





Abstract
Communication between the nervous and immune systems serves a key role in host-protective immunity at mucosal barrier sites including the respiratory tract. In these tissues, neuroimmune interactions operate in bidirectional circuits that can sense and respond to mechanical, chemical, and biologic stimuli. Allergen- or helminth-induced products can produce airway inflammation by direct action on nociceptive afferents and adjacent tissues. The activity of nociceptive afferents can regulate innate and adaptive immune responses via neuropeptides and neurotransmitter signaling. This review will summarize recent work investigating the role of neuropeptides CGRP, VIP, neuromedins, substance P, and neurotransmitters dopamine and the B2-adrenoceptor agonists epinepherine/norepinepherine, each of which influence type 2 immunity by instructing mast cell, innate lymphoid cell type 2, dendritic cell, and T cell responses, both in the airway and the draining lymph node. Afferents in the airway also contain receptors for alarmins and cytokines, allowing their activity to be modulated by immune cell secreted products, particularly those secreted by mast cells. Taken together, we propose that further investigation of how immunoregulatory neuropeptides shape respiratory inflammation in experimental systems may reveal novel therapeutic targets for addressing the increasing prevalence of chronic airway disease in humans.
INTRODUCTION
Chronic inflammatory disorders of the upper and lower respiratory tract are among the leading causes of mortality in the world.1 Airway diseases can be genetically inherited, such as cystic fibrosis or acquired at any point during life, as in asthma and allergic rhinitis (AR). Asthma and AR can develop as a consequence of maladaptive responses to innocuous substances, inflammatory diseases, infections, or other sensitizing events, the latter of which generally present with wheezing/coughing, mucus accumulation, itching, sneezing, nasal obstruction, and reversible airflow limitation.2 Although airway inflammation can be controlled with corticosteroids and β2-adrenoceptors agonists in many asthma and AR patients, an increasing percentage of patients do not respond to these treatments for reasons that remain unclear, which contributes to the prevalence of hospitalization and mortality due to respiratory disease.3
In asthma, chronic respiratory inflammation can cause airway hypersensitivity by increasing neural activity, which results in hypercontraction of smooth muscle, constricting small and large airways.4 This hypercontraction along with immune cell infiltration and mucus accumulation in the small airways are features of allergic asthma potentially damaging to the lungs, and sometimes fatal.5 The nature of the antigens that initiate allergic inflammation (fungal spores, pollen, air pollutants) has received considerable attention. It is becoming increasingly clear that inhaled environmental irritants, which could set off asthma attacks, are agonists of receptors found on sensory neurons that innervate nasal passages and upper airways.6–8 Given the importance of neural activity in controlling airway constriction, it is important that we better understand how the allergens, neurons, and immune cells interact in order to improve treatment for patients with intractable disease. Here, we discuss some of the neuromodulators involved in the pathophysiology of allergic airway inflammatory disease.
SENSORY INNERVATION OF THE MUCOSAL EPITHELIUM LINING THE RESPIRATORY TRACT
The airway epithelium forms a barrier to the external environment and the cell populations present in this microenvironment are key for sensing environmental factors and maintaining respiratory homeostasis. The respiratory tract consists of nasal epithelium in the nose/sinus cavity, larynx and trachea (upper airway), and alveoli and bronchioles in the lungs (lower airway) (Fig. 1). The nasal epithelium contains olfactory neurons that transmit odorant information to the brain via the olfactory bulb (Fig. 1, purple).9–12 Each level of the respiratory tract is also innervated by distinct sensory neuron populations with cell bodies in anatomically discrete ganglia. The nasal epithelium is also innervated by the trigeminal ganglia (TG, red),11–14 the upper airway by the superior (jugular) division of the vagal ganglia (green/blue), and the lower airways by the inferior (nodose) division (green/orange) (Fig. 1).15–17 The lower airway (i.e., distal lung) is also innervated by sensory neurons located in thoracic dorsal root ganglia (DRG) of the spinal cord (Fig. 1, inset),16 although the majority of lower airway innervation originates from the nodose ganglia.15
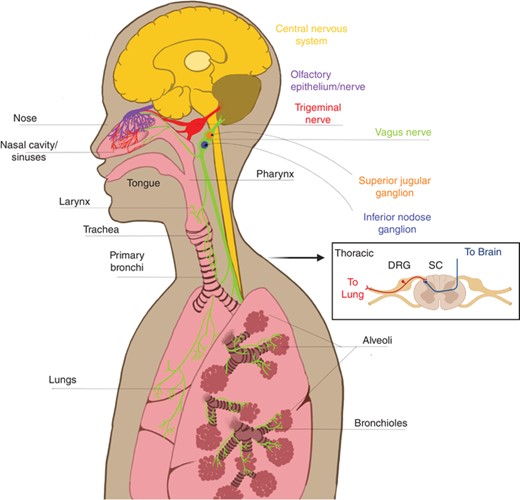
Sensory innervation of the upper and lower airways. The upper airways include the nose, nasal cavity, sinuses, and pharynx. The nasal cavity contains special olfactory neurons that extend axons into the nasal epithelium across the cribiform plate (purple). The nasal cavity and sinuses are also innervated by nociceptors that originate from the trigeminal nerve (red). The back of the nasal cavity and pharynx are also innervated by the superior division (jugular ganglion, orange) of the vagus nerve (green). Lower airways of the lungs, which the larynx and all structures below, are innervated by the inferior division (nodose ganglion, blue) of the vagus nerve and nociceptors originating in thoracic dorsal root ganglia (DRGs, inset). Arrow to inset: (cross-section spinal cord with DRG). Nociceptors have one end of their bifurcated axons in the periphery and the other end in the central nervous system (yellow), with cell bodies in the ganglia
The DRG, TG, and jugular ganglion arise developmentally from the neural crest and have sensory neurons that fall into similar categories based on single cell sequencing data.18,19 In contrast, the nodose ganglia arises developmentally from the epibranchial placodes, and while it does contain some of the same receptors and neuropeptides that will be discussed in detail below, single cell sequencing reveals different expression patterns in distinct populations compared with those found in the neural crest-derived neurons with their cell bodies in DRG/TG/jugular ganglion.18,19 The different contributions among these sensory neuron populations to airway inflammation is poorly understood and constitutes an important area for future study.
Sensory neurons are pseudounipolar, which means they have bifurcated axons, with one end in the peripheral tissue (e.g., respiratory tract) and the other end in the central nervous system (Fig. 1, inset). Airway-innervating neuronal afferents sense and respond to irritating and noxious environmental stimuli by altering their firing activity and secreting neuropeptides and neurotransmitters into the local respiratory environment (Fig. 2A). Firing and secretion into the periphery is commonly referred to as antidromic (Fig. 1, inset “to lung” and Fig. 2A). Antidromic signaling is an important process for neuroimmune crosstalk in the periphery,20,21 as well as actions of sensory afferents on parasympathetic efferent fibers that directly innervate smooth muscle and control airway contraction.4 Voltage-gated sodium channels expressed in neurons produce action potentials, or “firing,” in response to environmental stimuli that act on other channels and lower the local voltage. This action potential can also promote secretion of neuropeptides and neurotransmitters orthodromically to the central nervous system across synapses to other neurons in the spinal cord or brainstem nuclei that control breathing circuits14 (Fig. 1, inset “to brain”). Physiologically, this circuit can adjust breathing rates according to the qualitative nature of local stimuli. This review will focus on the neuroimmune cell interactions in the periphery.
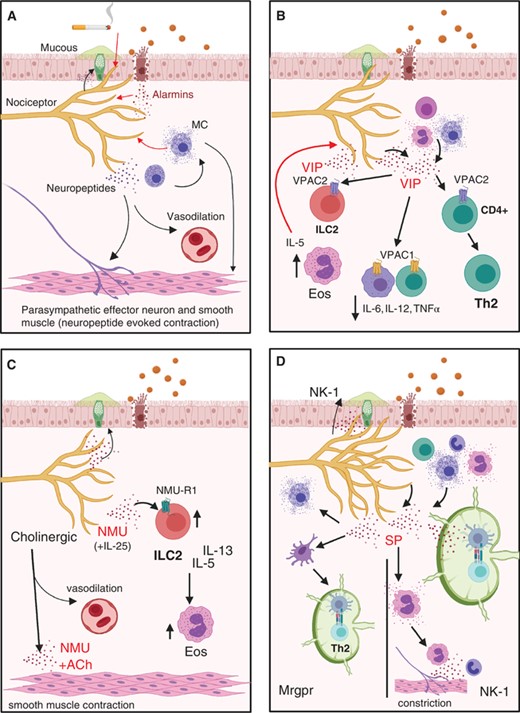
Nociceptors contribute to type 2 inflammation and airway constriction. (A) Nociceptors are able to respond to (red arrows) environmental irritants, alarmins, and mast cell products by secreting neuropeptides that can contribute to airway constriction (black arrows). (B) VIP to VPAC1 signaling suppresses release of proinflammatory cytokines from alveolar macrophages and T cells. VPAC2 signaling on ILC2s contributes to eosinophil (Eos) recruitment and a positive feedback loop of VIP release by nociceptors, as well as Th2 differentiation. (C) NMU-NMU-R1 signaling on ILC2s increases their proliferation and cytokine production. (D) SP+ afferents are highly branched in asthmatic lungs. SP can signal (left) through Mrgpr receptors on mast cells to evoke cytokine release and dendritic cells to evoke migration to the lymph node and antigen presentation instructing TH2 phenotype. SP can also signal (right) through NK-1 to evoke degranulation, recruit cells, and potentiate antigen presentation and T cell survival in the lymph node
AIRWAY NOCICEPTORS DETECT ENVIRONMENTAL IRRITANTS UNDER NORMAL CONDITIONS
All levels of the airway are innervated by neurons that respond to chemical, mechanical, and thermal stimuli.22 Pertinent to airway hypersensitivity and asthma are the chemoreceptor neurons referred to as nociceptors. Nociceptors are small in diameter, with little to no myelin lining their axons, and are often divided into 2 subgroups based on their fiber types: unmyelinated c fibers or lightly myelinated A-δ fibers.12 These fibers express key receptors on their terminal endings, which facilitates activation by specific stimuli and release of neuropeptides, such as the calcitonin gene-related peptide (CGRP).23–25 Two key receptors belong to the Transient Receptor Potential super family of nonselective cation channels, TRP ankyrin 1 (TRPA1) and TRP vanilloid 1 (TRPV1). TRPA1 is activated by a broad range of chemical irritants under normal conditions, including components of cigarette smoke (e.g., acrolein and/or formaldehyde), fungal compounds, and reactive oxygen species8,23 (Fig. 2A). TRPA1 specifically contributes to cigarette smoke-evoked release of CGRP in explant trachea preparations.24 TRPV1 also detects molecular constituents associated with environmental pollution or irritants.6,7 Capsaicin, the active component of chilli pepper, is an agonist for TRPV1 and is used to evoke cough in healthy human subjects and study this reflex.23 TRPA1 exhibits a high degree of overlapping expression with TRPV1, and both TRPA1 and TRPV1 contribute to CGRP release in the trachea evoked by irritating volatile anesthetics.25 Thus, under homeostatic conditions, TRPA1 and TRPV1 contribute to detection of harmful environmental factors that reach the airway epithelium.
AIRWAY NOCICEPTORS CONTRIBUTE TO ALLERGIC INFLAMMATION AND AIRWAY CONSTRICTION
C-fiber afferents bearing TRPV1 and TRPA1 are implicated in promoting the physiologic features of asthma and experimental models of allergic airway inflammation. C-fiber neuronal innervation is much denser in asthmatic patients and these neurons exhibit hyperexcitability.26,27 Further, neuronal TRPV1 expression can be increased following exposure to ozone in the ovalbumin (OVA)-induced murine asthma model27 and TRPV1+ c-fiber ablation alleviates house dust mite (HDM)-induced lung inflammation.28 Genetic deficiency or antagonism of TRPA1 can prevent immune cell infiltration and airway hypercontraction.29 It should be noted that these nociceptor populations express various receptors for innate immune cell mediators like histamine and cysteinyl leukotriene (cys-LT) and treatment with these molecules can also sensitize neurons30,31 and promote enhanced neuropeptide release (Fig. 2A). Preclinical evidence for TRPA1 has led to the development of a small molecule antagonist, GDC-0334, found to be effective at reducing immune cell infiltration due to airway instillation of OVA and reduces neurogenic inflammation in the skin of rodents and humans.32 Of note, there is evidence for TRPA1 expression in human lung fibroblast and smooth muscle cells that could contribute to nociceptor-mediated effects.32
In response to allergens or irritants, nociceptors secrete key neuropeptides such as substance P (SP), CGRP, and vasoactive intestinal polypeptide (VIP), which can selectively activate innate immune cells to induce neurogenic inflammation12,13,26 (Fig. 2). Cells other than nociceptors can also secrete neuropeptides (Figs. 2B, D, and 3B). Pulmonary neuroendocrine cells (PNECs), a rare type of airway epithelial cell reported to drive allergic inflammation, can release CGRP and, potentially, VIP.33–35 While loss of nociceptors can lead to attenuated immune responses, similar phenotypes have been reported when PNECs are absent.35 Moreover, PNECs have also been reported to have the same chemosensory receptors as nociceptive neurons suggesting they may also respond to noxious stimuli in a similar manner.26 SP can also be expressed by immune cells, including mast cells, eosinophils, neutrophils, and T lymphocytes.36 Irrespective of the source, neuropeptides including CGRP, VIP, Neuromedins (NM), and SP have been implicated in neurogenic inflammation. Understanding the specific contributions of these modulators may allow targeting these pathways in the treatment of airway inflammation.
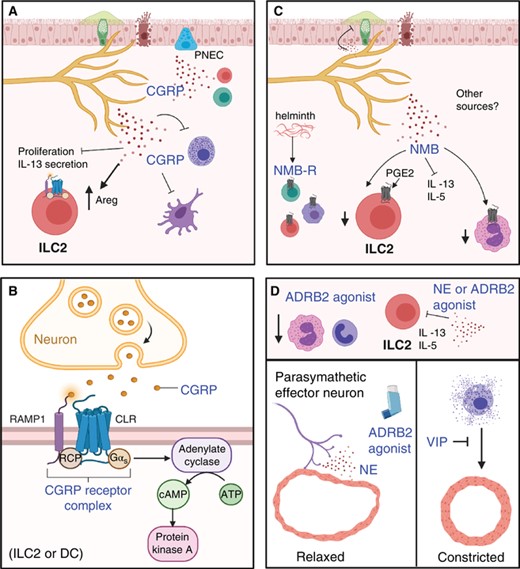
Some neuropeptides and neurotransmitters suppress type 2 inflammation (CGRP and NMB) and counter airway constriction (VIP, NE). (A) CGRP can be released by nociceptors, PNECs, and immune cells. CGRP suppresses type 2 immunity by inhibiting mast cell degranulation, ILC2 proliferation, IL-13 secretion, and DC migration. It also induces ILC2 into a regulatory phenotype (Areg+). (B) Signaling through the CGRP receptors leads to increased cAMP and PKA signaling. (C) The cellular source of NMB is unclear, but helminth infection induced the expression of its receptor (NMBR) in ILC2s, T cells, and alveolar macrophages. NMB suppresses ILC2 type 2 cytokine production, reduces mucous production and eosinophil accumulation. In the presence of PGE2, a suppressor of ILC2 proliferation, the NMB receptor expression is induced. (D) Norepinephrine (NE) and ARDB2 agonists suppress ILC2 production of IL-5 and IL-13. Additionally, both ARDB2 and VIP signaling contribute to relaxation of airways
The sensory neurons at each level of the respiratory tract are positioned to respond to inhaled substances or alarmins (e.g., IL-33, IL-25, and thymic stromal lymphopoietin; TSLP) released by damaged epithelium, which can lead to secretion of neuropeptides or neurotransmitters (Fig. 2A). These can act on several tissue-resident immune cell populations, including dendritic cells (DCs), alveolar and nonalveolar macrophages, mast cells, and innate lymphoid cells (ILCs)37 (Fig. 2). Coordination between the immune and nervous systems can promote defensive symptoms like coughing and sneezing, that involve central signaling of peripheral nociceptors to other neurons in the CNS. This can also mediate mucous secretion from goblet cells and smooth muscle contraction mediated by parasympathetic neurons adjacent to sensory neurons4 (Fig. 2A). Neurons can directly respond to secreted molecules from immune cells that lowers the threshold for action potential generation26 (Fig. 2A). A growing body of evidence has identified an array of secreted molecules including neurotransmitters, neuropeptides, cytokines, and chemokines that mediate bidirectional neuroimmune cell crosstalk. This review focuses on the regulation of airway inflammation by the neuropeptides CGRP, VIP, NMU, NMB, SP, neurotransmitters such as dopamine, norepinephrine, and exogenous ARDB2 agonists. We discuss their implications in Type 2 inflammation of the upper and lower respiratory tract.
CONTRIBUTION OF OLFACTORY INNERVATION TO INFLAMMATION IN THE NASAL EPITHELIUM AND RHINOSINUSITIS
In the nasal tract (Fig. 1, purple), the olfactory neuro-epithelium comprises a small portion of the nasal mucosa and is located in the high nasal vault beneath the cribriform plate.13 Olfactory receptor neurons detect odorants and their axons are found in cranial nerve I and synapse with second order neurons in the olfactory bulb to transmit odorants to the brain.12 The olfactory neuroepithelium has a specialized ability to regenerate due to an established reservoir of basal cells sub-classified into horizontal basal cells (HBCs) and globose basal cells (GBCs).10 HBCs are generally considered to be relatively quiescent, and primarily function to differentiate into GBCs—a multipotent cell type that can give rise to neuronal or nonneuronal cell types in the olfactory epithelium.9–11,38,39 In response to acute mucosal damage, TNF-α drives HBCs toward neuronal differentiation to promote regenerative processes.11 In contrast, chronic inflammation and excessive TNF-α secretion impairs the regenerative function of HBCs. Under high TNFα conditions, HBCs maintain their precursor phenotype and contribute to inflammatory signaling by secretion of chemokines that recruit macrophages such as C-C motif Ligands 19 and 20 (CCL19, CCL20). In this scenario, the HBCs no longer give rise to mature olfactory neurons, which likely contributes to the loss of smell and the persistent inflammation associated with chronic rhinosinusitis.40
NOCICEPTOR INVOLVEMENT IN TYPE 2 INFLAMMATION IN THE AIRWAYS AND LYMPH NODE
Type 2 inflammatory responses are widely thought to be an evolutionary adaptation to expel parasitic helminths and/or repair the tissue damage caused by their infection.41 Maladaptive inflammation due to protease-mediated damage to epithelia may explain how some allergic responses are initiated. Type 2 inflammatory responses are driven by eosinophils, mast cells, ILC2, TH2, M2 cells, combined with the release of IL-4, 5, 9 and 13, 25, 33, and TSLP in response to allergic antigen exposure and parasitic helminth infections42–44 (Fig. 2A). ILC2s express the Th2-associated transcription factor GATA3 and induce robust secretion of IL-5 and IL-13 at mucosal surfaces following helminth infection or allergic disease.42–44 Sensory neurons in the skin can express diverse cytokine receptors including those for IL-4, IL-5, IL-33, and TSLP,45–48 indicating that their activity can be modulated by type 2 cytokines. Nociceptors are in close proximity to goblet cells and can increased mucus production, which further exacerbates Type 2 inflammation due to narrowing of the airways (Fig. 2A).
Epithelial breakdown releases factors that can activate DCs for migration to local lymph nodes where they can present helminth- or allergen-derived antigens to naïve T helper cells and drive their polarization into a Th2 phenotype (Fig. 2D). Within the lymph node, nodose-derived nociceptors, marked by the expression of the voltage gated sodium channel 1.8 (Nav1.8), innervate the portal and vascular regions of the popliteal and inguinal lymph nodes. The density of these nociceptors could be increased by agonists for TLR4 and the TLR1/2 heterodimer.49 Optogenetic stimulation of lymph node nociceptors could evoke gene expression changes in a number of lymphoid cell types including LEC2s, NECs, neutrophils, and NK cells.49 Based on their findings, the authors suggest that nociceptors may contribute to trafficking of antigen presenting cells into and out of the lymph node.49 Previous studies indicated that neuropeptide-bearing afferents expressing CGRP or SP could be localized to airway-associated lymph nodes, and that capsaicin increased trafficking of FITC-dextran from the lung mucosal tissues to the draining lymph node.50,51 Further study of neuronal innervation of airway-associated lymph nodes is needed under normal and pathologic conditions to fully understand the role of nociceptors in antigen presentation and T cell differentiation.
After Th2 cells exit the lymph node to return to the site of damage, they secrete type 2 cytokines that, along with other chemokinetic mediators, recruit and activate a number of innate immune cells (mast cells, macrophages, neutrophils, and eosinophils), promoting inflammation. Mast cells are associated with allergic airway inflammation and constriction via their interactions with nociceptors, parasympathetic effectors, and smooth muscle cells (Figs. 2A, B, D, and 3D). Notably, mast cells expressing histamine and IL-4 have been reported to be enriched in the airways of asthmatic patients.52 As noted above, nociceptors have receptors for both of these factors.30,46 In addition to histamine, mast cells secrete a number of soluble mediators that can directly stimulate smooth muscle contraction and airway remodeling including cys-LT and serotonin. Moreover, mast cell-derived serotonin was proposed to activate neural circuits implicated in antigen-mediated bronchoconstriction.52 Mast cell-derived neurotrophic factors are overexpressed in the lungs of infants infected with respiratory syncytial virus,53 suggesting that mast cells may promote airway inflammation by releasing neuromodulators that directly act on lung afferents (Fig. 2A). The following will highlight some of the mechanisms whereby immune cells involved in type 2 immunity can be modulated by neuropeptides (Figs. 2 and 3).
NEUROPEPTIDES AS IMMUNOREGULATORS
CGRP as an anti-inflammatory neuropeptide
CGRP is a neuropeptide found in both the central and peripheral nervous systems. Originating from the Calca gene, which encodes procalcitonin or CGRP depending on splicing. CGRP exists in 2 isoforms: alpha-CGRP (CGRP 1) and beta-CGRP (CGRP 2).54 Particularly, α-CGRP a 37-amino acid peptide has been recently recognized for its neuromodulator effects.54 This neuropeptide acts as a ligand of a heteromeric receptor complex comprised of the G Protein-Coupled Receptor (GPCR) transmembrane receptor Calcitonin Receptor-Like Receptor and the accessory protein Receptor Activity Modifying Protein 1, which confers specificity for CGRP and is rate limiting for trafficking of the receptor to the plasma membrane55 (Fig. 3B). Binding of CGRP to its receptor leads to phosphorylation and internalization of the receptor, which can decrease the cellular ability to respond to CGRP autocrine and paracrine signaling.54
C-fibers nociceptor neurons in particular are the primary source of CGRP and expression exists along the continuous length of these sensory neurons.56 Increases in CGRP+ neurons have been observed in response to airway inflammation.57,58 There are also other cellular sources of CGRP, including PNECs,35 and potentially ILC2s and T cells59–61 (Fig. 3A). The significance of these multiple sources of CGRP to airway inflammation is not entirely clear, but the presence of CGRP receptor on many immune cell types including mast cells, ILC2s, T and B cells, macrophages, and DCs26 indicates that CGRP may produce both autocrine and paracrine effects important to the regulation of local immune responses. PNECs are disproportionately elevated in asthma patients62 and are densely innervated by neurons and found closely localized with airway ILC2s,42 suggesting that these cells maybe a relevant contributor to the modulatory effects of CGRP (Fig. 3A).
Recent studies into airway hypersensitivity have mostly revealed CGRP to possess anti-inflammatory activity. CGRP stimulation of ILC2s was shown to induce a unique transcriptional profile and reversed some gene expression profiles normally mediated by proinflammatory mediators such as NM U, IL33, and IL25.58,60 Further, CGRP treatment resulted in reduced eosinophil responses following helminth infection or allergic challenge, suggesting that CGRP inhibits type 2 inflammation by targeting ILC2s58,60 (Fig. 3A). Evidence also shows that CGRP can inhibit DC responses in the lung, suggesting that CGRP can limit type 2 inflammation by acting on different cellular targets.56,63 Moreover, CGRP can promote wound healing in the lungs by increasing ILC2 expression of AREG.56 All together, these studies suggest a role for CGRP to suppress type 2 inflammation and promote wound healing (Fig. 3A).
In contrast, OVA-induced allergic inflammation combined with ozone exposure exacerbated airway hypersensitivity and inflammation, which was associated with elevated CGRP levels.27 Critically, this synergistic activity between OVA and ozone was reversed when TRPV1 neurons were inhibited with capsazepine, which was associated with reduced CGRP and TSLP content in lung tissue.27 This study did not examine any other neuropeptides, such as SP or NMU (Figs. 2C and D), which can have opposing effects with CGRP (Fig. 3A). CGRP can also trigger mast cell degranulation and CGRP+ neurons are reported to be localized with mast cells, which could further enhance allergic inflammation due to neuron sensitization56,60 (Fig. 2A). Although CGRP has been proposed as a potential therapeutic in exacerbated airway inflammation,26 a better understanding of its release mechanisms and some clarity regarding conflicting reports of its effects on type 2 immunity will be imperative if CGRP treatment is to become a viable target for clinical treatment of inflammatory diseases.
VIP has inflammatory and suppressive effects on immunity
VIP is a small 28-amino acid neuropeptide that is one of the most abundant, with primary expression in the gut and lung.42 The primary source of VIP is reported to be Nav1.8+ nociceptors in the central and peripheral nervous systems.33 VIP+ neurons, like CGRP+ neurons, were found juxtaposed to immune cells in the periphery suggesting their ability to influence immune cell functions. This neuropeptide is expressed in immune cells such as lymphocytes, eosinophils, and mast cells.33 VIP acts as a promiscuous ligand for a number of receptors that are members of the GPCR family of transmembrane proteins including Vasoactive Intestinal Peptide Receptors 1 and 2 (VPAC1, VPAC2), Chemoattractant Receptor Homologous Molecule Expressed on Th2 cells (CRTH2), and the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor 1 (PAC1), the latter receptor named for its higher affinity neuropeptide ligand PACAP.33
At mucosal barrier sites, VPAC1 is enriched in the lungs, whereas VPAC2 is expressed predominantly in the stomach and intestines.33 Further, VPAC1 is expressed constitutively on macrophages, especially in tissue-resident alveolar macrophages, and T cells, whereas VPAC2 expression can be induced upon activation33,64 (Fig. 2B). Eosinophils, mast cells, basophils, and Th2 cells can also express the VIP receptor CRTH2 and reports have found that blocking this receptor reduces airway hypersensitivity in animal models of allergic inflammation.33 CRTH2 receptor was found to be elevated on eosinophils in AR patients,33 emphasizing the therapeutic potential of targeting this pathway in allergic disease. VIP may work in a positive feedback loop to increase the expression of VIP receptors such as CRTH2.33 Nonetheless, CRTH2 is also the receptor for prostaglandin D265 therefore the biologic nuances between cellular sources of prostaglandin D2 and VIP in airway hypersensitivity deserves further investigation.
VIP was originally characterized by its ability to promote intestinal blood flow through vasodilating activity. In addition, VIP regulates contractility and heart rate of cardiac muscle, bronchodilation, and mediates anti-inflammatory effects.66 Notably, VIP antagonizes the constrictor effect of mast cell effector products such as histamine, prostaglandin F2a, and leukotrienes66 (Fig. 3D). Increased expression of VIP and its receptors was reported in the context of mast cell inflammation.67 However, the extent to which VIP regulates mast cell responses needs to be further evaluated. VIP interaction with its GPCR receptors stimulates phospholipase C (PKC), MAPK, and PKA-adenylate cyclase pathways that increase intracellular cAMP.
VIP may be a relevant target for both the upper and lower respiratory tract in patients with pulmonary hypersensitivity and allergic asthmatic responses. In direct contrast to CGRP, asthmatic patients have decreased VIP levels as opposed to unaffected participants.33 Moreover, patients with AR have significantly elevated expression of VPAC receptors compared with healthy individuals.68 VIP+ adrenergic fibers innervate bronchial and tracheal smooth muscle possibly explaining its ability to modulate airway hypersensitivity.33 Moreover, VIP+ nerve fiber and VPAC mRNA expression have been identified in airway mucosal glands66 indicating that VIP may play a role in regulating airway mucus production (Fig. 2B). However, the definitive role of VIP in mucus secretion has been controversial since results vary depending on the species of model organism and the markers being assessed for validation.66
In the context of type 2 inflammation, TCR stimulation can up-regulate VPAC2 expression contributing to Th2 polarization of CD4+ T cells64 (Fig. 2B). Further, the VPAC2 receptor is also highly expressed on ILC2s from lung as compared with from intestine64 (Fig. 2B). It has been shown that VIP enhances the production of IL-5 and eosinophil recruitment through ILC2 effector function specifically by signaling through VPAC2 receptor47,64(Fig. 2B). Critically, IL-5 was found to provoke sensory neuron release of VIP, suggesting a positive feedback loop that promoting airway inflammation47 (Fig. 2B). These findings suggest that VIP may be an important driver of lung inflammation and potential therapeutic target.
NMs regulate type 2 responses
NMU, from the Nmu gene on chromosome 4, originates as a large precursor which is cleaved into 2 biologically active forms, NmU-25 (which is 25 amino acids in size) and NmU-8 (which is 8 amino acids in size).69 Like many of the neuromodulators discussed here, it is highly conserved amongst a variety of mammalian species.69 NMU is highly expressed in cholinergic neurons,70–72 as well as in the dorsal horn of the spinal cord and somatosensory cortex, suggesting NMU is involved in sensory perception.73 NMU signals through GPCR family proteins NMU-R1 and NMU-R2, on chromosomes 2 and 5 respectively, with the former being more widely expressed in the periphery and the latter being restricted to the CNS.69 NMU receptors seem to be expressed on immune cells involved in type 2 inflammation. For example, only ILC2s express the NMU-R1 receptor as opposed to T cells or group 1 or group 3 ILCs70–72,74 (Fig. 2C). Further, mast cells and eosinophils also express this receptor and may be subjected to NMU modulation of immune responses74 (Fig. 2C). How eosinophils biologically respond to NMU remains unclear.
NMU interaction with mast cells leads to degranulation releasing early inflammatory mediators that contribute to smooth muscle contraction and vasodilation74 (Fig. 2A). Further, effector ILC2s, IL13+ and IL5+, expanded in the presence of NMU and cultured ILC2s from the lung responded synergistically to NMU and IL25 in producing effector cytokines IL5 and IL1370 (Fig. 2C). This suggests the cooperative function of pro-type 2 cytokine pathways and nociceptive neuropeptides. NMU activation of ILC2s promoted the recruitment of eosinophils and mucus production in the context of allergic airway inflammation and helminth infection71,72,74 (Fig. 2C). In contrast to its impact on the lower airway how NMU contributes to inflammation in the nasal cavity in the context of AR remains unknown.
Similar to NMU, a recent study identified NMB, another member of the neuromedin family of neuropeptides, as a potent regular of airway inflammation.75 NMB is a decapeptide that binds to the GCPR family member NMBR, which was found to be induced in ILC2s, T cells, and alveolar macrophages following helminth infection.75 Contrary to NMU, treatment with NMB suppressed IL-5 and IL-13 secretion by ILC2s upon helminth infection75 (Fig. 3C). Consequently, NMB administration resulted in reduced eosinophil recruitment and mucus production in the lungs of helminth-infected mice75 (Fig. 3C). While NMB was sufficient to directly suppress ILC2 secretion of type 2 cytokines, it did not alter their proliferation, suggesting that NMB may target specific ILC2 functions75 (Fig. 3C). Critically, PGE2, a known suppressor of ILC2 proliferation,76 was found to promote NMBR expression in ILC2s,75 suggesting a model in which PGE2 limits ILC2 proliferation and promotes their ability to respond to NMB-induced suppression of cytokine secretion (Fig. 3C). Despite the potent regulatory effects of NMB to regulate ILC2 functions, the effects of this neuropeptide on other lung-resident immune cells remain to be elucidated. Further, whether lung-innervating neurons are the cellular source of NMB and whether they release it upon airway inflammation is unknown (Fig. 3C). Nonetheless, these studies highlight neuromedins as potential pharmacologic targets for the treatment of united airway diseases.
β2-Adrenergic receptors are classical targets in asthma treatments
β-Adrenergic receptors are GPCR transmembrane protein receptors that include β2-adrenoreceptors (ARDB2), primarily expressed in the airway smooth muscle77 and on a variety of immune cell types involved in allergic responses such as Th2, ILC2, and eosinophils78 (Fig. 3). ARDB2 is the receptor for epinephrine/norepinephrine released by sympathetic nerves42 (Fig. 2A). Synthetic agonists that target ARDB2 have been used historically to treat asthmatic symptoms due to their ability to induce relaxation of the airway smooth muscle77,78 (Fig. 3D). Importantly, PET scans have shown that children with asthmatic symptoms have a reduction in ARDB2 expressing cells.77 Further, tissue from patients who died from asthmatic complications have shown reductions in their ability to respond to bronchodilation when stimulated with β2-agonists.78
Engagement of ARDB2 has been shown to reduce neutrophils and eosinophils in mucosal tissues and blood from asthmatic patients, suggesting that this receptor contributes to anti-inflammatory functions78 (Fig. 3D). In an acute lung injury mouse model, β2-agonists reduce the expression of proinflammatory chemokines, cytokines, and neutrophils in lung tissue.79 Type 2 cytokines such as IL-13 can impair the cAMP generation that normally follows β2-agonist treatment of human epithelial cells, suggesting that proinflammatory signals contradict the functions of ARDB2.78 Importantly, ARDB2 knockout mice exhibit exacerbated inflammation following helminth infection, LPS treatment and respiratory viral infection, associated with increased susceptibility and exacerbated lung pathology.80,81 Further, ARDB2-deficient mice, present with increased ILC2 responses and lower intestinal worm burdens following helminth infection suggesting that ADRB2 activation suppress ILC2 activation.81 In support of this possibility, helminth-infected mice treated with the β2-agonist clenbuterol presented with reduced ILC2 proliferation.81
Despite the availability and proven function of ARDB2 agonists in asthma relief, the endogenous activation of these pathways in the context of airway inflammation is less defined. In one of the few studies that focused on the endogenous ARDB2 ligand norepinephrine, it was found that norepinephrine mitigates the immune response by lowering the transcript levels of inflammatory cytokines and chemokines in alveolar macrophages and ILC2s80 (Fig. 3D). In addition to the anti-inflammatory effects of ARDB2 engagement, contradictions in the literature describing β2-adrenoceptor signaling in immune cells seems to be dependent on the context of stimulation.82 That is, which agonist is being used, the differentiation state of the immune cell population, and what type of airway hypersensitivity model is being assessed. If ARDB2 is an important player in neuroimmune crosstalk, there should be further investigation into naturally expressed ARDB2 ligands and how they act in comparison with available β2-agonists.
Repeated exposure to ARDB2 agonists can lead to desensitization, which is an autoregulatory process wherein β2-adrenoceptor signaling is attenuated to protect against overstimulation.77 This process is thought to contribute greatly to poor clinical outcomes in long-term β2-agonist treatment in asthmatic patients. Desensitization occurs through phosphorylation of ARDB2 by proximal kinases.77 In respiratory viral infection, evidence suggests that desensitization of β2-adrenoreceptor leads to reduced broncho protection by β2-agonists in asthma patients.78 In the face of a global pandemic caused by a respiratory virus in which excessive inflammation leads to deleterious and sometimes fatal pulmonary damage, understanding how anti-inflammatory mediators such as ARDB2 agonists function in viral immunity is imperative not only for patients at risk for comorbidities in asthma but also for those patients who experience exacerbated immune responses to the novel SARS-CoV2 virus. It is possible that asthmatic patients who have been on β2-agonist treatment for long periods of time may be at greater risks for COVID-19-induced comorbidities.
Dopamine and early life allergic airway inflammation
Allergic asthma may develop at any age, but it is appreciated that children are more prone to this condition than adults.83 This may be due in part to greater dopaminergic innervation of young lung than adrenergic innervation, observed in both mouse and human tissue.84 Recent work indicates that dopamine released by sympathetic nerves in the young lungs acts on dopamine receptor D4 (DRD4) on CD4+ T cells to induce a Th2 phenotype.84 This DRD4-mediated mechanism was not effective in the adult lung.84 Given that adrenergic innervation was lacking in lungs of young mice, this may suggest that certain pediatric patient populations may fail to respond to ARDB2 blockers, and perhaps additional work regarding dopamine in early life could point to improved therapies for young patients.
Substance P mediated neurogenic inflammation
Substance P (SP) is a 10–12 amino acid neuropeptide present in both the central and peripheral nervous system.36 SP is a member of the tachykinin family encoded by the TAC1 gene in humans.36,85 While SP can signal through all 3 neurokinin receptors (NK-R1,2,3), which are also GPCR family members, SP is reported to be the preferred ligand of NK-R1 when acting on neurons.36,85,86 More recent work indicates that SP may interact with noncanonical receptors expressed by immune or other nonneuronal cell populations in proximity to neurons secreting SP. These receptors are members of the Mas-Related GPCR (MRGPR) family, which have been broadly implicated in mediating itch under normal and pathologic conditions and can be found on neuronal and nonneuronal cells.87,88 In particular, mast cells may utilize MrgprB2, or DCs may use MrgprA1 (in murine models)/MrgprX2 (in human tissues) to respond to SP released by adjacent afferents.87,88 We will first discuss canonical SP–NK1 receptor signaling in the context of allergic airway inflammation, then cover noncanonical SP–MRGPR signaling.
Canonical SP–NK1 receptor signaling in immunity
SP is synthesized and released by nociceptive nerve fibers in response to noxious stimuli, immune cell-mediated activation, and common allergens.36,86 Primary afferent nociceptive neurons that synthesize SP lie in close proximity to neighboring immune cells forming intricate clusters suggested to contribute to modulation of disease pathologies.86,89 However, SP and the NK receptors are also synthesized and expressed by immune cells, including those involved in allergic airway hypersensitivity such as mast cells, eosinophils, neutrophils, and T lymphocytes36 (Fig. 2D, right). SP and the NK-R1 receptor regulate T cell activation and survival.90 In particular, the NK-R1 receptor in T lymphocytes is suggested to promote cognate TCR activation by facilitating intracellular Ca2+ flux and downstream activation of key transcription factors (NFAT1, NFAT2, and NFkb-p65) required for synthesis and secretion of IL-2, which enhances T cell survival.90 Cognate T cell activation in the lymph nodes is classically driven by antigen presenting DCs. In the context of a triggered immune response, SP expression at the protein level was identified at the site of antigen-presenting DC-T cell synapse during cognate activation90 (Fig. 2D, right). Such evidence provides novel insight for T cells as an endogenous source of SP secretion and signaling during immune responses.
In the peripheral nervous system, SP is localized to primary afferent sensory nerve fibers and released in response to peripheral inflammation and noxious stimuli.91 Synergistic crosstalk between the nervous and immune system is proposed to regulate SP production and peripheral inflammatory responses36 (Fig. 2A). SP can induce degranulation of eosinophils and the release of reactive oxygen species, as well as activate neutrophils by triggering superoxide production36 (Fig. 2D, right). SP signaling through the NK-1 receptor regulates airway smooth muscle constriction and mucous secretion2 (Fig. 2D, right). Importantly, SP and the NK1 receptor expression are increased in patients with asthma, and positively correlate with airway obstruction.2,92 Bronchopulmonary C fibers that synthesize SP are required to produce neurogenic inflammation, airway constriction, and airway remodeling that characterizes allergic airway inflammation28 (Fig. 2A). In particular, in an allergic-like asthma model induced by HDM exposure, elevated levels of pulmonary SP are detected, however denervation of bronchopulmonary C fibers profoundly reduces evoked AHR, correlating with a significant reduction in pulmonary SP.28
In addition to modulating immune cell function, SP also asserts chemotactic properties that have been implicated in eosinophil recruitment to the lung93 (Fig. 2D, right). Interestingly, recent evidence has provided a new perspective for the contribution of eosinophils in altering neural circuitry and substance P expression in the lung.94 Type 2 high asthmatic patients with increased blood eosinophil levels display longer airway nerves and increased nerve branch points compared with control airways of type-2 low asthmatics94 (Fig. 2D). Moreover, moderate to persistent asthmatics display increased nerve length in addition to increased levels of SP expression.94 In one study, mice deficient in eosinophils show a reduction in airway hyperresponsiveness and do not display increased nerve length and branch point projections compared with their eosinophil-sufficient counterparts.94 Such evidence of the structural remodeling of sensory nerves facilitated by SP and eosinophils offers additional insight into neuromodulation events experienced by type 2 asthmatic patients. However, the mechanisms by which eosinophils and SP can effectively modify neural circuitry remains unclear.
Noncanonical signaling of SP via the MRGPR family members on mast and DCs
Connective tissue MCs in skin were found to respond to SP through the MRGPRX2 receptor in humans, and Mrgprb2 in mice, over the classic NK-R1.95,96 In a HDM-induced type 2 atopic dermatitis model, TRPV1+ nociceptor activation triggered the release of neuronal derived SP that in turn stimulates MC activation through the Mrgprb2 receptor to drive skin inflammation.86 Similarly, recent work indicates that intradermal treatment of the cysteine protease papain evokes the release of SP, and suppresses CGRP release.88 SP released by TRPV1+ afferents induced migration of CD301b+ DCs bearing the Mrgpra1 receptor to the draining lymph nodes to instruct T cells into a TH2 phenotype88 (Fig. 2D, left). It was unclear why or how CGRP release was suppressed, given that TRPV1+ neurons are capable of releasing both neuropeptides. This may be an important avenue of future study given CGRP's suppressive effects on type 2 immunity. Both neuropeptides can be released by TRPV1+ afferents, so some additional cellular markers are likely to play a role. Furthermore, in vitro stimulation of a human mast cell line with SP triggers the release of proinflammatory cytokines and chemokines (Fig. 2D, right) and this response is effectively ablated by siRNA targeted knockdown of MRGPRX2 receptor.95
These experimental lines of evidence supporting SP-Mrgpr signaling in mast or DCs to contribute to type 2 immunity are at the skin barrier. Recent evidence has confirmed MRGPRX2 expression in lung biopsies of both nonasthmatic and asthmatic patients, with increased levels in asthmatic patients,96 supporting the idea that this signaling may contribute to type 2 immunity in the allergic airway. Therefore, the direct implications of SP-induced activation of the MRGPRX2/b2 receptor and its contribution to asthma will be important to understand in the context of allergic airway disease. While the clinical effectiveness of these antagonists can be appreciated in animal models of allergic asthma, the inability to translate such outcomes in humans, given species differences in Mrgpr orthologs,87 has generated considerable discussion for the different mechanisms by which SP can influence inflammatory responses.97
CONCLUSION
There is a particular dearth of research on sensory neuron function(s) in the upper airways during AR. Although the innervation and architecture of neurons in the nasal epithelium is well-documented, whether these neurons respond to allergenic substances directly and/or soluble inflammatory mediators requires further investigation. Currently, the tools we have to treat asthma/AR (e.g., beta 2 agonists) are not effective in all patients. In those patients where these drugs are effective, they treat the constrictive and mucous production symptoms of asthma that arise downstream of triggering events, which likely require lung sensory neurons. A better understanding of the stimuli and pathways underlying these cascades could lead to the development of treatments designed to block the initiation of type 2 responses that drive the bronchoconstriction and mucus accumulation. In this review, we have highlighted the ability of neuropeptides to modulate type 2 inflammation that drives airway diseases. The neuropeptides CGRP and NMB, as well as ARDB2 agonists (nor)epinephrine, attenuate type 2 inflammation, whereas VIP, NMU, and SP promote these responses. Many of these neuropeptides originate from the same class of C fiber nociceptors, as well as other cells residing in the airway mucosal epithelium. Understanding how neurons regulate the expression of pro- and anti-inflammatory mediators will be an important aspect of future study.
AUTHORSHIP
E.E.J. wrote this manuscript and designed the anatomical illustration in Fig. 1 and initial concepts of the other figures, which were modified by H.R. following peer review. O.G., J.I.R., H.R., and D.R.H. wrote and revised this manuscript.
ACKNOWLEDGMENTS
NIH grants (AI095289, GM083204, UO1AI125940) and the Burroughs Wellcome Fund supported this work. We used Biorender to generate the graphical abstract, Fig. 1 inset, and Figs. 2 and 3. The original anatomical illustration in Fig. 1 was drawn by EEJ using Procreate and edited by HR using Adobe Illustrator Draw, then imported into a Biorender layout.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
Abbreviations
- AR
allergic rhinitis
- ARDB2
beta-2 adrenergic receptor
- CGRP
Calcitonin gene-related protein
- CRTH2
chemoattractant receptor-homologous Molecule expressed on Th2 cells
- DC
dendritic cell
- GPCR
G protein-coupled receptor
- HDM
house dust mite
- ILC2
Innate lymphoid cell group II
- MRGPR
Mas-related G protein receptor
- Nav1.8
voltage-gated sodium channel 1.8
- NK-R1
neurokinin receptor 1
- NMB
neuromedin B
- NMBR
neuromedin B receptor
- NMU
neuromedin U
- NMU-R1
neuromedin U receptor 1
- OVA
ovalbumin
- PAC1
pituitary adenylate cyclase-activating polypeptide type I receptor
- PACAP
pituitary adenylate cyclase-activating polypeptide
- PET
positron emission tomography
- PNEC
pulmonary neuroendocrine cells
- SP
substance P
- TRP
transient receptor potential channel
- TRPA1
transient receptor potential channel ankyrin member 1
- TRPV1
transient receptor potential channel vanilloid member 1
- TSLP
thymic stromal lymphopoietin
- VIP
vasoactive intestinal peptide
- VPAC1/2
vasoactive intestinal peptide receptor 1 and 2.
Author notes
Summary sentence: Review of how neural afferents initiate immune responses via neuropeptides, and resolve inflammation and airway constriction via CGRP, NMB, VIP, and ARDB2 signaling.

{kind=link}
{kind=link}
{kind=link}