brain MRI at 4 months of age was interpreted as normal (A: sagittal, B: coronal, and C: axial); the white matter signal was developmentally appropriate. By 2 years of age (D: sagittal, E: coronal, and F: axial), a repeat T2-weighted MRI showed white matter changes (* in E and F) with only subtle structural abnormalities (arrow in E). MRI, magnetic resonance imaging.
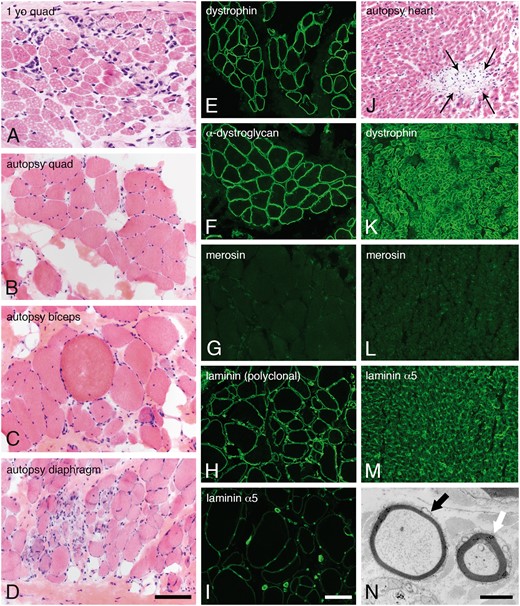
Skeletal muscle, heart, and sural nerve pathology. Dystrophic pathology was evident in H&E-stained cryosections from muscles evaluated as a diagnostic biopsy at age 1 year (A, quadriceps) or autopsy (B, quadriceps; C, biceps; D, diaphragm). Immunofluorescence studies performed in quadriceps obtained at autopsy showed normal expression of dystrophin (E) and α-dystroglycan (F, IIH6). In contrast, merosin was absent (G, 5H2 80 kDa antibody). Other isoforms of laminin were expressed in skeletal muscle basement membranes (H, polyclonal laminin antibody; I, laminin α5). Cardiac muscle evaluated at autopsy showed multifocal sites of lymphocytic aggregation, consistent with myocarditis (J, H&E). Immunofluorescence studies performed in the heart showed normal expression of dystrophin (K) but the total absence of merosin (L). Laminin α5 was strongly expressed (M). Abnormally thin myelin sheaths were noted by electron microscopy of the sural nerve (black arrow in N). The white arrow points to an axon with normal thickness myelin sheath. The scale bar in panel D is 100 µm for panels A–D and J–M; the scale bar in panel I is 100 µm for panels E–M. The scale bar in panel N is 2 µm. H&E, hematoxylin and eosin.
He had normal language development but delayed motor milestones. He sat without support at 1.5 years and rolled over at 2 years, but was never able to get into sitting position, stand or walk. He had chronic growth failure despite extensive GI evaluation and intervention, and a gastrostomy tube was placed at 5 years of age. He underwent spinal fusion for neuromuscular scoliosis at 13 years. Nocturnal BiPAP was initiated at age 14 years at which time his forced vital capacity was 0.28 L, 17% of predicted. Echocardiograms and EKGs were normal. He attended regular high school classes where he did well academically and socially. At age 15 years LAMA2 sequencing confirmed the diagnosis of MDC1A, identifying heterozygosity for these 2 frameshift pathogenic variants: c.2049_2050delAG, p.Arg683Serfs*21 and c.8669dupT, p. Leu2890Phefs*16.
At age 17, he presented with chest pain and difficulty breathing and was admitted to the pediatric intensive care unit with a pericardial effusion causing tamponade. While recovering from this he had an acute massive gastrointestinal hemorrhage, went into cardiorespiratory arrest and could not be resuscitated. Postmortem examination revealed an esophageal erosion as the cause of his GI bleed. There were mild, multifocal changes in the heart suggestive of myocarditis (Fig. 2J).
Postmortem Neuropathology
Muscle samples obtained from quadriceps, diaphragm, and biceps show features of a chronic, necrotizing myopathy (Fig. 2B–D). Merosin was absent by immunofluorescence, while dystrophin and dystroglycan appeared normal. Other laminins, most likely 111 and 511, were expressed at the sarcolemma of all muscle fibers (Fig. 2H, I). Immunofluorescence studies of cardiac muscle were similar (Fig. 2K–M).
The sural nerve was also evaluated at autopsy. Electron microscopy and teased fiber analysis revealed a mild demyelinating neuropathy (Fig. 2N and Supplementary DataFig. S1, respectively).
Gross evaluation of the cerebrum suggested the presence of diffuse, symmetric cobblestone malformation (Fig. 3A, B). Widespread, multifocal, superficial gliosis, and disruptions of the glia limitans with a minor degree of leptomeningeal heterotopia was prominent (Fig. 3C, D). In addition to the surface cobblestone appearance, adjacent gyri were focally fused within the depths of sulci (Fig. 3E, F). Microscopic studies confirmed multifocal regions of fusion between adjacent gyri, sometimes with bridging gliosis and axons (not shown). NeuN and neurofilament immunostains revealed cerebral cortical neuronal migration abnormalities that include forme fruste polymicrogyria and neurons within layer I that had atypical morphology (Fig. 3G). The extensive cortical disarray and glioneuronal leptomeningeal heterotopia associated with the severe cobblestone lissencephaly seen in dystroglycanopathies such as Walker-Warburg syndrome was not observed. Instead, there was an overall hexalaminar cerebral cortical structure in all lobes that also manifested architectural abnormalities of varying severity in every region sampled for histology. The abnormalities included heterotopion-like clusters of layer I neurons (Supplementary DataFigs S2 and S3B), crowding of layer II neurons (Supplementary DataFigs S2–S4), other subtle irregularities in the organization of layers II–VI (Supplementary DataFigs S2–S5), and increased numbers of white matter neurons (Supplementary DataFig. S5).
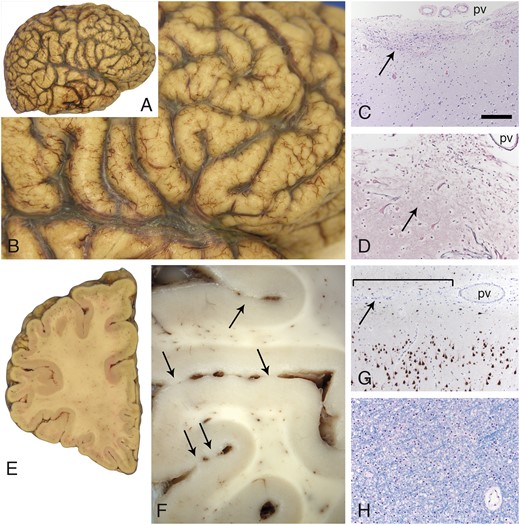
Cobblestone pathology in the cerebral cortex. The surface contour of the cerebral cortex was pebbled in a diffuse, relatively symmetric distribution that affected all lobes in both hemispheres (A, B). Multifocal sites of dense gliosis were noted along the cerebral cortical surface (C, arrow, H&E stain; pv). A reticulin stain (D) revealed that the pial surface basement membrane was disrupted at these sites of gliosis (arrow). Large, dysmorphic neurons (either side of the arrow, D) were often present at these sites. Coronal sections further revealed that adjacent gyri were partially fused across the intervening sulci (E and F, arrows; left frontal lobe). Superficially displaced neurons were also evident with neuN immunoperoxidase staining (G); subtle abnormal clustering of neurons was also often present within upper layers of the cortex. The bracket in G marks a region of fusion between adjacent gyri; the arrow points to the sulcus. A pv is present within the sulcus. White matter from multiple sites was unremarkable (H, LFB stain; frontal lobe centrum semiovale). The scale bar in panel C is 200 µm for panels C and G, and 100 µm for panels D and H. H&E, hematoxylin and eosin; LFB, luxol fast blue; pv = pial surface vessels.
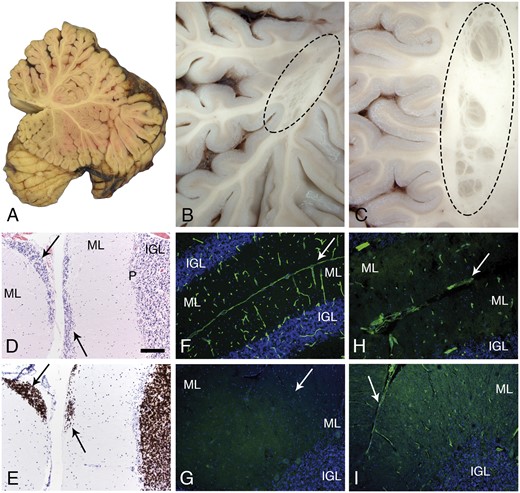
Cerebellar pathology. A mid-sagittal section of the cerebellum showed normal gross morphology (A). In contrast, several examples of subcortical, heterotopia was observed in the hemispheres (B, C; oval dashed lines). Microscopic evaluation revealed sites of failed external granule cell migration (arrows in D, H&E and E, neuN; IGL, P, ML). Immunofluorescence staining of cryosections revealed merosin at the pial surface basement membrane and perivascular basement membranes of control cerebellum (F); merosin was absent from the patient’s cerebellum (G; arrow marks the position of a sulcus). Other laminins were present at the patient’s pial surface and perivascular basement membranes (H, polyclonal laminin, α1 subunit on laminin-111; I, laminin a5). Arrows in H and I mark the position of sulci. The pial surface basement membrane was interrupted at some sites (H). The scale bar in panel D is 100 µm for panels C–I. H&E, hematoxylin and eosin; IGL, internal granule cell layer; ML, molecular layer; P, Purkinje cell layer.
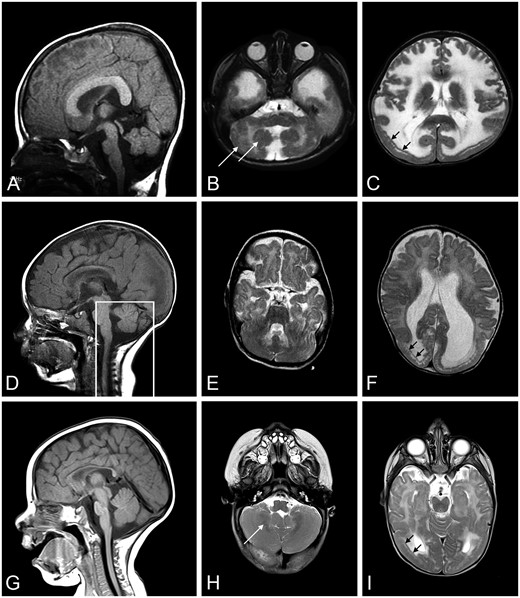
Brain MRIs from Patients 2, 3, and 4. A T2-weighted brain MRI was done for Patients 2 (A: sagittal, B, C: axial), 3 (D: sagittal, E, F: axial), and 4 (G: sagittal, H, I: axial). MRI at 18 months of age showed short brain in the antero-posterior direction (A), pontine hypoplasia and cerebellar vermis with cerebellar cortical dysplasia (B, white arrows), and temporal-occipital cobblestone malformation (C, black arrows) with frontal polymicrogyria in Patient 2. MRI at 8 months of age illustrate posterior-occipital malformation (D–F) in Patient 3. The box outlining posterior fossa structures designates a mid-sagittal image of these structures; they were not mid-sagittal in the image shown for the remainder of the brain. MRI at 6 months of age in Patient 4 showed occipital malformation (G–I), cerebellar cortical dysplasia (H), and T2 hyperintensity in the periventricular white matter (I). MRI, magnetic resonance imaging.
The cerebellum showed a few, multifocal sites of fusion between folia deep within the vermis (Fig. 4A) or in cerebellar hemispheres (Fig. 4C). Several examples of subcortical nodular heterotopia were present (Fig. 4B, C). Focal cerebellar cortical dysplasia and multifocal sites of failed granule cell migration were noted microscopically (Fig. 4D, E). The latter change is characteristic of cerebellar neuronal migration defects reported in mouse models of dystroglycanopathy (26–28). Similar to skeletal and cardiac muscle, laminin α2 was absent from brain (Fig. 4G), while other laminins were detected at a discontinuous glia limitans and in vascular basement membranes (Fig. 4H, I).
Interestingly, no white matter pathology to correspond with the MRI abnormalities was seen in cerebrum (Fig. 3H), cerebellum, brainstem, or spinal cord. However, circumferential subpial gliosis was prominent in both brainstem and spinal cord (not shown).
Patient 2 (also see Table)
Patient 2 was a girl born at term to consanguineous parents (first cousins). There were no neonatal complications. By 3 1/2 months of age, she had normal growth parameters, left esotropia, spontaneous movements in all 4 extremities, severe head lag, diffuse hypotonia, and serum CK at 5513 IU/L. Deep tendon reflexes were present in the lower extremities and decreased in the upper extremities. She achieved head control at 6 months, sat at 6–8 months with assistance, vocalized at 9 months, and had 2–4 single words by 2 years of age. A nasogastric tube was placed at 9 months. She had occasional voluntary movements of her arms but never achieved walking. Complex partial seizures started at 15 months that were treated with lamotrigine and topiramate. She had myopia and esotropia but no other ocular abnormalities. She developed scoliosis, severe laryngomalacia resulting in upper airway obstruction and recurrent aspiration pneumonias. She died at 11 1/2 years of age. An autopsy was not performed.
At age 6 months, a quadriceps femoris biopsy showed severely dystrophic, necrotizing myopathy (Supplementary DataFig. S6A). Merosin was completely absent using 2 different antibodies, monoclonals 5H2, and Mer3/22B2. The 80 kDa, 5H2 antibody is shown in Supplementary DataFigure S6C. Reflective of extensive myonecrosis and regeneration, dystrophin, α-dystroglycan, and β-dystroglycan immunofluorescence was variably intense; anti-α-dystroglycan IIH6 is shown in Supplementary DataFigure S6B. LAMA2 sequencing showed a novel, apparently homozygous, pathogenic variant in exon 13, predicted to result in premature protein termination (c.1823_1824delAT, p.Tyr608*). Four additional LAMA2 variants of unknown significance were identified: c.381C>A, c.3174 + 38A>G, and c.7760C>T and c.7830G>C.
![Proposed pathogenesis of cobblestone malformation. These schematics represent the normal interface of astrocytes with brain basement membranes and the developmental pathophysiology underlying cobblestone malformation. (A) Location of astrocytes within the mature brain parenchyma relative to the meninges (pia in blue and arachnoid [A] in black). The astrocyte foot processes abut the BM at the pial surface, forming the glia limitans. A glia limitans surround blood vessels as they enter the brain parenchyma (not shown here), maintaining the same astrocyte-basement interface as is present at the brain surface. (B) At the level of intraparenchymal capillaries, astrocyte foot processes directly abut the cerebral endothelial basement membranes. (C) In normal brain development, laminin α2 is expressed in the surface BM where radial glia (cerebrum; blue cells) or Bergmann glia (cerebellum; blue cells) anchor their foot processes and form a scaffold for immature neurons (green) to migrate to their respective destinations within the brain parenchyma. In the cerebrum, the predominant neuronal migration is inside-out. In the cerebellar cortex, granule cell neurons migrate outside-in. (D) In MDC1A, the loss of laminin α2 leads to loss of integrity of the glia limitans BM and over-migration of neurons through glia limitans defects in the cerebral cortex or failed migration of granule cell neurons in the cerebellum. The abnormal migration in either site results in heterotopia. BM, basement membrane; CP, cortical plate; EGL, external granule layer; IGL, internal granule layer; IZ, intermediate zone; ML, molecular layer; MZ, marginal zone; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnen/79/9/10.1093_jnen_nlaa062/1/m_nlaa062f6.jpeg?Expires=1750287120&Signature=tQCRe8L7WwFCLwyDmWC8M3S683A8KmW8tG4ZpuksY7rLeYW484ZD3q9CO82T8iI5y6SFz4M~v8nJTMcPzP9Qfma0j53M9syz6bhcnXjkSKy5cS1vBFhReJrJjXiBWEhRQDFwMco-6TVPvlObJA1xTIfDdkoStmLRSOhMXn6Z4uzN26h4HkrDLQmo90~FXgbnoNyzyrqJS9QaEamr9yXt0yIPZLcKlzbyjRVis3txf6Ssa-u7xZnpjQW3smONgNTCKhr379CeYgKLnsYHkylRnLiJ-pGR0UmPlJG~tt7xy3A3OySRD-3CXEG9e1ypYEsWokmo9V4Fy19dlN19-8EDQQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proposed pathogenesis of cobblestone malformation. These schematics represent the normal interface of astrocytes with brain basement membranes and the developmental pathophysiology underlying cobblestone malformation. (A) Location of astrocytes within the mature brain parenchyma relative to the meninges (pia in blue and arachnoid [A] in black). The astrocyte foot processes abut the BM at the pial surface, forming the glia limitans. A glia limitans surround blood vessels as they enter the brain parenchyma (not shown here), maintaining the same astrocyte-basement interface as is present at the brain surface. (B) At the level of intraparenchymal capillaries, astrocyte foot processes directly abut the cerebral endothelial basement membranes. (C) In normal brain development, laminin α2 is expressed in the surface BM where radial glia (cerebrum; blue cells) or Bergmann glia (cerebellum; blue cells) anchor their foot processes and form a scaffold for immature neurons (green) to migrate to their respective destinations within the brain parenchyma. In the cerebrum, the predominant neuronal migration is inside-out. In the cerebellar cortex, granule cell neurons migrate outside-in. (D) In MDC1A, the loss of laminin α2 leads to loss of integrity of the glia limitans BM and over-migration of neurons through glia limitans defects in the cerebral cortex or failed migration of granule cell neurons in the cerebellum. The abnormal migration in either site results in heterotopia. BM, basement membrane; CP, cortical plate; EGL, external granule layer; IGL, internal granule layer; IZ, intermediate zone; ML, molecular layer; MZ, marginal zone; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone.
Brain MRI at 18 months of age showed a short brain in the antero-posterior direction with anteriorly displaced Sylvian fissure, diffuse very irregular pebbled brain surface that is less severe frontally. There was a subtly abnormal gyral pattern in the frontal lobes and temporal-occipital more severe cobblestone malformation, as well as mild hypoplasia of pons and cerebellar vermis with striking cerebellar cortical dysplasia (Fig. 5A–C).
Patient 3 (also see Table)
Patient 3 is a boy born at term without complications. Generalized hypotonia was first recognized at 4 months of age. He was alert with no dysmorphic features. Deep tendon reflexes were present. His CK at 5 months was 2130 IU/L. Echocardiogram showed a small ventricular septal defect and small atrial septal defect with slight right ventricular dilation. At 6 months, he achieved head control and was able to sit with support but unable to roll over. He was completely nasogastric tube fed. He had normal hearing screen and normal ophthalmology exam. At last evaluation as a child, he was non-ambulatory with scoliosis, hip dysplasia, and speech problems due to weak oral motor muscles.
Muscle biopsy of quadriceps femoris is reported to have shown atrophic and regenerating fibers suggestive of a CMD, and immunostaining for merosin was inconclusive. The biopsy was not available for evaluation at the University of Iowa. However, LAMA2 gene sequencing revealed 2 heterozygous pathogenic variants, c.1580G>A, p.Cys527Tyr and c.7732C>T, p.Arg2578*; both have been reported to be causative of MDC1A (15, 29, 30). MRI at 8 months revealed cobblestone malformation in the posterior-occipital region (Fig. 5D–F).
Patient 4 (also see Table)
Patient 4 is a girl born at 34 weeks’ gestation. She was hospitalized for 10 days for oxygen supplementation and had several days of nasogastric tube feedings. Hearing, vision, and feeding were deemed to be normal at the time of discharge. During subsequent months, delays in motor milestones and hypotonia were recognized. At 14 months, she was alert and responsive, and was making noises. She only had distal finger movements spontaneously. She had a high arched palate, profound head lag, axial and limb hypotonia with poor head control, and contractures at the ankles. She had persistently elevated CK level greater than 1300 IU/L. EMG/NCS showed myopathic changes with normal nerve conduction.
Muscle biopsy showed a severe, chronic necrotizing myopathy (Supplementary DataFig. S6C). Immunostaining revealed nearly complete loss of merosin with mildly abnormal immunostaining for α-dystroglycan (Supplementary DataFig. S6D, E). LAMA2 sequencing showed 2 frameshift heterozygous pathogenic variants: c.4692_4695dupTGCA, p. Arg1566Cysfs*13 and c.7732C>T, p.Arg2578*. MRI at 6 months showed T2 hyperintensity in the periventricular white matter, occipital lissencephaly, and cerebellar dysplasia (Fig. 5G, H).
DISCUSSION
MDC1A is a CMD accompanied by peripheral neuropathy and abnormal white matter based on MRI; most patients have a structurally normal brain (4). Here, we describe postmortem observations of widespread, symmetric cobblestone malformation and cerebellar dysplasia in the brain of an adolescent male with MDC1A whose brain MRI showed only subtle structural abnormalities. Interestingly, there was no pathologic evidence of leukoencephalopathy corresponding to the white matter hyperintensities seen in MDC1A. This is only the second reported description of the brain pathology of MDC1A (21). We also describe 3 additional patients who had asymmetrical cobblestone malformation found on brain MRI. Together, this suggests that malformations of cortical development are more common than is currently recognized in individuals with MDC1A, raising the possibility that MDC1A patients with brain MRI showing only abnormal white matter signal might have diffuse histopathologic abnormalities such as bilateral symmetric cobblestone malformation.
Abnormal brain MRI in MDC1A was initially described in 1995, and the range of abnormalities has grown to include diffuse or focal white matter abnormalities, cerebellar cysts, neuronal migration abnormalities leading to polymicrogyria, focal cortical dysplasia, double cortex, and occipital pachygyria. White matter hyperintensities on T2 are the most common and are typically seen after the age of 6 months (18). Neuronal migration defects described as focal cortical dysplasia or polymicrogyria were reported in 9/175 (5%) of patients with MDC1A in one review of published cases (14). Most of these abnormalities were more prominent in the occipital cortex, which was not the case in our autopsy study of the aforementioned patient.
Clinical disorders of brain function are recognized as part of the MDC1A phenotype. Intellectual disability of varying severity is seen in 7% and seizures occur in 8%–20% of those with MDC1A (14, 15, 20). These are not easily explained by abnormal white matter signal alone. A small series correlating cognitive abilities and brain MRI suggested that a structural abnormality such as cerebellar hypoplasia is a risk factor to lower performance IQ although it is not invariably present (31, 32). Among 51 patients evaluated, Geranmayeh et al (15) reported only 2 patients had intellectual disability, and another one had communication difficulties although 15 patients in the series had structural changes in addition to white mater signal abnormalities. This suggests that MDC1A patients with significant MRI abnormalities can have normal neurological and intellectual functioning. Patient 1 in our series had normal intellectual function and never had clinical seizures although his brain showed cobblestone malformation at autopsy (Table).
While several reports have previously commented on the radiographic features of MDC1A, knowledge of MDC1A brain histopathology prior to the current report was limited to the autopsy of a single male infant (21). This was a child who from birth demonstrated global hypotonia, weak cry and suckling, club foot, and arthrogryposis. He had an elevated CK, and EMG/NCS showed motor unit potentials of short amplitudes and durations with normal common peroneal NCV, suggestive of a myopathic process. Histopathological examination of his quadriceps femoris biopsy showed complete deficiency of laminin α2, overexpression of laminin α5, and normal dystrophin and α-sarcoglycan levels. These findings were confirmed at autopsy at 4 months of age in evaluation of quadriceps and myocardium.
Gross examination of the cerebral cortex at autopsy showed right parieto-occipital cortical malformation with areas of complete fusion of adjacent gyri in the left occipital cortex. In addition, within the occipital cortex, there were areas of partial fusion across the intervening sulci between adjacent gyri. Bilateral symmetrical communicating hydrocephalus was noted in coronal sections. Analysis of paraffin-embedded brain sections stained with Heidenhain-Woelcke myelin stain showed normal myelination within the cerebrum, cerebellum, and brainstem. H&E staining revealed fusion of layer I between adjacent gyri that obliterated the sulci. GFAP immunostaining revealed clusters of large astrocytic cells with atypical morphology within the regions of fused adjacent gyri, suggestive of glioneuronal leptomeningeal heterotopia. In cerebral blood vessels and the glia limitans, laminin α2 immunostaining was negative with preservation of laminin α5 staining.
Our Patient 1 represents the second individual with postmortem evaluation of MDC1A brain pathology; in contrast to the first, this patient was a much older individual at the time of death (4 months compared to 17 years). In addition, Taratuto et al (21) did not report histopathological abnormalities within the cerebellum. Our Patient 1 had a normal brain MRI at 4 months of age, while repeat MRI at 2 years showed typical diffuse T2 white matter hyperintensities and only subtle structural abnormalities (Fig. 1). At the age of 17 years, postmortem evaluation of the brain showed diffuse, symmetric cobblestone changes (Fig. 3). The histopathology is best interpreted as forme fruste cobblestone malformation. Fusion of adjacent gyri and a minor degree of neuronal migration abnormalities were present; however, the extensive glioneuronal heterotopia of full-fledged cobblestone malformation (historically termed cobblestone lissencephaly) were not present (23, 33–35). Our patient manifested a mild degree of cerebellar neuronal migration abnormalities: focal failure of granule cell migration and subcortical heterotopia (Fig. 4). Thus, both postmortem examinations of MDC1A showed evidence of abnormal neuronal migration within the spectrum of cobblestone malformation.
It is also of interest that while the most striking brain MRI finding in MDC1A is white matter hyperintensity, there is no histopathologic evidence of leukoencephalopathy in either our case and in the previous case evaluated by autopsy (21). The pathophysiology underlying the abnormal white matter MRI signal in MDC1A remains unknown.
Cobblestone malformation has been described as an isolated entity but is more typically observed in the severe CMDs due to abnormal glycosylation of α-dystroglycan: dystroglycanopathies classically described as Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama CMD (23). Dystroglycanopathy-related genes that lead to cobblestone malformation include Protein O-Mannosyltransferase 1/2 (POMT1, POMT2), Protein O-Linked Mannose N-Acetylglucosaminyltransferase 1 (POMGNT1), Fukutin (FKTN), Fukutin-Related Protein (FKRP), and Like-Acetylglucosaminyltransferase (LARGE) (23, 36, 37). The neuropathology of cobblestone malformation is a neuronal migration disorder characterized by cerebral cortical dysplasia and glioneuronal heterotopia. Disruptions in the pial surface basement membrane lead to disorganization of radial glia processes and aberrant migration of Cajal Retzius cells and immature neurons from all layers of the cortex (Fig. 6) (24). In humans, this results in a “pebbled” surface appearance due to the heterotopia and abnormal cortical lamination (38, 39).
Recessive variants in non-dystroglycanopathy genes have been reported in patients who have cobblestone malformation by imaging: the genes include laminin β1 (LAMB1; [40]), laminin γ3 (LAMC3; [41]), tubulin β3 (TUBB3; [42]) and adhesion G protein-coupled receptor G1 (ADGRG1; formerly GPR56 [43]). Some of these latter genotypes have been modeled in mice where the developmental neuropathology appears to be similar to the dystroglycanopathies (44, 45). We note that a recent report implicating COL4A1 variants can cause neuronal migration deficits in humans has been questioned (46). The first of 2 patients reported in that analysis was shown earlier by Satz et al in Western blot analysis of brain tissue to have a dystroglycanopathy (see Figure 1 of Satz et al [47]). Using cultured cells from the same patient (patient 5 of their paper), Willer et al (48) found a homozygous c.1120-1G>T variant (IVS8 splice-site variant leading to the deletion of exon 9) in Isoprenoid Synthase Protein-Containing Domain (ISPD), which is now a well-established dystroglycanopathy gene. The ISPD variants are a more compelling explanation for the patient’s neuronal migration abnormalities than COL4A1.
The potential for pathogenetic similarities between MDC1A and the dystroglycanopathies is readily apparent, because α-dystroglycan is a high-affinity receptor for laminin α2 in muscle, brain, and peripheral nerve (26, 49). A disruption of binding between the cell surface and its basement membrane may occur when the receptor, α-dystroglycan, is hypoglycosylated or when laminin α2 is deficient (9, 50). Because laminins are extracellular matrix molecules, heterotrimers consisting of α, β, and γ chains from the LAMA, LAMB, and LAMC families, that mostly localize to basement membranes, it is not surprising to see cobblestone malformation pathology in patients with variants in LAMA2, LAMB1 or LAMC3.
Analysis of our series contributes to the understanding of the brain pathology associated with MDC1A (Fig. 6). Most importantly, our autopsy case suggests that MDC1A patients with structurally normal brain MRIs may have the pathology of cobblestone malformation on gross or histopathological examination. Interestingly, these changes were wide-spread and symmetrically distributed throughout the brain in our case and the single previously published case. The histopathological finding of cobblestone malformation and additional cases identified on brain MRIs underscore the fact that cobblestone malformation can be a clinically important feature of MDC1A in some patients. We suspect that at least some previous reports of pachygyria, and agyria in the occipital lobes of MDC1A patients may represent cobblestone malformation. These abnormalities, whether subtle or overt, likely underlie the seizures and intellectual disability that occur in a subset of patients with MDC1A.
Clinical Features of MDC1A Patients with Cobblestone Malformation.
| Patient | Clinical Findings | CK (U/L)* | LAMA2 Variants | Brain MRI Results |
|---|---|---|---|---|
| 1 | Hypotonia, development delay | 5951 |
| |
| 2 | Hypotonia noted at 3 ½ months, intellectual disability, seizures | 5513 |
| 18 months: diffuse pebbled brain surface, frontal polymicrogyria and temporal-occipital cobblestone malformation, mild hypoplasia of pons and cerebellar vermis, and striking cerebellar cortical dysplasia |
| 3 | Hypotonia at 4 months | 3130 |
| 8 months: disorder of neuronal migration in the posterior-occipital region |
| 4 | Hypotonia, development delay | >1300 | 6 months: diffuse white matter changes and minor cortical changes suggesting possible migration abnormalities |
| Patient | Clinical Findings | CK (U/L)* | LAMA2 Variants | Brain MRI Results |
|---|---|---|---|---|
| 1 | Hypotonia, development delay | 5951 |
| |
| 2 | Hypotonia noted at 3 ½ months, intellectual disability, seizures | 5513 |
| 18 months: diffuse pebbled brain surface, frontal polymicrogyria and temporal-occipital cobblestone malformation, mild hypoplasia of pons and cerebellar vermis, and striking cerebellar cortical dysplasia |
| 3 | Hypotonia at 4 months | 3130 |
| 8 months: disorder of neuronal migration in the posterior-occipital region |
| 4 | Hypotonia, development delay | >1300 | 6 months: diffuse white matter changes and minor cortical changes suggesting possible migration abnormalities |
LAMA2, laminin α2; MDC1A, congenital muscular dystrophy type 1A; MRI, magnetic resonance imaging.
Highest recorded serum creatine kinase.
Clinical Features of MDC1A Patients with Cobblestone Malformation.
| Patient | Clinical Findings | CK (U/L)* | LAMA2 Variants | Brain MRI Results |
|---|---|---|---|---|
| 1 | Hypotonia, development delay | 5951 |
| |
| 2 | Hypotonia noted at 3 ½ months, intellectual disability, seizures | 5513 |
| 18 months: diffuse pebbled brain surface, frontal polymicrogyria and temporal-occipital cobblestone malformation, mild hypoplasia of pons and cerebellar vermis, and striking cerebellar cortical dysplasia |
| 3 | Hypotonia at 4 months | 3130 |
| 8 months: disorder of neuronal migration in the posterior-occipital region |
| 4 | Hypotonia, development delay | >1300 | 6 months: diffuse white matter changes and minor cortical changes suggesting possible migration abnormalities |
| Patient | Clinical Findings | CK (U/L)* | LAMA2 Variants | Brain MRI Results |
|---|---|---|---|---|
| 1 | Hypotonia, development delay | 5951 |
| |
| 2 | Hypotonia noted at 3 ½ months, intellectual disability, seizures | 5513 |
| 18 months: diffuse pebbled brain surface, frontal polymicrogyria and temporal-occipital cobblestone malformation, mild hypoplasia of pons and cerebellar vermis, and striking cerebellar cortical dysplasia |
| 3 | Hypotonia at 4 months | 3130 |
| 8 months: disorder of neuronal migration in the posterior-occipital region |
| 4 | Hypotonia, development delay | >1300 | 6 months: diffuse white matter changes and minor cortical changes suggesting possible migration abnormalities |
LAMA2, laminin α2; MDC1A, congenital muscular dystrophy type 1A; MRI, magnetic resonance imaging.
Highest recorded serum creatine kinase.
This study was funded by the Iowa Wellstone Muscular Dystrophy Cooperative Research Center, NINDS (U54-NS053672 to K.D.M. and S.A.M.).
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
Author notes
Himali Jayakody and Sanam Zarei contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}