





Abstract
Dendritic epidermal T cells (DETCs) are the main source of insulin-like growth factor-1 (IGF-1) in epidermal tissue, which promote re-epithelialization and wound healing. In refractory wounds, IL-1β has been shown to activate NF-κB and suppress IGF-1 expression in DETCs. Nevertheless, the underlying mechanisms remain unclear. In this study, chromatin immunoprecipitation analysis revealed that IL-1β did not inhibit NF-κB binding to IGF-1 promoter, indicating that IL-1β/NF-κB may suppress IGF-1 expression by alternative mechanisms. MiRNAs negatively regulate gene expression predominantly by base pairing to the 3′ untranslation region (UTR) of target mRNAs. Let-7f-5p, miR-1a-3p, and miR-98-5p have been identified as IGF-1-specific miRNAs that can bind directly to the 3′UTR of IGF-1 mRNA and dysregulate IGF-1 mRNA and protein levels. In IL-1β-treated epidermis around wounds or DETCs in vitro, NF-κB promoted the expression of let-7f-5p, and IGF-1 expression was impeded via NF-κB/let-7f-5p pathway. As pre-let-7f-5p, let-7f-1 is located in the 3′UTR of LOC118568094, and let-7f-2 is located in the intron of HUWE1. We discovered that NF-κB p65 bound to the promoters of LOC118568094 and HUWE1 to accelerate let-7f-5p expression, but NF-κB p65 did not affect the methylation levels of LOC118568094 and HUWE1 CpG islands. Injections of Let-7f-5p antagomir into IL-1β-treated and ischemic wound margins restored IGF-1 secretion in DETCs and promoted wound healing. In conclusion, we demonstrated that NF-κB signaling pathway activated by IL-1β could increase let-7f-5p expression to inhibit IGF-1 production in DETCs and delay wound healing. And let-7f-5p antagomir utilized in wound margin could effectively promote refractory wound healing.
INTRODUCTION
Burns and trauma are typical accidental injuries that cause wounds by destroying the integrity of skin tissue.1 Wound repair confronts numerous hurdles, particularly the pathologic mechanism of the refractory wound, which has yet to be fully investigated. Studies have demonstrated that excessive wound inflammatory mediators such as IL-1β, IL-23, and IL-17A can inhibit the re-epithelialization of the wound and possibly lead to the establishment of the refractory wound.2–4 Dendritic epidermal T cells (DETCs) are 1 subset of γδ T cells and uniformly express an invariant Vγ5Vδ1 TCR (according to Heilig and Tonegawa's nomenclature).5,6 DETCs exclusively reside in the murine epidermis and comprise more than 90% of T cells in the epidermis.4 DETCs, unlike other T cells, perform their physiologic function primarily through secreting cytokines.7 DETCs are the main source of insulin-like growth factor-1(IGF-1) in epidermal tissue, which helps to maintain homeostasis, stimulate re-epithelialization, and expedite wound healing.7–9 DETCs can release IGF-1 rapidly and in large amounts after skin injury.7 In a proinflammatory environment, such as diabetic wounds, the production of IGF-1 in DETCs is impaired, which is one of the major causes of delayed wound healing.3,10,11
Our previous research has demonstrated that the expression of IL-1β was significantly promoted in wound margins after skin damage or skin graft transplantation.3,12 After skin injury, the administration of IL-1β cytokine or neutralizing antibody to wound margin significantly reduced or boosted IGF-1 synthesis by DETCs surrounding wounds.3 In addition, adding IL-1β to DETCs in vitro significantly reduced IGF-1 production.3 These findings showed that IL-1β inhibited IGF-1 synthesis in DETCs. NF-κB proteins are one of the key transcription factors in the inflammatory response.13,14 Our group showed that IL-1β promoted the intracellular phosphorylation of NF-κB p65 as well as the translocation of NF-κB p65 from the cytoplasm to the nucleus.3 The expression of IGF-1 in DETCs was dramatically enhanced when NF-κb signaling was blocked.3 Furthermore, it has been previously reported that blocking NF-κB by transgene or a pharmacologic inhibitor both produced hyperplastic epithelium in vivo and that NF-κB partially inhibited keratinocyte proliferation in vitro.15,16 As a result, we hypothesized that NF-κB, which is activated by IL-1β, could suppress IGF-1 production in DETCs. The mechanisms under which NF-κB suppresses IGF-1 expression are unknown. There has not been much investigation on the negative control of IGF-1 gene expression. We intend to explore whether IL-1β could prevent NF-κB from binding to the IGF-1 promoter. Besides, microRNAs (miRNAs) have recently come to light as a crucial new biologic process regulator in a number of disease-related processes, such as wound healing and hypertrophic scar formation.10,11 MiRNAs are small conserved RNA molecules of ∼18–25 nucleotides that negatively control gene expression predominantly by base pairing to the 3′ untranslated region of target mRNAs.17 Let-7f-5p, miR-1a-3p, and miR-98-5p have been identified as IGF-1-specific miRNAs that can bind directly to the 3′ untranslation region (UTR) of IGF-1 mRNA and dysregulate IGF-1 mRNA and protein levels.17–19 However, the relationship between NF-κB and IGF-1-specific miRNAs has not been reported. It is crucial to examine the mechanism by which NF-κB inhibits IGF-1 production, as well as if and how the NF-B signaling pathway regulates these IGF-1-specific miRNAs.
MATERIALS AND METHODS
Mice
C57BL/6 wild-type (WT) mice were obtained from the Animal Center of the 9th hospital of Shanghai Medical University (Shanghai, China). All experiments were carried out on sex-matched mice aged 6–8 weeks. All experiments were conducted under conventional animal raising conditions, in accordance with ethical guidelines, and with the approval of the Animal Ethics Committee of Shanghai Medical University.
Full-thickness excision wound model
Mouse model administrated with IL-1β was generated as previously reported.3 Excision incisions on the mouse back were cut down to the deep fascia using a sterile punch with a diameter of 6 mm. And then IL-1β (R&D Systems, Minneapolis, Minnesota, USA) was subcutaneously injected with a dose of 20 ng/wound in the wound margin for continuously 3 days after excision. Macroscopic digital pictures of the wound region were taken on indicated days. The wound area was calculated at each time point relative to day 0 until it was entirely closed by ImageJ. The percent wound area was calculated as area of actual wound/area of original wound × 100. The surgery to produce hindlimb ischemia was conducted out as reported previously.20 Hair was removed with a depilatory cream and the hindlimbs were lengthened and fastened. Under microscopic supervision at 20× magnification, the left hindlimb was visualized, and a 1-cm latitudinal skin incision was made at the inguinal ligament. The underlying femoral artery was revealed after the subcutaneous tissue was peeled away. The neurovascular bundle was exposed by piercing the membranous femoral sheath. The femoral artery and vein were isolated from the femoral nerve and ligated with 7-0 silk sutures before being cut with a single incision between sutures. Finally, a continuous 7-0 silk suture was used to close the skin incision. A 4-mm punch biopsy equipment was used to establish an ischemic full-thickness wound on the ventral surface of the thigh. The wounds were covered with a piece of transparent membrane (negative pressure wound therapy kit, Wuhan, Hubei, China) immediately after excision to avoid wound injury caused by friction when mice walked. Let-7f-5p antagomir (5 nM/wound; Ribobio, Guangzhou, Guangdong, China) was injected in wound margins on days 0, 1, and 2 after wounding.
Isolation of epidermal sheets
The whole skin at wound edge was gathered, which was less than 5 mm away from the border of wound. Skin tissue of 3 or more mice at wound edge was obtained for quantitative real-time PCR (qRT-PCR) and western blotting (WB) examination. The wound edge was wiped off of subcutaneous tissue, which was then cut into 0.5 × 0.5 cm2 pieces. Epidermal sheets were separated from the wound edge after incubation with 5 mg/ml dispase II for 1–2 h at 37°C.
Preparation, stimulation, and transfection of DETCs
DETCs were prepared as previous report.3 In briefly, skin was cut off from the back of hair-removed mice and subcutaneous tissue was wiped off. Epidermal sheets were separated as described above, then cut into pieces and soaked for 30 min in 0.3% trypsin/GNK solution at 37°C, then washed by PBS and filtered through a 70-μm strainer to isolate epidermal cells. DETCs were purified to 15–20% purity using Lympholyte-M (Cedarlane Laboratories, Burlington, Ontario, Canada). DETCs were expanded for 2–4 weeks in 48-well plates (1–2 × 106/ml) with RPMI containing 10% FBS, 1 μg/ml Con A, 1 mM Na pyruvate (Sigma–Aldrich, Saint Louis, Missouri, USA), 2 mM glutamine, 25 mM HEPES, 50 μM 2-ME (Biosharp, Hefei, Anhui, China), 100 M nonessential amino acids (Gibco, Carlsbad, California, USA), 10 ng/ml mouse IL-2, 1× penicillin–streptomycin solution. The purity of expanded DETCs was >95%, which was verified by flow cytometric analysis (Figure S1). DETCs were rested for 2 weeks without ConA before being employed in the subsequent tests. DETCs were treated for 24 h with anti-CD3 (5 g/ml) alone or in combination with murine IL-1β (100 ng/ml; R&D System) or NF-κB inhibitor (BAY11-7082, 10 μM; Selleck, Houston, Texas) for in vitro stimulation. Let-7f-5p mimic, control mimic, let-7f inhibitor, and control inhibitor were purchased from ambion of Thermo Fisher scientific (Waltham, Massachusetts, USA). MiRNA mimic or inhibitor and lipofectamine RNAiMAX transfection reagent (Thermo Fisher scientific) were prepared in transfection mixtures with Opti-MEM serum-free medium (Gibco) without antibiotic according the manufacturer's recommends. DETCs (1 × 105/well) were seeded in 24-well plates in antibiotic-free medium before transfection, and then transfected with let-7f-5p mimic (10 nM), mimic control, let-7f-5p inhibitor (100 nM), or inhibitor control according to the manufacturer's instructions. Then, DETCs were incubated with transfection mixtures for 24 h. RNA or protein were extracted from DETCs for following experiments.
Flow cytometry analysis
Cells were blocked with anti-CD16/32 Ab (clone 2.3G2; SunGene Biotech, Tianjin, China), and then their surface were stained with the following monoclonal Abs: FITC Hamster Anti-Mouse Vγ5 TCR (clone 536; BD, Franklin Lakes, New Jersey, USA), and PE-Cyanine5 Hamster Anti-Mouse γδ TCR (clone GL3; eBioscience, San Diego, California, USA) for 30 min at room temperature. Stained cells were measured on Attune Acoustic Focusing Cytometer (Life Technologies, Carlsbad, California, USA), and analyzed by FlowJo software (BD).
Quantitative real-time PCR
The primer sequence of mmu-let-7f-5p, mmu-miR98-5p, mmu-miR1-3p, mouse snoRNA202, IGF-1, and GADPH were synthesized by Sangon Biotech (Shanghai, China) and indicated in Table S1. RNAs were extracted by TRIzol reagents or a Qiagen RNeasy kit (Invitrogen, Carlsbad, California, USA). cDNA was synthesized by TaqMan reverse transcription kit (Life Technologies) following the manufacturer's instructions. QRT-PCR was performed on Bio-Rad RT-PCR system using SYBR Green Supermix. The expression levels of mRNA or miRNA were respectively normalized to GAPDH or snoRNA202 and calculated as fold differences by the method where F = 2−ΔΔCt, ΔCt = Cttarget gene − CtGAPDH and ΔΔCt = ΔCtinduced − ΔCtreference.
Western blot
Tissue protein extracted from wound margin tissue was as previously report.3 Briefly, the epidermal sheets of wound margin were grinded into powder in liquid nitrogen, and then lysed in lysis buffer with protease inhibitor, PSMF and phosphatase inhibitor (KeyGEN BioTECH, Nanjing, Jiangsu, China). The concentration of extracted protein was tested by BCA protein assay kit (Thermo Fisher Scientific). Protein samples (25 μg/well) were separated by 10 or 12% SDS-PAGE and then transferred to polyvinylidenedifluoride (PVDF) membranes (Merck, Darmstadt, Germany). PVDF membranes were incubated with 3% bull serum albumin (Biosharp) at room temperature for 2 h, and then with following Abs at 4°C overnight: anti-IGF-1 (1:1000; Abcam, Cambridge, UK), anti-NF-κB p65(D14E12), anti-phospho-NF-κB p65 (Ser536, 93H1) (1:1000; Cell Signaling Technology, Boston, Massachusetts, USA). Anti-GAPDH Ab (1:2000; SunGene Biotech) was as control. PVDF membranes were visualized using goat-anti-rabbit/mouse/rat-HRP IgGs (1:5000; SunGene Biotech) at room temperature for 1 h, and followed by chemiluminescence liquid for seconds (Solarbio, Beijing, CHN). Protein expression intensity was analyzed by ChemiDoc™ XRS detection system (Bio-Rad, Hercules, California, USA).
H&E staining and immunohistochemistry
Wound edge samples were embedded in paraffin by routine methods. The slices were stained with H&E and the neoepithelial length was measured by blinded manner under microscope. The length of the neoepithelium was determined as the length of the epithelial tongue, or as the distance that between the advancing edges of the epidermal keratinocytes and the existence of hair follicles in nonwounded skin. The expression levels of indicated cytokines in wound margin were detected by immunohistochemistry. Skin sections were deparaffinized and hydrated in xylene and graded alcohol series. Antigen retrieval was performed by heating sections in 10 mM citric acid (pH 6) at 95°C for 15 min. Sections were blocked with goat-anti-rabbit serum or 3% BSA with 0.3% Triton X-100 (Sigma–Aldrich) at room temperature for 1 h, and then stained by primary antibodies IL-1β and IGF-1 (1:100–200; Abcam) at 4°C overnight. After washed by PBS, slices were incubated with secondary antibodies at room temperature for 1 h. Slides were dealt with drops of diaminobenzidine solution (Boster, Wuhan, Hubei, China) and counterstained with hematoxylin (Beyotime, Shanghai, China). Stained sections were detected under Olympus BX51 microscope (Tokyo, Japan).
Chromatin immunoprecipitation
DETCs were treated with IL-1β or control PBS for 24 h, and then were fixed in 1% formaldehyde (final concentration) for 10 min to crosslink nucleoprotein complexes. 1/20 volume glycine solution (2.5 M) was used to stop crosslinking reaction. Samples were then washed in prechilled PBS for 3 times. Cells were collected and centrifuged at 444 g for 5 min. Then, cells were lysed and transferred to lysis buffer. The sheared DNA complex was immunoprecipitated by incubating the lysates with anti-NF-κB p65 (D14E12, 1:100; Cell Signaling Technology) or IgG antibodies overnight. The antibody-target protein–DNA complex was band and precipitate by Protein A. The precipitated complex was washed to remove unspecific binding. Then NF-κB–DNA complex was eluted and collected. DNA was separated by chromatin immunoprecipitation (ChIP) elution buffer and Proteinase K. The promoters of IGF-1, let-7f-1, and let-7f-2 (the precursors of let-7f-5p) were amplified by PCR reaction. Primers for the predicted NF-κB binding sites among the promoter regions of IGF-1, LOC118568094, and HUWE1 were displayed in Table S2. The obtained PCR products were detected by 1% agarose gel electrophoresis. QRT-PCR was performed on Bio-Rad RT-PCR system using SYBR Green Supermix. ChIP signal (% input) were calculated: %input = 25%*2ΔCt, ΔCt = Ctinput − CtChIP. The results were considered positive when the fold differences of ChIP signal (% input) between the 2 groups was 3 times or more.
Methylation-specific PCR
DETCs were stimulated with anti-CD3ε (5 μg/ml; SunGene Biotech) either alone or combined with murine IL-1β (100 ng/ml; R&D System) for 24 h. DNA was treated with Sodium bisulfite solution to convert unmethylated cytosine to uracil, while leaving methylcytosine unaltered. Then bisulfite converted DNA were subjected to methylation-specific PCR (MS-PCR) with primers for CpG methylation analysis, as shown in Table S3. T/A cloning and sequencing were performed subsequently.
Bioinformatics prediction analysis
The pre-miRNAs of let-7f-5p are let7f-1 and let-7f. Let-7f-1 is located at the 3′UTR of LOC118568094 and let-7f-2 is located in the intron of HUWE1. The DNA sequences of 2 kb upstream of IGF-1, LOC118568094, and HUWE1 as the promoter regions were searched respectively through the Gene database in PubMed. The binding sites of NF-κB to these promoter regions were predicted on CIS-BP website. The predicted binding sites were selected based on high scores and clearly distinguishable positions.
Statistical analysis
Data were shown as means ± standard error of standard deviation (SD). The two-tailed Student's unpaired t-test or one-tailed Mann–Whitney test was used to compare the data from the 2 groups. One-way ANOVA with Bonferroni's multiple comparison test was used to evaluate data with more than 2 groups. The p values and graphs were manufactured by GraphPad Prism software (GraphPad Software, San Diego, California, USA). p < 0.05 was considered significant.
RESULTS
NF-κB could not directly inhibit IGF-1 expression in DETCs, but could promote the transcription of miRNA let-7f-5p
By binding to promoters, NF-κB proteins act as important transcription factors that directly regulate gene transcription.21 Therefore, our group explored whether IL-1β could prevent NF-κB from binding to the IGF-1 promoter. The 2 kb DNA sequence upstream of IGF-1 is regarded as the promoter region, which was searched through the gene database in PubMed. The binding sites between NF-κB p65 and the promoter region of IGF-1 were predicted on CIS-BP, and then the predicted binding sites were selected based on high scores and positions that could be completely distinguished (Figure 1A). P1–P4 are the names of the 4 selected sites. In RT-PCR electrophoresis, the fluorescence intensity of the NF-κB group of P1 was considerably elevated in IL-1β stimulation compared with control (Figure 1B). QRT-PCR confirmed that the binding of NF-κB p65 to P1 was promoted in the IL-1β group compared with control (Figure 1C). These findings suggested that after IL-1β stimulation, NF-κB p65 was more likely to bind the IGF-1 promoter in DETCs, and may act as a negative regulatory transcription factor, competing for binding sites in the IGF-1 promoter with “the positive regulatory transcription factor.”

NF-κB did not directly inhibit DETCs to express IGF-1 but promoted the production of let-7f-5p and miR-98-5p. (A) The predicted binding sites between NF-κB p65 and IGF-1 promoter. Four sites named P1–P4 were selected according to predicted scores and positions. (B and C) DETCs were stimulated by IL-1β (100 ng/ml) for 24 h, and PBS as control. (B) Precipitated DNA obtained from ChIP was analyzed by RT-PCR electrophoresis. The first lanes represent DNA ladder. (C) Precipitated DNA obtained from ChIP was also analyzed QRT-PCR. (D) The expression of let-7f-5p, miR-1a-3p, and miR-98-5p in DETCs with IL-1β treatment versus control were detected by qRT-PCR. (E) Age- and sex-matched WT mice were treated with IL-1β (20 ng/wound) or PBS (as control) on days 0, 1, and 2 after wounding (n = 3/group). The expression of let-7f-5p, miR-1a-3p, and miR-98-5p were detected by qRT-PCR on day 3 postexcision. (F) DETCs were pretreated with BAY 11–7082 (10 μM) for 1 h before the addition of IL-1β (100 ng/ml). The expression of let-7f-5p and miR-98-5p were detected by qRT-PCR. The values were calculated as the mean ± sd. All data represent at least 3 independent experiments. p Value was analyzed between control and IL-1β by two-tailed Student's unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001)
Moreover, NF-κB may increase the production of several regulatory genes, such as miRNAs, to reduce the posttranscriptional expression of IGF-1 mRNA. Let-7f-5p, miR-1a-3p, and miR-98-5p can directly target the IGF-1 3′UTR, decreasing IGF-1 expression at both the mRNA and protein levels.17–19 In IL-1β-stimulated DETCs in vitro, let-7f-5p and miR-98-5p were up-regulated compared with control according to QRT-PCR analysis (Figure 1D). The expression levels of let-7f-5p and miR-98-5p were also increased in the epidermis of wound margin with IL-1β injection compared with control (Figure 1E). The expression level of miR-1a-3p was not affected by IL-1β both in vivo and in vitro (Figures 1D and E). To investigate the role of NF-κB p65 in miRNA expression, BAY 11–7082(BAY) was used as NF-κB inhibitor by specifically blocking the phosphorylation of IκBα.22 The expression level of let-7f-5p and miR-98-5p were suppressed when DETCs were treated with IL-1β and BAY compared with IL-1β treatment alone (Figure 1F). These results indicated that NF-κB enhanced the expression of let-7f-5p and miR-98-5p in DETCs. NF-κB was found to enhance the expression of let-7f-5p and inhibit the expression of IL-10 in macrophages in the osteolysis model, resulting in excessive bone destruction, implying that NF-κB could regulate the expression of let-7f-5p in macrophages.23 As a result, we focused on let-7f-5p and investigated whether and how NF-κB could enhance let-7f-5p expression in DETCs to reduce IGF-1 production.
Let-7f-5p has been shown to bind directly to the 3′UTR of IGF-1 mRNA.17 Figure 2A depicted the binding locations. The effects of let-7f-5p on IGF-1 expression in DETCs were also observed by our study. DETCs were transfected with let-7f-5p mimic or control and incubated for 24 h. QRT-PCR study revealed that let-7f-5p mimic considerably increased the level of let-7f-5p in DETCs when compared with the control mimic (Figure 2B). Furthermore, when compared with control, the expression levels of IGF-1 mRNA and protein were declined in let-7f-5p mimic group (Figures 2C and D). In addition, DETCs were transfected with let-7f-5p inhibitor or inhibitor control and cultured for 24 h. Chemically synthesized miRNA inhibitors are single-stranded RNA molecules that, following transfection into cells, hinder miRNA processing or transport from the nucleus to the cytoplasm, hence limiting miRNA's regulatory function. MiRNA inhibitors do not always result in miRNA degradation, but the effects of miRNA inhibitors can be determined by assessing the expression levels of target genes. The expression level of let-7f-5p in inhibitor group was slightly lower than in inhibitor control (Figure 2E), but let-7f-5p inhibitor significantly improved the expression levels of IGF-1 mRNA and protein in the presence of IL-1β stimulation (Figures 2F and G).
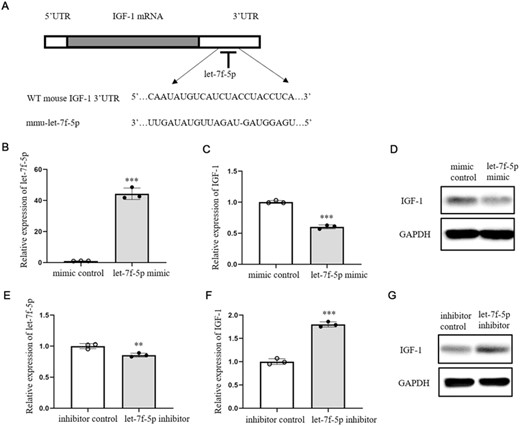
Let-7f-5p down-regulated its target IGF-1 in DETCs. (A) The binding site between WT IGF-1 3′ UTR and let-7f-5p. (B-D) let-7f-5p mimic or mimic control (10 nM) was transfected into DETCs with lipofectamine™ RNAiMAX transfection reagent (lipofectamine RNAiMAX) and then cultured for 24 h. The expression of let-7f-5p was tested by qRT-PCR (B). The expression of IGF-1 was detected by qRT-PCR (C) and WB (D). (E–G) let-7f-5p inhibitor or inhibitor control (100 nM) was transfected into DETCs with lipofectamine RNAiMAX and then stimulated by IL-1β for 24 h. Let-7f-5p expression was analyzed by qRT-PCR (E). The expression of IGF-1 was detected by qRT-PCR (F) and WB (G). The data represent the mean ± sd of 3 independent experiments. p Value was assessed by two-tailed Student's unpaired t-test (B, C, E, and F) (**p < 0.01, ***p < 0.001)
IGF-1 expression was impeded via NF-κB/let-7f-5p pathway
IGF-1 expression was suppressed by IL-1β via activating NF-κB in DETCs, as previously reported.3 And NF-κB was verified above to promote let-7f-5p expression in DETCs. It was required to explore whether NF-κB down-regulated IGF-1 expression by let-7f-5p. DETCs were randomly divided into 5 groups and received the following treatments: blank control group, IL-1β group, IL-1β+ BAY, IL-1β+ BAY+ control mimic, and IL-1β+ BAY+ let-7f-5p mimic. The phosphorylation levels of NF-κB p65 and IκBα were elevated in the IL-1β stimulation group, whereas the relative expression levels of IGF-1 mRNA and protein were decreased when compared with control (Figures 3A–D). When DETCs were incubated with BAY combined with IL-1β, the phosphorylation levels of NF-κB p65 and IκBα were reduced, but the relative expression levels of IGF-1 mRNA and protein were increased (Figures 3A–D), indicating that NF-κB was required for IL-1β to suppress IGF-1 expression. In addition, the phosphorylation levels of NF-κB p65 and IκBα in the IL-1β+ BAY+ let-7f-5p mimic group was similar to that in IL-1β+ BAY+ mimic control group, while the relative expression levels of IGF-1 mRNA and protein in the IL-1β+ BAY+ let-7f-5p mimic group were lower than that in IL-1β+ BAY+ mimic control group (Figures 3A–D), suggesting that let-7f-5p mimic could directly down-regulate the expression level of IGF-1 when NF-κB was inhibited. The let-7f-5p inhibitor was also used to confirm this occurrence. DETCs were randomly divided into these 5 groups with indicated treatments: blank control group, IL-1β group, IL-1β+ BAY, IL-1β+ inhibitor control and IL-1β+ let-7f-5p inhibitor. The difference in phosphorylation levels of NF-κB p65 and IκBα between groups IL-1β+ let-7f-5p inhibitor and IL-1β+ inhibitor control was not as substantial as the difference between groups IL-1β and IL-1β+ BAY (Figures 3E and F). However, IL-1β+ let-7f-5p inhibitor significantly increased IGF-1 mRNA and protein expression compared with IL-1β+ inhibitor control, which was similar to the up-regulated trend of IL-1β+ BAY relative to IL-1β (Figures 3G and H), indicating that let-7f-5p inhibitor could restore IGF-1 expression similarly to BAY when DETCs were treated with IL-1β. These results demonstrated that let-7f-5p may be downstream of IL-1β/NF-κB to regulate IGF-1 expression.
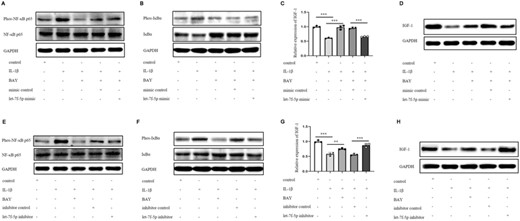
NF-κB impeded IGF-1 expression via let-7f-5p. (A–D) DETCs were allocated into 5 groups according to different treatments: control, IL-1β (100 ng/ml), IL-1β+BAY 11–7082 (10 μM), IL-1β+BAY 11–7082+mimic control (10 nM), IL-1β+BAY 11–7082+let-7f-5p mimic (10 nM). DETCs were pretreated with BAY 11–7082 for 1 h before the addition of IL-1β and/or let-5p mimic/ mimic control. (A and B) Cells were stimulated for 30 min. (A) The expression of phospho-NF-κB p65 (Ser536) and total NF-κB p65 in DETCs were analyzed by WB. (B) The expression of phospho-IκBα (Ser32) and total IκBα in DETCs were analyzed by WB. (C and D) Cells were stimulated for 24 h. The expression level of IGF-1 was detected by qRT-PCR (C) and WB (D). (E–H) DETCs were divided into 5 groups according to different treatments: control, IL-1β, IL-1β+BAY 11–7082 (10 μM), IL-1β+inhibitor control (100 nM), IL-1β+let-7f-5p inhibitor (100 nM). DETCs were pretreated with BAY 11–7082 for 1 h before the addition of IL-1β and/or let-5p inhibitor/ inhibitor control. (E and F) Cells were stimulated for 30 min. The expression of phospho-NF-κB p65, total NF-κB p65 (E), phospho-IκBα and total IκBα (F) in DETCs were analyzed by WB. (G and H) Cells were cultured for 24 h. (G) The expression of IGF-1 mRNA in DETCs was determined by qRT-PCR. (H) The protein expression level of IGF-1 was monitored by WB. Bars show the mean ± sd. p Values in C and G were calculated using one-way ANOVA with Bonferroni's multiple comparison test between the 2 groups marked under the horizontal lines (ns no statistical significance, *p < 0.05, **p < 0.01, ***p < 0.001). All data represent at least 3 independent experiments
NF-κB promoted the expression of let-7f-5p
Let-7f-5p has 2 pre-miRNAs, let-7f-1 and let-7f-2, whose positions in the genome were found using the UCSC Genome Browser Gateway (Figures 4A and B). Let7f-1 is only located in the 3′UTR of LOC118568094. LOC118568094, also known as translation initiation factor IF-2-like, may affect the initiator tRNA moving into the translation mechanism and the initial step in the peptide chain elongation cycle.24 In theory, LOC118568094, which shares the same promoter as let-7f-1, is considered the let-7f-1 host gene. Let7f-2 is located in the HUWE1 intron. HUWE1, an E3 ubiquitin ligase, promotes host defense against bacterial infection by mediating inflammasome assembly and caspase-1 activation.25 Furthermore, miR-98-5p (miR-98) pre-miRNA is identified in the same intron of HUWE1 as let-7f-2 (Figure 4B). HUWE1 has been correlated to miR-98 expression and is thought to be the miR-98 host gene.26 Therefore, we assumed that HUWE1 is likewise the let-7f-2 host gene. In this research, the expression levels of LOC118568094 and HUWE1 were increased by IL-1β in DETCs (Figure 5A), while they were declined when BAY was added combined with IL-1β to DETCs (Figure 5B), suggesting LOC118568094 and HUWE1 were positively related with let-7f-5p. As a result, we investigated whether NF-κB p65 could bind to the LOC118568094 and HUWE1 promoters. The same strategy was used to predict and choose the binding sites between NF-κB p65 and promoters. Figures 5C and F show 3 NF-κB p65 binding sites to LOC118568094 (P1, P2, P3) and 2 NF-κB p65 binding sites to HUWE1 (P1, P2). However, input RT-PCR electrophoresis revealed that LOC118568094 P1 was invalid (Figure S2). Therefore, the predicted binding site was excluded from the following experiments. In RT-PCR electrophoresis, there was no discernible band of NF-κB groups of LOC118568094 P2 (Figure 5D). Furthermore, the cycle thresholds of these NF-κB groups were not detected. In DETCs treated with IL-1β, the fluorescence intensity of the NF-κB groups of LOC118568094 P3 in RT-PCR electrophoresis was significantly increased compared with control (Figure 5D). QRT-PCR confirmed that NF-κB p65 binding to LOC118568094 P3 was increased in IL-1β than in control (Figure 5E). Furthermore, ChIP and qRT-QCR analysis revealed a significant difference in HUWE1 P2 between IL-1β group and control (Figures 5G and H). These findings showed that NF-κB p65 could directly bind to the promoters of LOC118568094 and HUWE1.
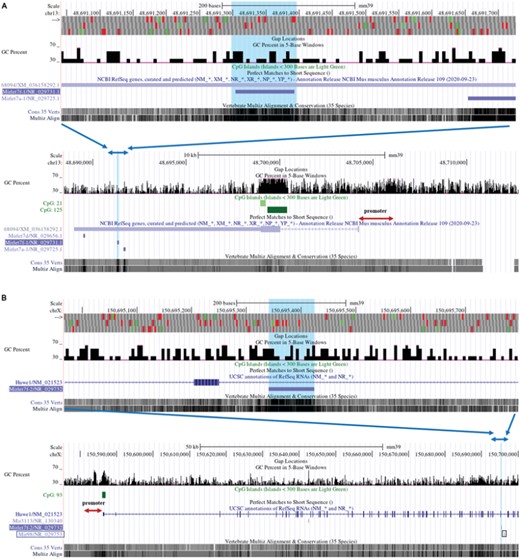
The locations of pre-miRNAs in Genome Reference Consortium Mouse Build 39 (GCA_000001635.9) GRCm39 were obtained from UCSC. (A) The purple line segment in the upper lane indicates let-7f-1, which is located in the 3′UTR region of LOC 118568094. The blue arrows indicate the position of the upper lane in the lower lane. (B) The purple line segment in the upper lane indicates let-7f-2, which is located in the intron region of Hawe1. The blue arrows indicate the position of the upper lane in the lower lane. MiR-98 is circled by the black box. The promoter's location is shown with red arrows. CpG islands are represented by green line segments
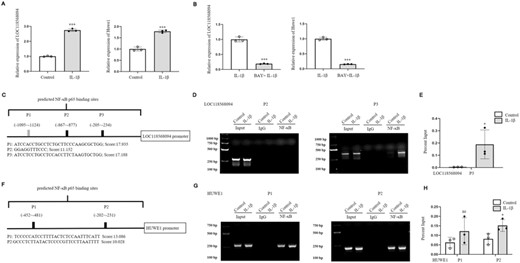
NF-κB p65 could directly bind the promoter of LOC118568094 and facilitate the transcription of pre-let-7f-5p. (A) DETCs were stimulated by IL-1β (100 ng/ml) for 24 h. Control cells were treated with equivalent PBS. The mRNA expression levels of LOC118568094 and HUWE1 were detected by qRT-PCR. (B) DETCs were stimulated by IL-1β (100 ng/ml) for 24 h. Inhibition group of DETCs was pretreated with BAY 11–7082 for 1 h before the addition of IL-1β. QRT-PCR was conducted to analyze the expression levels of LOC118568094 and HUWE1. (C and F) Schematic illustrations of predicted NF-κB binding sites in the promoter of LOC118568094 or HUWE1, which were named as P1–P3 or P1–P2, and labeled with sequences and scores. (D and G) RT-PCR analysis of the precipitated DNA. First lanes represent DNA ladder. (E and H) QRT-PCR analysis of the precipitated DNA. The data represent the mean ± sd of 3 independent experiments. p Values were calculated using two-tailed Student's unpaired t-test (A, B, and H), and one-tailed Mann–Whitney test (E) (*p < 0.05, ***p < 0.001)
DNA methylation patterns in CpG-rich regions of promoter, called as CpG islands, regulate gene expression in mammalian cells. Excessive methylation of CpG dinucleotides in promoter represses gene expression.27 By detecting the methylation levels of CpG islands in LOC118568094 and HUWE1, we intended to explore whether IL-1β could influence the expression of these 2 genes. Methylation-specific PCR is a method for analysis of DNA methylation patterns in CpG islands. The CpG islands on LOC118568094 and HUWE1were separated into 3 segments for methylation analysis (Figure S3). The results showed no significant variation in methylation levels between IL-1β and control groups (Figure 6), indicating that IL-1β did not affect the methylation levels of CpG islands in the promoter regions of LOC118568094 and HUWE1.

NF-κB did not influence the methylation levels of CpG islands in LOC118568094 and HUWE1. DETCs were stimulated by IL-1β (100 ng/ml) for 24 h. Control cells were treated with equivalent PBS. CpG islands on LOC118568094 (A) and HUWE1 (B) were divided into 3 segments respectively for methylation analysis
Let-7f-5p antagomir promoted the wound healing in IL-1β-treated mice
In IL-1β-treated mouse model, the effects of let-7f-5p antagomir on wound healing were investigated. IL-1β+ let-7f-5p antagomir or IL-1β+ control was injected in the wound margin on days 0, 1, and 2 after wounding (Figure 7A). On the days following excision, the wound area was measured, as shown in Figure 7B. Let-7f-5p antagomir enhanced wound healing as compared with control (Figure 7C). The wounds were stained with H&E on day 3 postexcision (Figure 7D). The length of neoepithelium was much longer in mice treated with let-7f-5p antagomir than in control animals (Figure 7E). When compared with control, the expression of IGF-1 in the epidermis around wounds treated with let-7f-5p antagomir was increased (Figures 7F–H). These results indicated that let-7f-5p antagomir rescued wound healing by facilitating IGF-1 expression.
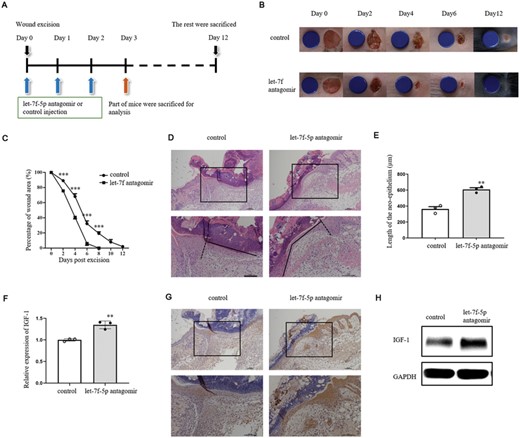
Let-7f-5p antagomir facilitated wound healing in IL-1β-treated mice. (A) Age- and sex-matched WT mice were treated with IL-1β (20 ng/wound) and let-7f-5p antagomir or control (5 nM/wound) on days 0, 1 and 2 after wounding. Skin biopsies at wound sites were obtained on day 3 postoperation for the following experiments (n = 5/group) (D–L). Wound healing on the rest mice was observed until day 12 after excision (n = 10/group). (B) Representative wound photographs on days 0, 2, 4, 6, and 12 postexcision. The blue discs indicate the initial wound size (diameter = 6 mm). (C) Wound closure rates were calculated on days 0, 2, 4, 6, 8, 10, and 12 postwounding. (D) Re-epithelialization of full-thickness skin wound margin was analyzed by H&E. (E) Length of the neoepithelium of skin wound margin were measured in groups treated with control or let-7f antagomir. (F–H) Expression of IGF-1 was analyzed by qRT-PCR (F), immunohistochemical detection (G), and WB (H). The data represent the mean ± sd of 3 independent experiments. p Values were calculated by one-way ANOVA with Bonferroni posttests (C) or two-tailed Student's unpaired t-test (E and F) (**p < 0.01, ***p < 0.001)
Let-7f-5p antagomir promoted the wound healing in ischemic mouse model
We also explored the role of let-7f-5p in wound healing of ischemic mouse model. On day 3 postoperation, the wound area of ischemic wounds was bigger than control (Figure 8A). The wound healing was declined in ischemic wounds compared with control (Figure 8B). H&E staining of wounds was conducted on day 3 postexcision (Figure 8C). The length of neoepithelium was longer in control than ischemic group (Figure 8D). Let-7f-5p expression was facilitated in the epidermis around wounds of ischemic group compared with control (Figure 8E), while IGF-1 expression was decreased in the epidermis around wounds of ischemic group compared with control (Figures 8F and G). Besides, IL-1β was verified to be accelerated in ischemic wound margins (Figure 8H). Phosphorylation levels of NF-κB p65 and IκBα in ischemic wound margins were up-regulated compared with control (Figures 8I and J). These results demonstrated that the delayed wound healing and the increased expression levels of IL-1β/NF-κB were positive relatives with let-7f-5p and negative relative with IGF-1 in ischemic wound margins, which was similar to the phenomenon in IL-1β-treated wound margins, suggesting that up-regulated let-7f-5p may also be responsible for the inhibited expression of IGF-1 in ischemic wound model. To test the effect of let-7f-5p on wound healing, antagomir was injected into the wound margins of ischemia mice on days 0, 1, and 2 after surgery (Figure 9A). Figure 9B depicts the wound area 3 days after the excision. When compared with control, wound healing was improved in let-7f-5p antagomir group (Figure 9C). The wounds were stained with H&E on day 3 postexcision (Figure 9D). The length of the neoepithelium was significantly longer in let-7f-5p antagomir group than in control (Figure 9E). In the epidermis of ischemic wound margins treated with let-7f-5p antagomir, IGF-1 expression was increased relative to control (Figures 9F and G), indicating that let-7f-5p antagomir could improve ischemic wound healing by improving IGF-1 expression.
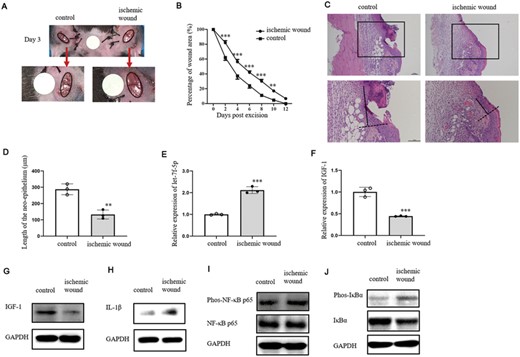
The expression of let-7f-5p was increased in ischemic wound margin. (A) Representative wound healing images on day 3 after excision of ischemic and control wounds. Red arrows indicate that the lower pictures are an enlargement of the upper pictures. Wound edges are marked by black circles. The white discs indicate the initial wound size (diameter = 4 mm). (B) The percentage of wound area on days 0, 2, 4, 6, 8, 10, and 12 postoperation of ischemic and control wounds (n = 10/group). (C–J) Skin biopsies were collected on day 3 postexcision for the following analysis (n = 3–5/group). (C) Re-epithelialization of full-thickness skin wound margin was analyzed by H&E. (D) Length of the neoepithelium of ischemic and control wound margin. (E and F) QRT-PCR analysis of let-7f-5p (E) and IGF-1 (F) expression in the wound margin of ischemic and control groups. (G and H) WB analysis of IGF-1 (G) and IL-1β (H) in the wound margin of ischemic and control groups. (I and J) The expression level of phospho-NF-κB p65 (Ser536), total NF-κB p65 (I), phospho-IκBα (Ser32), and total IκBα (J) were measured by WB. The data represent the mean ± sd of 3 independent experiments. p Values were calculated by one-way ANOVA with Bonferroni posttests (B) or two-tailed Student's unpaired t-test (D, E, and F) (**p < 0.01, ***p < 0.001)
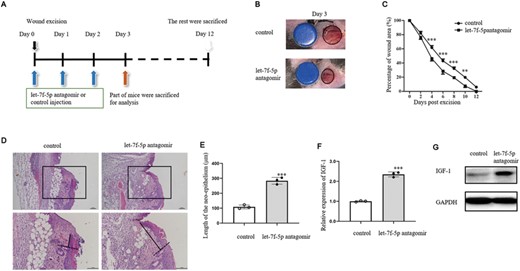
Wound healing in ischemic model was promoted by let-7f-5p antagomir injection in wound margin. (A) Ischemic wounds were treated with let-7f-5p antagomir or control antagomir (5 nM/wound) on days 0, 1, and 2 after wounding. Wound margins for the following analysis were collected on day 3 postwounding (n = 3–5/group). Wound healing was observed until day 12 after excision (n = 10/group). (B) Representative wound healing images on day 3 after excision of ischemic wound treated with let-7f-5p antagomir or control. The blue discs indicate the initial wound size (diameter = 4 mm). (C) Wound closure rates were calculated on days 0, 2, 4, 6, 8, 10, and 12 postwounding. (D) Ischemic wound margins of let-7f-5p antagomir and control was stained with H&E. (E) Length of the neoepithelium in wound margins of let-7f antagomir and control. (F and G) Expression of IGF-1 was analyzed by qRT-PCR (F) and WB (G). The data represent the mean ± sd of 3 independent experiments. p Values were calculated by one-way ANOVA with Bonferroni posttests (C) or two-tailed Student's unpaired t-test (E and F) (*p < 0.05, **p < 0.01, ***p < 0.001)
DISCUSSION
DETCs are the only source of IGF-1 in epidermal tissue to maintain epidermal tissue homeostasis, promote re-epithelialization, and facilitate wound healing.7–9 However, the secretion of IGF-1 by DETCs was damped in the proinflammatory environment such as ischemic wound and diabetic wound, and led to delayed wound healing.10,28,29 Our group found that proinflammatory cytokine IL-1β can activate NF-κB and suppress IGF-1 expression.3 However, how NF-κB suppresses the production of IGF-1 remains unknown. MiRNAs have recently identified as a crucial negative regulator of mRNA expression. IGF-1-specific miRNAs let-7f-5p, miR-1a-3p, and miR-98-5p have been proven to bind to the 3′UTR of IGF-1 mRNA and suppress IGF-1 posttranscriptional expression.17–19 Among these 3 miRNAs, the expression level of let-7f-5p was facilitated in the epidermis of IL-1β stimulated wound margins in vivo or DETCs in vitro compared with controls. When NF-κB was suppressed by BAY, let-7f-5p mimic could directly down-regulate the expression level of IGF-1. When DETCs were activated by IL-1β, let-7f-5p inhibitor could restore IGF-1 expression similarly to the effect of BAY. These data suggested that let-7f-5p may be downstream of IL-1β/NF-κB to regulate IGF-1 expression. We investigated how NF-κB controlled the expression of let-7f-5p. The host genes for let-7f-1 and let-7f-2 are LOC118568094 and HUWE1, respectively. After DETCs triggered by IL-1β, ChIP analysis revealed that NF-κB p65 bound to the anticipated locations on the promoters of LOC8568094 and HUWE1. But the activation of NF-κB by IL-1β seemed to not influence the methylation levels of CpG islands in LOC118568094 and HUWE1. Therefore, NF-κB supported the expression of let-7f-5p in DETCs by promoting transcription of LOC118568094 and HUWE1. To restore IGF-1 secretion in DETCs and promote wound healing, we utilized let-7f-5p antagomir administered at the wound edge of IL-1β-treated and ischemic wounds. Let-7f-5p antagomir was observed to promote IGF-1 expression and re-epithelialization. Based on above results, we regarded that NF-κB signaling pathway, which was triggered by IL-1β during wound healing, dramatically increased the expression of IGF-1-specific let-7f-5p and impeded posttranscriptional expression of IGF-1 mRNA in DETCs.
Except for the negative regulation of miRNAs on IGF-1 expression, Nocturnin and FoxO1 have been reported to inhibit IGF-1 expression. Nocturnin, a deadenylase protein, interacts with the 3′-untranslated region of IGF-1 mRNA to decrease gene expression in mouse bone.30 Furthermore, Foxhead box O1 (Fox O1), a member of Fox family transcription factors, has been demonstrated to suppress IGF1-luciferase reporter activity and to negatively regulate IGF-1 expression in mouse neonatal cardiomyocytes.31 Future research will be required to determine whether Nocturnin and FoxO1 interact with NF-κB and suppress IGF-1 expression in DETCs.
The positive regulation of IGF-1 gene expression has been explored in a variety of cell types from different species, such as human hepatoma cells, bovine hepatocytes, rat osteoblasts, and mouse cardiomyocytes. The results indicate that IGF-1 gene expression is regulated by several transcription factors in a tissue- and developmental stage-specific manner. CCAAT/enhancer binding protein α (C/EBP α), liver-enriched activating protein, hepatocyte nuclear factor 1 α (HNF-1 α), and HNF-3β are shown to activate IGF-I promoter in human hepatoma cell line.32–34 C/EPB-δ has been shown to regulate IGF-1 transcription in rat osteoblasts by binding to an element in the proximal promoter.35 Besides, HNF-3γ is reported to activate IGF-1 promoter and mediate IGF-I gene expression in the bovine liver.36 Foxhead box M1 (Fox M1) binds to IGF-1 promoter in mouse neonatal cardiomyocytes.31 Signal transducer and activator of transcription 5b (Stat 5b) is responsible for driving IGF-1 transcription by binding to enhancers in mouse liver.37–39 However, Stat5-binding domains have not been experimentally verified to interact with any of the IGF-1 promoters. In this article, NF-κB p65 binding to the IGF-1 promoter was enhanced in response to IL-1β stimulation, implying that NF-κB may operate as a negative regulatory transcription factor to compete with the positive regulatory transcription factors for binding domains in IGF-1 gene. In the future, it will be necessary to determine whether and how these positive transcription factors interact with NF-κB in DETCs.
Wound healing is a complex process including 4 phases of hemostasis, inflammation, proliferation, and remodeling. This procedure involves a variety of cell types such as neutrophils, macrophages, mast cells, monocytes, Th1 cells, Th17 cells, and regulatory T cells which also have impacts on wound healing.40–42 Although DETCs are the only T cell subgroup found in the epidermis, Vγ4 T cells residing at the dermis can enter the epidermis after skin injury.3,43 Whether let-7f-5p regulates these cells in wound healing needs to be clarified. MiR-98-5 expression was also increased in IL-1β-treated DETCs, and it is important to figure out what impact miR-98-5p plays in wound healing. IL-1β has been identified as an upstream regulator of inflammasome activity in the wound environment, attracting neutrophils and macrophage to wound margins, and preventing macrophage polarization to an anti-inflammatory phenotype.42 Therefore, IL-1β injected into wound margin in this manuscript may also accelerate inflammation to delay wound healing, apart from impairing IGF-1 production, which needs to be investigated further.
ACKNOWLEDGMENT
This work was supported by National Natural Science Foundation of China (32000645).
AUTHORSHIP
Y. L., J. W., W. H., Y. Z., and Y. L. contributed to conception and design of the study. Y. L., J. W., and Y. W. performed the experiments. Y. L. and J. W. collected data and performed the statistical analysis. Y. L. and J. W. wrote the first draft of the manuscript. Y. W., W. H., Y. Z., and Y. L. wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version. Y. L. and J. W. contributed equally to this work.
DISCLOSURE
The authors declare no conflict of interest.
REFERENCES
Abbreviations
- ChIP
chromatin immunoprecipitation
- DETC
sdendritic epidermal T cells
- H&E
hematoxylin-eosin staining
- IGF-1
insulin-like growth factor-1
- miRNA
microRNA
- MS-PCR
methylation-specific PCR
- qRT-PCR
quantitative real-time polymerase chain reaction
- UTR
untranslation region
- WB
western blot

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}