





Abstract
Trained immunity induces antigen-agnostic enhancement of host defense and protection against secondary infections, but inappropriate activation can contribute to the pathophysiology of inflammatory diseases. Tight regulation of trained immunity is therefore needed to avoid pathology, but little is known about the endogenous processes that modulate it. Here, we investigated the potential of interleukin (IL)-10, a prototypical anti-inflammatory cytokine, to inhibit trained immunity. IL-10 induced tolerance and inhibited trained immunity in primary human monocytes at both functional and transcriptional levels. Inhibition of STAT3, a signaling route that mediates IL-10 signals, induced trained immunity. IL-10 downregulated glycolytic and oxidative metabolism in monocytes but did not impact the metabolic effects of β-glucan–induced trained immunity. Furthermore, IL-10 prevented increased reactive oxygen species production in Bacillus Calmette–Guérin (BCG)–induced training but did not influence phagocytosis upregulation. In a cohort study of healthy volunteers vaccinated with BCG, genetic variants that influenced IL-10 or its receptor modulated BCG-induced trained immunity. Furthermore, circulating IL-10 concentrations were negatively correlated with induction of trained immunity after BCG vaccination in a sex-specific manner. In conclusion, IL-10 inhibited several, albeit not all, immunological functions amplified after induction of trained immunity. Follow-up studies should explore the precise molecular mechanism that mediates the effects of IL-10 on trained immunity. Addressing these knowledge gaps is an important step toward optimizing IL-10's potential as a therapeutic target in diseases characterized by inappropriate induction of trained immunity.
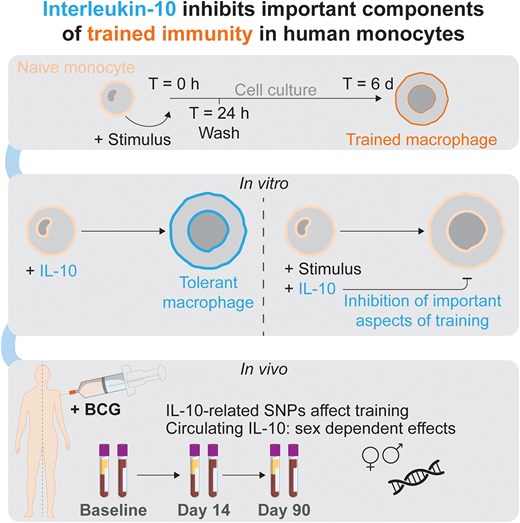
1. Introduction
Until recently, immunological memory was attributed solely to the antigen-specific responses of the adaptive immune system. However, in recent years, a growing body of evidence has shown that the innate immune system can also mount heterologous memory responses, a process called “trained immunity.”1,2 Trained immunity is mediated by long-lasting epigenetic and metabolic rewiring of innate immune cells, such as monocytes and natural killer cells, as well as myeloid progenitors in the bone marrow, by a variety of microbial and endogenous ligands.2 Innate immune memory is beneficial to host defense by boosting host defense against infections, but when trained immunity is inappropriately activated, this can lead to pathological inflammatory processes such as in hyper IgD syndrome, gout, or arterial wall inflammation in atherosclerosis.3–5 Therefore, induction of trained immunity needs to be finely tuned as not to cause damaging hyperinflammation.
Knowledge regarding the mechanisms that both induce and modulate trained immunity is needed to provide novel therapeutic targets for diseases in which trained immunity plays an important role in pathophysiology.6–9 Induction of trained immunity has been studied extensively: metabolic changes upon an initial cellular activation (such as by PRR-ligand recognition) provide energy for the immune response as well as metabolites that serve as cofactors for epigenetic enzymes, contributing to lasting modification of chromatin structure. In turn, these epigenetic alterations enable more efficient upregulation of cellular metabolism.2,10 The nature and strength of the stimulation is thought to determine the precise program of trained immunity or tolerance.2,11 In contrast, relatively little is known about the regulatory pathways that can modulate trained immunity. One of the most important cytokines that has modulatory effects on innate immune responses is interleukin (IL)-10.12 IL-10 is expressed together with proinflammatory genes and can be induced by proinflammatory factors, inducing resolution of inflammation.13 Subsequently, mice and humans deficient in IL-10 or its receptor are prone to developing inflammatory bowel diseases (IBDs).14,15 IBD inflammatory gene signatures overlap considerably with those associated with trained immunity, suggesting a role of trained immunity in IBD.16 Macrophage-specific overexpression of IL-10 suppresses atherosclerosis in a mouse model, whereas a murine IL-10 knockout model was more susceptible to developing atherosclerosis.17–19 Finally, in the context of trained immunity itself, IL-10 is upregulated upon restimulation of in vitro “trained” macrophages.20
We therefore hypothesized that IL-10 has regulatory properties and inhibits trained immunity induction. In line with this, we present proof-of-concept evidence that transient exposure to IL-10 induces tolerance and blunts induction of trained immunity in primary human monocytes. Cytokine responses during acute stimulation and trained immunity induction were inhibited by IL-10, at both the transcriptional and protein levels. IL-10 prevented upregulation of reactive oxygen species (ROS) production following Bacillus Calmette–Guérin (BCG) training but not the increase in phagocytosis observed after β-glucan training. Genetic variants near IL10 or its receptor that affected their expression, as well as circulating IL-10 concentrations at baseline, were associated with regulatory effects on BCG-induced trained immunity in humans.
2. Methods
2.1 Monocyte isolation and cell culture
Buffy coats were obtained from the Sanquin blood bank. Alternatively, fresh whole blood was drawn from healthy volunteers after obtaining written informed consent (approval NL32357.091.10 of the Arnhem-Nijmegen ethical committee). The material was diluted with calcium/magnesium-free phosphate-buffered saline (PBS). Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation over Ficoll-Paque (GE Healthcare). The cells were washed 3 to 5 times with PBS and resuspended in Dutch-modified RPMI 1640 (Invitrogen) supplemented with 50 μg/mL gentamicin, 2 mM GlutaMAX (Thermo Fisher Scientific), and 1 mM pyruvate (hereafter referred to as RPMI+). Monocytes were negatively selected using MACS technology (Pan Monocyte Isolation Kit; Miltenyi Biotec) unless otherwise indicated. The monocytes were counted using a Sysmex XN-450 hemocytometer and brought to 1 × 106/mL RPMI+ before seeding into culture plates. Monocytes were cultured under standard cell culture conditions (37 °C, 5% CO2) in RPMI+ further supplemented with 10% human pooled serum, unless otherwise indicated.
2.2 24-h Stimulation assays and trained immunity in vitro model
For all stimulation assays and trained immunity experiments, we used a previously published in vitro model.20,21 Briefly, monocytes were adhered for 1 h and washed to ensure maximum purity. The number of monocytes and size/type of culture plate depended on the type of experiment. The cells were then stimulated (also referred to as “trained”) for 24 h using the reagents specified in Supplementary Table 1. Following a wash with warm PBS to remove training stimuli, the cells were left to rest and differentiate into macrophages during 5 d. The medium was refreshed intermittently on day 3. On day 6, the macrophage phenotype was determined to assess the effect of the day 0 stimulation. The cells were either harvested immediately (in the case of chromatin immunoprecipitation analysis) or restimulated with RPMI or lipopolysaccharide (LPS) for 4 h (for RNA) or 24 h (for cytokine production).
2.3 Enzyme-linked immunosorbent assay measurements in cell culture supernatants
Concentrations of tumor necrosis factor (TNF) and IL-6 were measured in cell culture supernatants using R&D Systems DuoSet enzyme-linked immunosorbent assay kits, according to the manufacturer's instructions. Sample concentrations were interpolated from a standard curve and adjusted for dilution factors using a combination of Gen5 software (version 3; BioTek) and Microsoft Excel.
2.4 Seahorse metabolic analysis
Monocytes were trained and cultured into macrophages as described previously. The macrophages were harvested using EDTA and gentle scraping for cell dissociation. A total of 1 × 105 macrophages were seeded in each well of a 96-well Seahorse plate (Agilent) and adhered for 1 h (in RPMI+, standard cell culture conditions). The medium was changed to “assay medium” 45 to 60 min prior to running the assay (cells in assay medium were incubated in ambient CO2 concentration, 37 °C). For glycolysis stress tests, the assay medium consisted of nonbuffered Dulbecco’s Modified Eagle Medium (pH 7.35 to 7.45) supplemented with L-glutamine (1 mM). For mitochondrial stress testing, the assay medium consisted of nonbuffered Dulbecco’s Modified Eagle Medium (pH 7.35 to 7.45) supplemented with L-glutamine (2 mM), D-glucose (11 mM), and pyruvate (1 mM). Glycolysis and Mito Stress Tests were used to measure glycolytic and mitochondrial parameters on a Seahorse XF96 analyzer (Agilent). The used inhibitors and their final concentrations are described in Supplementary Table 2.
2.5 RNA extraction and complementary DNA synthesis
Monocytes were trained and cultured into macrophages as described previously. On day 6, the macrophages were restimulated with RPMI or LPS for 4 h. The cells were lysed with Buffer LBP (Macherey-Nagel), snap-frozen in liquid nitrogen, and stored at −80 °C.
RNA extractions were performed using NucleoSpin RNA plus kits (Macherey-Nagel), according to the manufacturer's instructions. RNA concentration and purity were assessed using NanoDrop spectrophotometry (Thermo Fisher Scientific). Equal amounts of RNA were reverse-transcribed into complementary DNA (cDNA) using the Bio-Rad iScript cDNA synthesis kit. cDNA was stored at 4 °C (short term) or −20 °C (long term) until analysis.
2.6 Quantitative polymerase chain reaction analysis
The SYBR Green method was used to perform quantitative polymerase chain reaction analysis on a StepOne Real-Time quantitative polymerase chain reaction apparatus (Thermo Fisher Scientific). The ΔΔ-Ct method was used to compare gene expression relative to HPRT. The primer sequences are available in Supplementary Table 3.
2.7 Phagocytosis assay
Percoll-enriched monocytes were trained as done previously with BCG or β-glucan, or medium as a control. On day 6, 25 µg/mL pHrodo Red Zymosan BioParticles were added to the cells and the plate was then centrifuged. The measurements were performed in the IncuCyte S3, at 37 °C for 24 h, with acquisition every 15 min for the first 2 h and then 1 acquisition per hour (2 images per well with 10× objective, green acquisition on 300 ms). Two sets of data were generated: 1 for Total Green Object Integrated Intensity (GCU × µm²/Image) and 1 for Phase Object Count. The results are expressed as GCU/cell number.
2.8 ROS assay
Adherent monocytes from PBMCs were trained as described previously but in a white opaque plate. On day 6, ROS levels were measured following opsonized zymosan (10 mg/mL) restimulation. Luminol (5-Amino-2,3-dihydro-1,4-phthalazinedione; Sigma Aldrich) was added to each well in order to start the chemiluminescence reaction. Each measurement was carried out in at least 4 repetitions. Chemiluminescence was determined every 145 s at 37 °C for 1 h. Luminescence was expressed as relative light units per second and as area under the curve.
2.9 300BCG cohort information
The 300BCG study, from which we used data for this study, included a total of 325 adults from the Netherlands. For this study, data were used from 324 individuals for whom genetic data and/or cytokine and circulating IL-10/IL-10RB data were available. The Arnhem-Nijmegen Medical Ethical Committee approved of this study (NL58553.091.16). Blood was drawn before BCG vaccination (a standard dose of 0.1 mL BCG [BCG-Bulgaria; InterVax]), and 2 wk and 3 mo after vaccination.
2.10 300 BCG genetic analyses
DNA isolation from EDTA whole blood samples was performed using the Gentra Puregene blood kit (Qiagen).22 The DNA samples were genotyped and the resulting data were processed as previously described.22 Quantitative trait locus mapping was performed as previously described.22
2.11 300BCG PBMC stimulation, cytokine measurements, and Olink-targeted proteomics
PMBCs were isolated from EDTA whole blood as described previously.23 Briefly, Ficoll-Paque density gradient centrifugation was used and the cells were cultured in RPMI with Dutch modifications and supplemented with 50 μg/mL gentamycin, 2 mM GlutaMAX, and 1 mM pyruvate. For stimulation experiments,23 the cells were incubated in the presence of heat-killed Staphylococcus aureus (1 × 106 colony-forming units/mL, nonspecific stimulus) for 24 h. Cytokines were measured in the supernatants by enzyme-linked immunosorbent assay. Plasma-targeted inflammatory proteomics were measured by Olink proximity extension assay and data were processed as previously described.23 The Olink data were corrected for sex and age using a linear regression approach.
2.12 Statistical analyses
Unless otherwise indicated, all data are presented as mean ± SD. All statistical testing was performed in R (version 4.4.0; R Foundation for Statistical Computing). Unless otherwise indicated, Wilcoxon matched-pairs signed rank tests were used to compute P values. P values below 0.05 (2-tailed) were regarded as statistically significant. When exact P values are not provided in the figures, statistical significance is indicated in the caption.
2.13 Other software and R packages
The following R packages were used: Tidyverse core packages (v2.0.0),24 magrittr (v2.0.3),25 openxlsx (v4.2.5.2),26 viridis (v0.6.2),27 janitor (v2.2.0),28 ggpubr (v0.6.0),29 ggside (0.2.2),30 corrplot (v0.92),31 readxl (v1.4.2),32 ggthemes (v4.2.4),33 psych (2.3.3),34 conflicted (v1.2.0),35 patchwork (v1.2.0),36 rstatix (v0.7.2),37 smplot2 (v0.2.4),38 and ggprism (v1.0.5).39 Illustration and finalization of figures was performed using Adobe Illustrator.
3. Results
3.1 IL-10 inhibits cytokine production in acute inflammation and trained immunity
In a first set of experiments, we validated the anti-inflammatory effects of IL-10 on the stimulation of acute inflammation. Primary human monocytes were stimulated with LPS for 24 h with or without addition of IL-10 (Fig. 1A). As expected, the presence of IL-10 significantly reduced LPS-induced proinflammatory cytokine production (Fig. 1B). We then investigated the long-term effects of transient exposure to IL-10 (Fig. 1C). After exposing monocytes to IL-10 for 24 h, followed by a resting period of 5 days, restimulation on day 6 with LPS induced lower production of TNF and IL-6 compared with restimulated control cells (Fig. 1D).
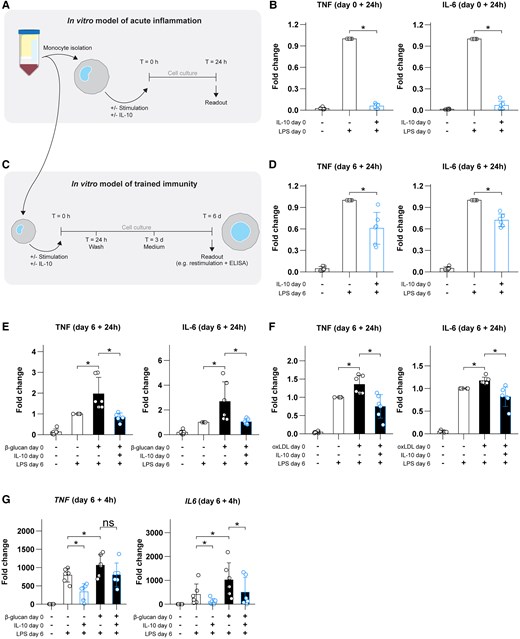
IL-10 dampens acute inflammation, induces tolerance, and inhibits trained immunity in primary human monocytes. (A) Schematic overview of the acute inflammation model. (B) Fold changes in TNF and IL-6 responses during acute LPS-induced monocyte responses, with or without IL-10 present. (C) Schematic overview of the in vitro trained immunity model used throughout the rest of the present work. (D) TNF and IL-6 responses on day 6 following exposure to IL-10. (E, F) TNF and IL-6 responses on day 6 following stimulation with β-glucan or oxidized low-density lipoprotein (oxLDL), respectively, with or without addition of IL-10. (G) Transcriptional responses to LPS of the genes TNF and IL6 (using HPRT as a reference gene) on day 6 after exposure to β-glucan and/or IL-10. Data are presented as mean ± SD, with individual points representing biological replicates. *P < 0.05. ns = not significant; T = time.
Additionally, induction of trained immunity by β-glucan was prevented when IL-10 was added during the initial stimulation of the cells (Fig. 1E). Similarly, IL-10 inhibited oxidized low-density lipoprotein–induced trained immunity, suggesting a broad modulatory effect independent of the stimulus used to induce trained immunity (Fig. 1F). Thus, IL-10 dampens monocyte inflammatory responses both during acute induction of inflammation and after long-term stimulation of trained immunity.
It is well known that activation of the transcription factor STAT3 is required for the anti-inflammatory effects of IL-10.12,13 We therefore hypothesized that STAT3 might be a molecular pathway that endogenously regulates innate immune memory. Inhibition of STAT3 phosphorylation by the small molecule inhibitor STATTIC40 increased the production of TNF on day 6 (Supplementary Fig. 1), indicating that this pathway is indeed important for negative regulation of trained immunity. STATTIC also had a tendency to enhance IL-6 production, but this did not reach statistical significance.
3.2 IL-10 inhibits trained immunity at the transcriptional level
One of the mechanisms through which IL-10 acutely suppresses immune responses is by restricting transcription of proinflammatory mediators.41 We hypothesized that a similar mechanism is at play during its long-term suppressive activities on trained immunity. We therefore performed quantitative reverse-transcription polymerase chain reaction to assess the transcriptional activation of several genes during LPS restimulation of trained macrophages. The transcription of TNF and IL6 messenger RNA in response to LPS following prior induction of trained immunity with β-glucan was reduced by previous exposure to IL-10 (Fig. 1G). Together, IL-10 exerts transcriptional repression in an enduring manner.
3.3 IL-10 induces metabolic anergy but does not impact the enhanced glycolysis induced by β-glucan
Metabolic reprogramming is an important mechanism for the induction of innate immune memory, and IL-10 can modulate the metabolic pathways.2,12,42 To investigate whether IL-10 impacts glycolytic and oxidative metabolic rates, we assessed glycolysis and oxidative phosphorylation in trained monocytes using Seahorse technology on day 6. Addition of IL-10 on day 0 appeared to cause an overall lowering of the glycolysis and oxidative phosphorylation on day 6 (Fig. 2A, B). We subsequently calculated several metabolic parameters to statistically test this observation. For all calculated parameters except glycolytic reserve, IL-10 alone on day 0 significantly downregulated glycolytic metabolism on day 6 (Fig. 2C). As previously described,43 β-glucan induced an upregulation of glycolytic metabolism; interestingly however, this effect of β-glucan was not influenced by IL-10 (Fig. 2C).
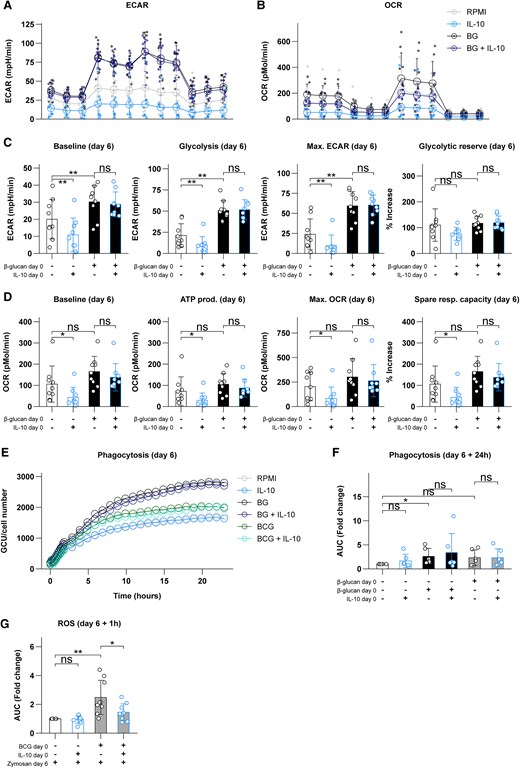
IL-10 inhibits glycolysis and oxidative phosphorylation but does not affect metabolic effects of β-glucan (BG). (A) Overview of the extracellular acidification rate (ECAR) during glycolysis stress testing. (B) Overview of the oxygen consumption rate (OCR) during mitochondrial stress testing. (C) Calculated glycolytic parameters on day 6 following exposure to IL-10, BG, or a combination. (D) Calculated oxidative parameters on day 6 following exposure to IL-10, BG, or a combination. (E) Phagocytosis of zymosan particles during 24 h on day 6 following initial exposure to BG or BCG with or without IL-10 (expressed as mean of biological and technical replicates). (F) Relative change in area under the curve of the data presented in panel E. (G) Production of ROS upon restimulation with opsonized zymosan for 1 h on day 6 following initial exposure to BCG with or without IL-10. Data are presented as mean ± SD, with individual points representing biological replicates. *P < 0.05, **P < 0.01. ATP = adenosine triphosphate; AUC = area under the curve; ns = not significant.
Exposure to IL-10 alone on day 0 also lowered oxidative metabolism on day 6, with the exception of spare respiratory capacity (Fig. 2D). In contrast, β-glucan did not significantly impact oxidative metabolism44 in these experiments (Fig. 2D). IL-10 appeared to slightly decrease oxidative parameters in β-glucan–trained cells (Fig. 2B), but this did not reach statistical significance (Fig. 2D). These results indicate that IL-10 on day 0 modulates glycolysis and oxidative phosphorylation on day 0 in naïve (“nontrained”) macrophages but does not impact the upregulated glycolytic capacity observed in β-glucan–induced trained immunity.
3.4 IL-10 prevents upregulation of ROS, but not phagocytosis, during induction of trained immune responses
We performed additional experiments to assess other aspects of the trained immune response that may be influenced by IL-10. We first assessed phagocytosis, a defining function of macrophages. During 24 h of observation using IncuCyte technology, we observed that training with β-glucan, but not BCG, significantly upregulated phagocytosis on day 6 (Fig. 2E, F). However, addition of IL-10 on day 0 did not appear to prevent the increased phagocytosis capacity observed in β-glucan–trained cells (Fig. 2E, F). To gain additional insights into the effects of IL-10 on BCG training as well, we measured ROS production. BCG training increased ROS production, and addition of IL-10 on day 0 inhibited this significantly (Fig. 2G). Altogether, it appears that IL-10 selectively inhibits different facets of trained immunity.
3.5 The effect of genetic variants of IL-10 genes on BCG-induced trained immunity
To validate the role of IL-10 in the modulation of trained immunity, we used data collected from a large cohort of BCG vaccinees (the 300BCG study) (Fig. 3A).22,23 We assessed the impact of single nucleotide polymorphisms (SNPs) around IL10 and IL10RB (in a window of 150 kb up- and downstream of the genes of interest) on trained immunity induction by BCG vaccination in healthy volunteers (Fig. 3B). For this analysis, we correlated the changes in S. aureus–induced ex vivo IL-1β production capacity 14 or 90 d after BCG vaccination with the genetic data of 278 healthy volunteers. The fold change in cytokine production was used as a measurement of the magnitude of the trained immunity response. The top hit near IL10, intronic variant (G > A) rs61815626, was associated with BCG-induced trained immunity, with allele G decreasing IL-1β production (P = 5.1 × 10−3) (Fig. 3B, left). The association of this SNP with systemic IL-10 expression was inverse to its relationship with trained immunity: the G allele increased circulating IL-10 (Fig. 3C, left). The strongest association for IL10RB, rs2834169, was also intronic (C > T) but was located in the IL10RB gene itself (encoding for the receptor subunit responsible for signal transduction).45 Allele T of this SNP was associated with decreased trained immunity induction (P = 6.2 × 10−4) (Fig. 3B, right), while the circulating levels of IL10Rβ were increased (Fig. 3C, right). Together, these data show that genetic variation influencing IL-10/IL-10R pathway influence BCG-induced trained immunity in vivo in human volunteers.
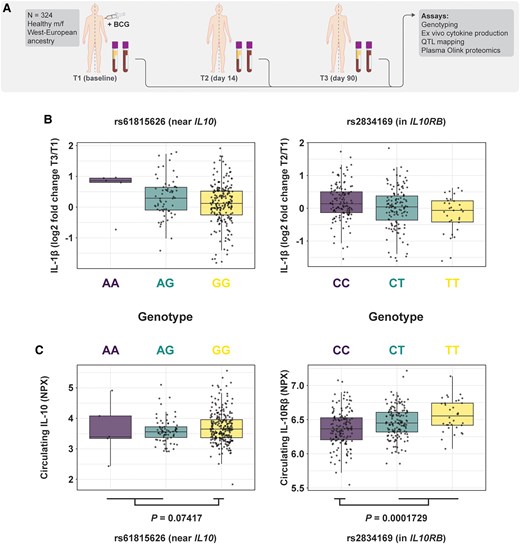
SNPs influencing expression of IL10 or its receptor subunits impact induction of in vivo trained immunity in humans. (A) Schematic overview of the cohort used for mapping of cytokine quantitative trait loci (QTL). (B) The SNPs in close proximity to IL10 (left) and IL10RB (right) that were most significantly associated with trained immunity (P = 5.1 × 10−3 and P = 6.2 × 10−4, respectively) were used to stratify trained immunity induction, as measured by fold changes in IL-1β production following stimulation with Staphylococcus aureus. (C) The circulating levels of IL-10 and IL10Rβ, stratified by genotype of the previously mentioned SNPs (Student's t test was performed between indicated groups to test statistical significance). f = female; m = male; NPX = normalized protein expression.
3.6 Circulating IL-10 concentrations are negatively associated with trained immunity induction in a sex-specific manner
We further explored the association between circulating IL-10 concentrations immediately prior to BCG vaccination and induction of trained immunity by this vaccine. To that end, we performed correlation analyses between the levels of circulating IL-10 and fold changes in the ex vivo PBMC cytokine response to S. aureus. Because trained immunity effects of the BCG vaccine are different in men and women,23 we performed theses analyses separately for each sex. Circulating IL-10 concentrations had a weak negative correlation with changes in ex vivo cytokine production 14 d after vaccination in men but not in women (Fig. 4A). This association was not present at 90 d after vaccination (Fig. 4B).
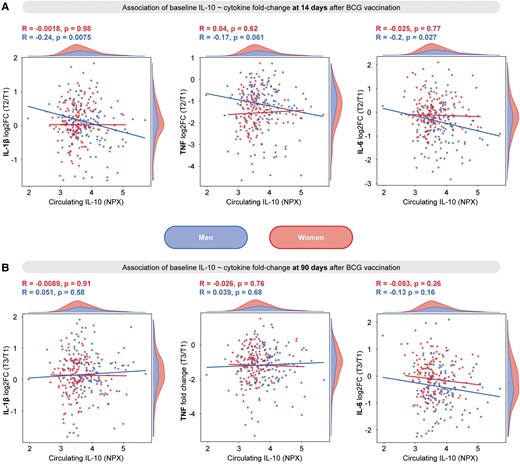
Circulating IL-10 at baseline is negatively associated with changes in ex vivo cytokine production in men but not in women in a trained immunity model with BCG vaccination. (A) Pearson correlation between IL-10 prior to vaccination and log2 fold change (log2FC) in IL-1β, TNF, and IL-6 production by PBMCs (following 24-h stimulation with Staphylococcus aureus) at 14 d after vaccination. (B) Pearson correlation between IL-10 prior to vaccination and log2FC in IL-1β, TNF, and IL-6 production by PBMCs (following 24 h stimulation with S. aureus) at 90 d after vaccination. NPX = normalized protein expression; T = time.
4. Discussion
In the present study, we demonstrate that IL-10 induces tolerance and inhibits trained immunity responses in primary human monocytes. While metabolic upregulation in trained cells was not inhibited by IL-10, the cells were functionally impaired in their capacity to upregulate cytokine gene transcription and protein secretion upon stimulation with LPS after training induced by β-glucan or oxidized low-density lipoprotein. The importance of IL-10 as a modulator of trained immunity was validated by showing that several SNPs that influence expression of IL-10 or the β-subunit of its receptor also impact induction of trained immunity after in vivo BCG vaccination. Circulating IL-10 concentrations were negatively associated with in vivo trained immunity in a sex-specific manner.
The data presented in this study thus indicate that IL-10 is a negative regulator of trained immunity. However, we found that not all facets of trained immunity are impacted equally by IL-10. While IL-10 alone induced metabolic downregulation, it did not affect the upregulated glycolysis elicited by β-glucan training. Furthermore, while IL-10 inhibited the increase in ROS production after BCG training, it did not prevent the increase in phagocytosis observed after β-glucan training. The precise nature of IL-10's effects should be the subject of follow-up investigations. However, it is tempting to speculate that IL-10 may specifically inhibit those aspects of trained immunity that influence inflammation (cytokine production, ROS), but not cell-intrinsic parameters (upregulated metabolism, phagocytosis). This would align well with the concept that IL-10 and its cytokine family members are important for tissue protection.12,13,15 Along that line, the upregulation of IL-10 itself in trained macrophages, which was previously shown,20 is likely to represent a feedback mechanism that controls this process and limits the danger of hyperinflammation. Thus, IL-10 could impose an upper limit on the magnitude of trained immunity that could otherwise be induced following repeated immune insults, protecting against tissue damage.
IL-10 is not the only immunological factor with inhibitory effects on trained immunity. Previous work has shown that anti-inflammatory members of the IL-1 family can also inhibit trained immunity.46 IL-1 receptor antagonist, that blocks the biological activity of both IL-1α and IL-1β, impairs β-glucan–induced trained immunity in mice.47 IL-37 and IL-38, 2 relatively recently discovered IL-1 family members, also inhibit β-glucan training in mice.48,49 These are likely redundant mechanisms that complement each other, with each of these modulators having a relatively minor impact, as shown by the strength of the correlation between circulating IL-10 and trained immunity induction in vivo. Future research into the complete network of factors that inhibit (or indeed reverse) trained immunity is warranted to increase our understanding of this phenomenon.
It is important to discern between the effects of IL-10 on acute inflammation vs. trained immunity. Not all anti-inflammatory mediators necessarily inhibit trained immunity. IL-4 inhibits acute monocyte responses toward LPS but simultaneously induces trained immunity and does not inhibit β-glucan training.50 In the current study, we demonstrate that IL-10 can inhibit both acute innate immune activation and long-term trained immunity effects. Interestingly, not all immune functions upregulated during induction of trained immunity have been impacted equally by IL-10: while IL-10 inhibited trained immunity effects on cytokine production and ROS release, it did not modulate phagocytosis or cellular metabolism. Future studies to distinguish the pathways that differentially regulate these processes would be important for a more in-depth understanding of trained immunity and inflammatory processes.
Upstream of mTOR (mammalian target of rapamycin) inhibition, STAT3 is an important transducer of IL-10R signaling,12,13 and inhibition of STAT3 pathway by itself induced trained immunity. This strengthens the argument that IL-10/IL-10R is a negative regulatory pathway of trained immunity, but from these data alone it is not possible to draw hard conclusions on the role of STAT3 specifically for IL-10–mediated suppression of trained immunity. This warrants more in-depth future studies, which should also consider the clinical relevance of the findings presented here. Patients have been described with defects in the IL-10R or STAT3 signaling,15,51 who we expect (based on our results) to display inappropriately strong trained immunity responses. Indeed, defects in IL-10 pathway have been associated with IBD, while patients with hyper IgE syndrome and STAT3 signaling defects sometimes also display allergic and autoimmune characteristics. If such defects in the regulation of trained immunity are confirmed, this immunological process would become an important potential therapeutic target in these diseases.
An additional potential area of future investigation is the possibility to use either recombinant IL-10 or derivates to modulate trained immunity in vivo. First, mouse models would enable studying IL-10's effects on trained immunity in the natural context of tissues. In humans, IL-10 therapy has been attempted in clinical trials,52 but this has not resulted in any approved therapies thus far. However, potential IL-10–based therapeutics in inflammatory diseases must overcome several challenges. First of all, finding the therapeutic window in which a potent anti-inflammatory cytokine such as IL-10 is effective, but does not cause systemic immunosuppression, is bound to be a delicate process. Second, potential applications that aim to boost trained immunity by blocking IL-10 may also encounter similar side effects as checkpoint blockade in cancer immunotherapy. Third, systemically administered cytokines are rapidly cleared from the circulation and must therefore be applied in large quantities. This is costly and would cause broad nonspecific effect, which may be associated by significant side effects. Elongating the circulation half-life of IL-10 would make it easier to reach desired systemic concentrations, but, as mentioned previously, this might come with unacceptable side effects. Therefore, advanced therapeutic approaches that would target IL-10 specifically to the trained myeloid cells, such as the recently described nanobiological particles that permit effective myeloid cell targeting,53,54 could be a viable alternative.
In conclusion, we demonstrate the capacity of IL-10 to inhibit trained immunity induction in human monocytes. IL-10 exposure of these cells leads to long-term tolerance and inhibition of several key aspects of trained immunity. Future research should be performed to confirm (and potentially utilize) these findings in in vivo human diseases.
Acknowledgments
The authors are very grateful for the participants of the 300BCG study and the study team, in particular Charlotte de Bree, Vera Mourits, Simone Moorlag, Heidi Lemmers, Helga Dijkstra, and Reinout van Crevel. They thank Heidi Lemmers for preparing the lipopolysaccharide used in this study. The oxLDL preparation team of the authors’ lab kindly prepared the oxidized low-density lipoprotein used for this study. Professor David Williams (East Tennessee State University) provided the β-glucan that the authors used in the training experiments.
Author contributions
Conceptualization: R.J.R., L.C.d.J., L.A.B.J., AZ, M.G.N. Data curation: V.K., V.M., R.J.R. Formal Analysis: R.J.R., F.S., L.C.d.J., V.M., V.K. Funding Acquisition: R.J.R., L.A.B.J., M.G.N. Investigation: R.J.R., F.S., L.C.d.J., L.A.G., V.M., V.K., A.Z. Methodology: R.J.R., F.S., L.A.G., V.M., V.K., A.Z. Project Administration: R.J.R., F.S., V.M., V.K. Resources: L.A.B.J., M.G.N. Software: V.M., V.K., R.J.R. Supervision: V.K., V.M., L.A.B.J., A.Z., M.G.N. Visualization: R.J.R., F.S., L.C.d.J. Writing—original draft: R.J.R., L.C.d.J., M.G.N. Writing—review and editing: R.J.R., F.S., L.C.d.J., L.G., V.M., V.K., L.A.B.J., A.Z., M.G.N.
Supplementary material
Supplementary material is available at Journal of Leukocyte Biology online.
Funding
R.J.R. was supported by a Radboudumc personal PhD grant. M.G.N. was supported by a Spinoza grant from the Dutch Research Council (NWO) and an ERC Advanced Grant (#833247).
Data availability
Data are available upon reasonable request to the corresponding author.
References
Author notes
Conflict of interest statement. M.G.N. and L.A.B.J. are scientific co-founders of and have equity in Trained Therapeutix Discovery. The other authors report no conflicts of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}