





Abstract
Fas ligand (FasL) is best known for its ability to induce cell death in a wide range of Fas-expressing targets and to limit inflammation in immunoprivileged sites such as the eye. In addition, the ability of FasL to induce a much more extensive list of outcomes is being increasingly explored and accepted. These outcomes include the induction of proinflammatory cytokine production, T cell activation, and cell motility. However, the distinct and opposing functions of membrane-associated FasL (mFasL) and the C-terminal soluble FasL fragment (sFasL) released by metalloproteinase cleavage is less well documented and understood. Both mFasL and sFasL can form trimers that engage the trimeric Fas receptor, but only mFasL can form a multimeric complex in lipid rafts to trigger apoptosis and inflammation. By contrast, a number of reports have now documented the anti-apoptotic and anti-inflammatory activity of sFasL, pointing to a critical regulatory function of the soluble molecule. The immunomodulatory activity of FasL is particularly evident in ocular pathology where elimination of the metalloproteinase cleavage site and the ensuing increased expression of mFasL can severely exacerbate the extent of inflammation and cell death. By contrast, both homeostatic and increased expression of sFasL can limit inflammation and cell death. The mechanism(s) responsible for the protective activity of sFasL are discussed but remain controversial. Nevertheless, it will be important to consider therapeutic applications of sFasL for the treatment of ocular diseases such as glaucoma.
INTRODUCTION
Apo-1/Fas/CD95 was initially identified as a death receptor over 30 yr ago when a monoclonal antibody raised against a cell surface protein of a human B lymphoblast cell line was shown to kill a variety of malignant lymphocytes both in vitro and in vivo.1 A monoclonal antibody specific for the same antigen, raised by immunization with the human FS-7 fibroblast cell line, was also shown to be cytolytic for a variety of cell types, including T cells, myeloid cells, and fibroblasts.2 The natural ligand (Fas ligand [FasL]/CD95L) was subsequently identified in 1993,3 and Fas/FasL interactions were further implicated in the maintenance of self-tolerance when an inactivating retrotransposon insertion into the Fas gene and a loss of function point mutation in the FasL gene, respectively, were shown to be the cause of the autoimmune lymphoproliferative disease associated with the lpr and gld mutations.4–7 FasL is mainly expressed by cytotoxic effector cells although expression by macrophages, dendritic cells, inflamed endothelial cells, and even microglial cells and astrocytes have been described.8–13 Apart from its apoptotic function, FasL has also been shown to induce the production of numerous proinflammatory cytokines and to play a key role in T cell activation and differentiation.14–17 Although FasL is clearly important for host defense, excessive expression of FasL can have severe pathologic consequences.18–21 It follows that a number of mechanisms are involved in the rapid and effective modulation of FasL activity. As described in the following text, cleavage of the FasL extracellular domain by metalloproteases can severely limit FasL function and likely serves as a critical mode of regulation, especially in the eye.
FASL STRUCTURE, CLEAVAGE, AND THE REGULATION OF FASL ACTIVITY
FasL structure
FasL is a type II transmembrane protein that consists of an extended N-terminal cytoplasmic domain, a transmembrane domain, a stalk region, a homotrimerization or self-assembly domain, and a C-terminal Fas binding domain, as depicted in Figure 1. The cytoplasmic region includes a casein kinase I (CKI) phosphorylation site, also present in other TNF family members, that is associated with an increase in cytosolic Ca++ levels upon phosphorylation.22 It also contains an extended proline-rich region (PRR), unique to FasL, which interacts with a variety of proteins containing SH3 or WW domains.23 PRR binding proteins serve a range of functions that include the trafficking of FasL to secretory lysosomes where FasL can be retained until further activation signals direct FasL exportation to the plasma membrane.24 Sequestration of FasL in secretory lysosomes is an efficient mechanism for priming effector T cells and NK cells while limiting their cytotoxic activity.
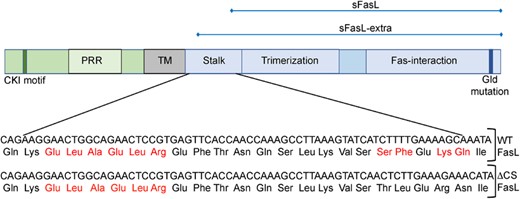
Murine FasL protein structure. The cytoplasmic, transmembrane, and extracellular domains are depicted in green, gray, and blue, respectively. The location of the casein kinase I motif (CKI), proline-rich region (PRR), and the inactivating gld mutation are indicated, as are the stalk, trimerization, and Fas-interaction regions of the extracellular domain. Potential cleavage sites are indicated by the red font in the expanded stalk region and the inactivating mutations found in ΔCS mice are indicated in the lower panel in blue highlight. Regions corresponding to the soluble fragments soluble FasL fragment (sFasL) and sFasL-extra are shown at the top of the figure
The stalk region of both human and mouse membrane-associated FasL (mFasL) can be cleaved (shed) by metalloproteinases to release a soluble FasL fragment (sFasL). The cleaved fragment can still form FasL trimers but has little to no capacity to induce apoptosis or inflammation,25–28 functions that depend on multimerization of mFasL.29,30 Moreover, a number of studies have shown that sFasL can actively block the activity of mFasL,31,32 either by sterically interfering with the ability of mFasL to bind Fas,27 or by activating a distinct pro-survival program.33–35 Cleavage is therefore a rapid mechanism for limiting and potentially reversing the damage caused by FasL engagement of Fas+ target tissues.
FasL cleavage
The murine FasL cleavage site was initially identified as murine Ser124/Leu125 and/or Lys127/Gln128 based on N-terminal amino acid sequences of the naturally cleaved product.27,28 MMP3 and MMP7 were subsequently implicated in this process, based on their in vitro capacity to cleave FasL from the surface of a mouse thymoma cell line.36 However, in an in vitro proteolysis assay, MMP7 was shown to cleave FasL at leucine residues within the ELAELR sequence (Glu107-Arg112), located right outside the transmembrane (TM) domain, and thereby generate a fragment corresponding to almost the entire extracellular domain (sFasL-extra) (Fig. 1).37 To what extent this fragment is ever generated in vivo remains to be determined. Gene targeting of the Ser124/Leu125 and Lys127/Gln128 cleavage sites to encode Thr124/Leu125 and Arg127/Asn128 results in an uncleavable proapoptotic form of FasL. This line is designated ΔCS for deleted cleavage site (Fig. 1).13 Similar results were obtained by the replacement of residues 124–128 with GluGluAlaAlaAla.38 Together these data point to the 124/125 and/or 127/128 sites as necessary and sufficient for the natural cleavage process and the disintegrin and metalloprotease, ADAM10, has been shown to generate this shorter natural cleavage product.39,40
Importantly, in contrast to naturally cleaved murine sFasL, in vivo and in vitro assays indicate that murine sFasL-extra retains modest cytotoxic and proinflammatory activity.32 The structural basis for this difference remains unresolved because both the long and short fragments contain the trimerization domain and can readily form trimers.28 Expression of sFasL-extra as a transgene on the B6 background, in the absence of mFasL, resulted in a gld/gld (FasL loss of function mutation) systemic lupus erythematosus-like phenotype, revealing a critical role for uncleaved FasL in the maintenance of self-tolerance. However, these sFasL-extra transgenics were reported to develop histiocytic sarcomas,38 tumors that have not been described in gld/gld mice. This observation suggested a role for weak FasL engagement in tumorigenesis. It should be kept in mind that experimental crosslinking of sFasL, common for commercial reagents, can restore apoptotic activity and the use of recombinant sFasL-extra and crosslinked sFasL have led to some confusion over the in vivo role of the natural cleavage product.
In addition to generating the shorter sFasL cleavage product, ADAM10 cleavage also generates a 17 kDa membrane-embedded N-terminal fragment that can then be further processed by the signal peptide peptidase-like molecule SPPL2a to release a fragment derived from the intracellular domain. This intracellular fragment can then translocate to the nucleus and regulate gene transcription,40 a process referred to as reverse signaling. The CKI binding motif, identified in the cytoplasmic domains of both FasL and other TNF family members, has been implicated in the regulation of this process.22,41 Additional pathways downstream of molecules binding to the PRD also contribute to reverse signaling.42 Experimental evidence for reverse signaling has been documented in CD8+, and to a lesser extent, CD4+ T cells.41,42
Factors that enhance proapoptotic/proinflammatory activity of sFasL
The activity of both sFasL and mFasL can also be regulated by post-translational modifications. For example, sFasL accumulates in the bronchioalveolar lavage fluid of patients who develop acute respiratory distress syndrome (ARDS). In ARDS patients, infiltrating neutrophils can promote aggregation of sFasL through oxidation of a pair of methionine residues in the C-terminal end of the stalk region, leading to increased cytotoxic activity.43 In addition, the cytotoxic activity of human sFasL, isolated from transfected CT26 cells, can also be significantly increased when bound to extracellular matrix proteins such as collagen or fibronectin.44 Human sFasL has also been reported to play a key role in the keratinocyte death responsible for the blister lesions of pemphigus vulgaris patients,45 perhaps by binding to matrix proteins. Additional modifications include phosphorylation of serines within the CKI motif that are likely to impact reverse signaling and N-linked glycosylation of residues in the extracellular domain that may impact the solubility and function of sFasL.46,47
Decoy receptor DcR3
The soluble decoy receptor DcR3 can bind Fas as well as the receptors for TNF family members LIGHT and TL1A and thereby prevent engagement of the receptors by the functional ligands.47 DcR3 is overexpressed by a wide range of cancers and elevated expression is associated with poor prognosis,48,49 presumably due to its ability to block cytotoxic T cell activity. However, DcR3 has decoy independent proinflammatory functions that likely contribute to its impact on cancer immunity as well as other systemic autoimmune diseases. Moreover, DcR3 can neutralize the activity of sFasL and its capacity to modulate inflammatory responses in this context remains unexplored.
FUNCTIONS OF FASL
Cell death
Most early descriptions of FasL focused on the capacity of FasL to induce apoptosis.50 Two distinct apoptotic pathways were then defined. The first depended on the assembly of the now classical Fas/Fas-associated death domain (FADD)/caspase 8 death inducing signaling complex (DISC) and the ensuing rapid activation of caspase 8 in Fas+ Type I cells.51 Subsequent studies identified a second pathway, present in Fas+ Type II cells, that is less dependent on DISC formation and more dependent on the cleavage of BID, mitochondrial depolarization, and activation of caspase 9, where apoptosis can be rescued by overexpression of bcl-2.52,53 Fas can be expressed on a range of target cells, including activated lymphocytes and antigen-presenting cells, virally infected cells, and tumors. By contrast, the expression of FasL is much more restricted and mainly associated with common CD4, CD8, and NK cytotoxic effector populations. It follows that FasL cytotoxicity plays a role in viral immunity as shown for influenza, mouse hepatitis virus, and West Nile virus.54–56 Although FasL was originally identified for its ability to induce tumor cell death, especially by CD4+ effector cells,1,57 many Fas+ tumors are resistant to FasL-mediated cytotoxicity, and therefore the role of FasL in tumor immunity is controversial.16,58–60 Co-expression of Fas and FasL by T cells, following engagement of the T cell receptor, can result in the phenomenon of activation-induced cell death (AICD). The role of Fas in AICD was first described in T cell hybridoma cell lines61,62 and then shown to play a role in limiting T cell expansion, particularly in previously activated T cell populations.63,64 Constitutive FasL expression was also reported at sites of immune privilege, where it has been assumed that expression of FasL by Sertoli cells in the testis or corneal epithelial and endothelial cells in the eye protects these tissues from infiltrating Fas+ effector cells by inducing apoptosis.65–68 However, FasL can also trigger an extensive range of nonapoptotic functions, as summarized in the following text.
Proinflammatory cytokine production
The association between constitutive FasL expression and sites of immune privilege suggested that FasL might be useful therapeutically as a means to protect tissue transplants. However, this assumption proved incorrect, as shown by the accelerated rejection and massive neutrophil influx of transplanted FasL transgenic or FasL-transfected islet cells.19,69 Subsequent in vitro studies utilizing recombinant sFasL-extra suggested that soluble FasL per se had neutrophil chemotactic activity.70,71 However, fibrosarcoma cells expressing only a noncleavable membrane-bound form of FasL were also found to induce a potent neutrophil-associated inflammatory response.14 In vivo studies from our own group, using i.p. injections of tumor lines expressing different isoforms of FasL, showed that cells expressing wild-type (WT) FasL induced neutrophil extravasation into the peritoneal cavity, cells expressing a membrane-restricted form of FasL induced a much more robust response, and cells expressing sFasL-extra induced only modest inflammation. By contrast, cells expressing sFasL (natural cleavage product) completely failed to induce any evidence of inflammation.32 Moreover, the presence of sFasL-expressing cells, and not vector controls, blocked the neutrophil response elicited by cells expressing mFasL, indicating that sFasL functions as an mFasL antagonist. Inflammation most likely reflects FasL engagement of Fas+ macrophages, monocytes, and dendritic cells and the ensuing transcription, processing, and release of IL-1β and IL-18 [14, 72], as well as the induction and release of additional neutrophil/macrophage chemokines such as MIP1a/CCL3, MIP1b/CCL4, MIP2, and MCP1, KC/CXCL1, and IL8.15,17,73 Cytokine release in this context is not dependent on cell death as Fas+ antigen-presenting cells can be resistant to Fas-mediated apoptosis if they express the FasL inhibitory protein (FLIP).74–76 Importantly, FasL-induced processing of IL-1β and IL-18 is independent of the canonical inflammasome/caspase 1 pathway and instead depends on caspase 8.72
FasL in autoimmune disease
Both Fas-deficient lpr mice and FasL-deficient gld mice develop a lymphoproliferative disease associated with many of the clinical manifestations of systemic lupus erythematosus (SLE). In humans, Fas or FasL deficiency can result in a related autoimmune lymphoproliferative syndrome. These conditions have been attributed to an inability to eliminate Fas+ autoreactive lymphocytes that are normally kept in check by FasL-mediated cell death. However, FasL can play a proactive role in autoimmune disease pathogenesis and actually promote tissue inflammation or destruction. For example, Fas and FasL-deficient mice are protected in a myelin oligodendrocyte glycoprotein (MOG) model of experimental autoimmune encephalomyelitis (EAE),77 where Th17 cells play a key role in EAE pathogenesis. To better understand the factors responsible for skewing T cell differentiation to the Th17 lineage, Kuchroo and colleagues used a combination of transcriptional profiling, advanced computational analysis, and siRNA blockade to identify key regulators of this process. These studies identified Fas as a major positive regulator of Th17 differentiation. Because the in vitro cultures used purified T cells, it is likely that T cells both expressed the Fas receptor and FasL. They went on to show that a lower proportion of Th17 and a greater proportion of Th1 cells accumulated in the CNS of MOG-immunized Fas–/– mice when compared to MOG-immunized WT mice.78 Remarkably Il17a-Cre Fasfl/fl mice, had the same phenotype, indicating that T cell Fas expression is required for Th17 differentiation.79 Subsequent studies pointed to a role for Fas in Th17 stabilization through a mechanism that involved direct interaction of STAT1 with Fas, which in turn prevented STAT1 phosphorylation and nuclear translocation; in the absence of Fas, excessive levels of STAT1 block Th17 differentiation.79 Intriguingly, Legembre and colleagues have further shown that engagement of Fas on Th17 cells by sFasL can promote Th17 motility and confer the ability to cross endothelial barriers.80 Together, these studies provide important evidence for nonapoptotic functions of Fas in T cell differentiation.
Another FasL-dependent mechanism that could contribute to EAE pathogenesis has been proposed by Pasare and colleagues. They first showed that the proinflammatory cytokine IL-1 played a necessary role in the MOG model of EAE and then demonstrated that T cells producing TNF and FasL were potent inducers of IL-1 in Fas+ dendritic cells; TNF induced the transcription of pro-IL-1 and FasL activated caspase 8-dependent cleavage of the induced pro-IL-1.81 T cell expression of FasL is also required for skin disease in a recently described inducible model of cutaneous lupus. In these studies, adoptive transfer of WT but not FasL-deficient T cells induced skin disease in Fas+ recipients.82 Transcriptional analysis of skin revealed a significant increase in many of the proinflammatory cytokines previously associated with FasL activation of myeloid cells. Fas/FasL interactions also contribute to a murine model of alopecia areata, another model of autoimmune skin disease.83 These studies point to a key proinflammatory function of FasL+ T cells in certain autoimmune diseases.
T cell activation and differentiation
Although mFasL can initiate apoptosis in Fas+ thymocytes, naïve CD4+ cells, and restimulated Fas+ peripheral T cells61,63,64,84,85 a number of factors such as FLIP, expression of bcl-2 family members, and membrane location can lead to a wide range of alternative outcomes,86 as exemplified by the EAE studies described earlier. In fact, co-engagement of Fas and CD3 enhances proliferation through NFkB- and Erk-mediated induction of IL-2; this costimulatory activity depends on incorporation of FLIP in the Fas signaling complex and relatively low levels of FasL.87–89 Recent studies by Siegel and colleagues have shown that simultaneous Fas and TCR engagement on naïve CD8+ T cells can result in precocious differentiation. This Akt-dependent pathway results in the generation of highly active effector memory T cells (EMT)90 but limits the ability of these cells to expand and persist for an extended period in vivo.
Additional studies from the Siegel lab have shown that Fas-mediated apoptosis requires the assembly of Fas multimers into lipid rafts, and Fas accumulation in lipid rafts depends on palmitoylation of a membrane-proximal cysteine, C194.91 Cells expressing the palmitoylation-defective mutant C194V are resistant to FasL-mediated apoptosis. Unexpectedly, the C194V Fas mutation still rescues mice from the lymphoproliferative disease that occurs in Fas-deficient lpr mice, a syndrome initially attributed to a failure in T cell apoptosis. Even though C194V T cells do not undergo apoptosis following FasL engagement, they still retain the ability to undergo precocious differentiation and are much more likely to become EMTs.91 It follows that engagement of Fas in lipid rafts leads to a qualitatively different outcome (apoptosis) as compared to FasL engagement outside of lipid rafts (EMT cell differentiation).
Nonapoptotic FasL-induced T cell responses have also been described by Legembre and colleagues. Rather than manipulate Fas, this group used preprimed (PHA/IL-2) Fas+ T cells to compare the signaling events induced by mFasL vs. naturally cleaved sFasL. They first confirmed previous studies and showed sFasL did not induce caspase 8 cleavage or apoptosis. However, they found that sFasL induced Fas capping and the formation of a distinct Fas signaling complex that they termed the motility inducing signaling complex. The sFasL-treated cells further underwent morphologic changes leading to the formation of pseudopods and became more motile. This response depended on a direct interaction between Fas and PLCγ1, resulting in Ca+ influx, activation of the Src family kinase C-yes, and phosphorylation of Akt.33 Intriguingly, the same group reported increased levels of sFasL in the serum of SLE patients and found that the level of serum sFasL correlated with disease severity.33 In addition, patient serum was shown to induce endothelial cell transmigration of Th17 and Th1 cells.80 The Legembre lab also showed that sFasL activation of T cells depended on PLCγ1 binding to the calcium-inducing domain of Fas and that this interaction could be blocked by the protease inhibitor ritonavir. In follow-up studies, in vivo administration of ritonavir was shown to alleviate renal disease in lupus prone mice and this effect correlated with a significantly decreased Th17 signature.92 Overall, these studies demonstrate a critical role for sFasL in the activation of pathogenic T cells. From a broader perspective, they further establish a unique and active role for sFasL in immunomodulation.
FASL AND IMMUNE PRIVILEGE
Immunoprivileged sites
Several sites within the body are characterized as immunoprivileged, including the eye, brain, testis, and maternal-fetal interface, based on their attenuated and in some cases tolerizing immune reactivity.93,94 The strict regulation of inflammation within these sites is vital in maintaining both the integrity and function of the tissues involved. Left unregulated, inflammation in the eye has the potential to cause extensive ocular damage that results in impaired vision. To prevent inflammation-induced tissue damage, immunoprivileged sites have developed multiple mechanisms to regulate local immune responses. The Fas-FasL system is one such system that contributes to the maintenance of immune privilege in the ocular microenvironment.95,96 In 1995, Ferguson and colleagues demonstrated that FasL was constitutively expressed on cells in the cornea, retina, iris, and ciliary body. They went on to use a model of herpes simplex viral infection to demonstrate that FasL, expressed by the corneal endothelium and epithelium, induced apoptosis in Fas+ infiltrating inflammatory cells and thereby prevented virus-triggered inflammation and also induced HSV tolerance. In FasL-deficient gld mice, the infiltrating Fas+ inflammatory cells failed to undergo apoptosis, resulting in uncontrolled inflammation and extensive corneal tissue damage.66,67 These studies pointed to FasL-mediated cytotoxicity as an active mechanism of immune privilege. Subsequent studies went on to demonstrate that FasL-induced apoptosis of infiltrating immune cells also explained why corneal transplants are one of the most successful types of transplants with rodent studies demonstrating that FasL+ corneal allografts are accepted 45–50% of the time as compared to 0% survival of FasLneg allografts.68
The connection between FasL expression and immune privilege has been further supported by the demonstration that FasL contributes to the immunoprivileged status of the testis and the ability of FasL-transfected myoblasts to protect islet allografts from rejection.65,97 However, other studies led to conflicting results. For example, FasL transfection of islets per se triggered neutrophil infiltration and graft rejection,19 and numerous other reports documented the rapid rejection of FasL-expressing tumors and tissues.98,99 Moreover, the protective activity of FasL in the eye was challenged by examples of numerous FasL-associated ocular pathologies involving a range of cell types, partially illustrated in Figure 2. For example, FasL was shown to play a major role in animal models of glaucoma, where it contributes to the death of Fas+ retinal ganglion cells (RGCs).12
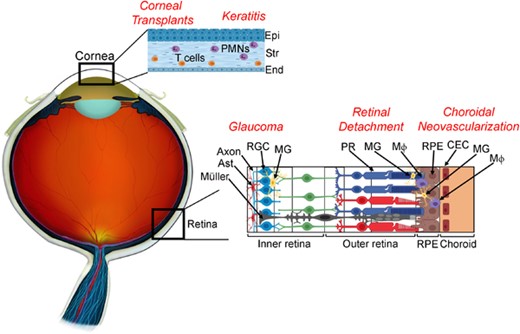
Cell types involved in Fas ligand (FasL)-mediated ocular pathologies. In corneal transplants, FasL expressed on donor corneal endothelium and epithelium promotes graft survival through apoptosis of Fas+ T cells. However, overexpression of membrane-associated FasL (mFasL) on corneal tissue results in accelerated graft rejection mediated by Fas+ neutrophils. In keratitis, FasL expressed on the corneal endothelium and epithelium is thought to limit the infiltration of Fas+ inflammatory cells. However, expression of mFasL-only exacerbates nonspecific tissue damage and corneal scarring through activation of infiltrating Fas+ neutrophils, whereas the soluble FasL fragment (sFasL) inhibits neutrophil-mediated inflammation and prevents nonspecific tissue damage. In Glaucoma, FasL expressed on microglia promotes neuroinflammation through activation of Fas+ astrocytes, Mueller glia, and microglia, as well as apoptosis of Fas+ RGCs. In retinal detachment, Fas activation plays an essential role in the apoptosis of Fas+ photoreceptors. However, activated microglia and/or infiltrating macrophages expressing both Fas and FasL also accumulate in the subretinal space and are thought to further contribute to the death of photoreceptors. In choroidal neovascularization, where abnormal choroidal vessels grow into the outer retina, FasL expressed on RPE is thought to prevent the ingrowth of vascular endothelial cells. However, activated microglia and/or infiltrating macrophages that express both Fas and FasL also contribute to local inflammation at the site of choroidal neovascularization (CNV) and have also been shown to play a prominent role in the development of CNV. Abbreviations: Epi, epithelium; Str, stroma; End, endothelium; Ast, astrocyte; RGC, retinal ganglion cell; MG, microglia; PR, photoreceptor; MΦ, macrophage; RPE, retinal pigment epithelium; CEC, choroidal endothelial cells
FasL in ocular tumor immunity
To explore the role of FasL in ocular tumor immunity, Nabel and colleagues used colon carcinoma cells transfected with WT FasL. They found these tumor cells failed to induce inflammation when injected into the anterior chamber of the eye and produced intraocular tumors comparable to the tumors that developed following the anterior chamber injection of FasLneg cells. These data indicate that tumor expression of WT FasL is not sufficient to trigger rejection in the immunoprivileged eye. However, the same WT FasL-expressing cells were proinflammatory and rejected following subcutaneous injection into the skin under conditions where FasLneg cells generated tumors.100 They attributed the disparate outcomes to the presence of TGFβ in the aqueous humor that dampened the proinflammatory effects of FasL in the eye. We obtained similar results following intraocular injection of transfected LY5178 T lymphoma cells; tumor cells expressing WT FasL or sFasL failed to perturb the immunoprivileged status of the eye and tumors grew progressively. However, LY5178 cells expressing (noncleavable) mFasL terminated immune privilege and induced a potent neutrophil-mediated inflammatory response resulting in tumor rejection.101 These studies demonstrate that mFasL can induce inflammation, even in an ocular environment. Although the difference between the rejection of WT FasL and mFasL-expressing tumors most likely reflects differences in the total level of FasL, a potential negative regulatory role of sFasL, generated from the WT FasL-transfected tumors, cannot be ruled out.
The role of FasL in tumor immunity is further complicated by the ability of tumor cells not only to release sFasL, but also to release microvesicles that incorporate full-length mFasL (FasL-V). A number of studies support the notion that FasL-V release enhances the capacity of the tumor to eliminate infiltrating Fas+ effector T cells before they come within striking range of the tumor.102–104 However, the ability of FasL-V to induce the production of proinflammatory cytokines may also impact tumor survival. In addition to the recruitment of neutrophils, as mentioned earlier, cytokines such as IL-6 and IL-1β can favor the outgrowth of Th17 cells over T regs, leading to more effective T cell-mediated antitumor immunity.60 Alternatively, FasL-V activation of the NFκB pathway may promote tumor metastasis.105 FasL-V can also be released by a variety of normal cell types, including ocular epithelial cells,106 but the role of FasL-V in the eye and the regulation of ocular tumors remains unexplored.
FasL isoforms in the eye
The apparent dichotomy, constitutive expression of FasL in nondiseased eyes as a mechanism of immune privilege and the association of FasL with numerous ocular pathologies (Fig. 2), can potentially be explained by the isoform of FasL expressed under different conditions. As reviewed earlier, mFasL is clearly proapoptotic and proinflammatory, whereas sFasL is incapable of mediating apoptosis or inflammation and can antagonize the activity of mFasL. Importantly, we have found that under homeostatic conditions, FasL in the murine eye is primarily cleaved to the soluble fragment, suggesting that it is the sFasL and not mFasL that contributes to the mechanisms responsible for immune privilege.107 In the human eye, early work by Wilson and colleagues actually demonstrated that FasL expression in normal corneal epithelium (ex vivo corneal epithelium removed at the time of excimer laser photorefractive keratectomy), was also primarily expressed as the soluble form.108 However, at the time of their study, the functional difference between mFasL and sFasL was not appreciated and it was proposed that cleaved sFasL-mediated apoptosis of Fas+ resident cells through paracrine mechanisms. Therefore, the sFasL may act to “modulate” the inflammatory effects of mFasL and only when the levels of mFasL exceed normal is termination of immune privilege and the development of destructive inflammation observed. Consequently, it becomes important to evaluate the role of FasL in ocular pathologies from the perspective of FasL isoforms.
FASL IN OCULAR PATHOLOGY
Corneal transplant
Corneal transplants enjoy the highest acceptance rate of all solid organ transplants.109,110 The remarkable success of corneal transplants is attributed to the immune privileged status of the eye111,112 and the constitutive expression of FasL on the cornea epithelium and endothelium has been shown to be essential in graft survival.68,113,114 In a murine model of corneal transplantation, FasL-positive grafts were accepted at a rate of 45–50%, whereas FasL-negative corneal grafts were universally rejected.68 T lymphocytes have been considered central to the process of corneal graft rejection115 and FasL expressed on the corneal graft is thought to promote graft survival through inducing apoptosis of infiltrating Fas+ effector T cells before they can destroy the graft.116 Moreover, blocking the cleavage of FasL on corneal grafts through the use of the antibiotic doxycycline, a broad spectrum matrix metalloproteinase inhibitor, increased the level of mFasL expressed on the corneal graft and improved corneal graft survival.116 However, doxycycline has effects beyond metalloproteinase inhibition that could promote engraftment and corneas transfected with an mFasL-only expression vector were quickly rejected via a neutrophil-mediated response, following both syngeneic and allogeneic transplantation.117 This response was similar to what we observed in our ocular tumor studies described earlier, where upon injection into the anterior chamber of the eye, LY5178 cells expressing (noncleavable) mFasL terminated immune privilege and induced a potent neutrophil-mediated inflammatory response resulting in tumor rejection.101 Whether the rejection of the mFasL-transfected corneas reflects an abnormally high level of FasL expression, a response to the adeno-associated viral (AAV vector), or the absence of sFasL remains to be determined, but these issues could be addressed by monitoring the fate of transplanted corneas obtained from transgenic ΔCS mice, where FasL expression is regulated by its endogenous promoter and expressed at physiologic levels, but resistant to cleavage and therefore only expressed as a membrane-bound protein.13
Keratitis
Corneal infections are a leading cause of vision loss resulting from corneal tissue damage and scarring. In both viral and bacterial keratitis, neutrophils are essential for pathogen clearance.118,119 However, it is the persistence of these neutrophils and their release of proteolytic enzymes that often leads to tissue damage and corneal opacity.120,121 In experimental models of viral and bacterial keratitis, FasL-deficient gld mice present with more extensive corneal tissue damage, most likely reflecting the need for FasL in pathogen clearance.122–124 In 2015, Stuart and colleagues demonstrated that topical treatment with a proapoptotic form of sFasL reduced corneal inflammation and neovascularization in acute and recurrent herpetic stromal keratitis and proposed that sFasL worked by augmenting the mFasL-induced apoptosis of Fas+ inflammatory cells.125 However, in a model of sterile corneal injury, epithelial debridement, intrastromal injection of plasmid DNA encoding cleavage-resistant mFasL severely exacerbated the inflammatory response and the extent of keratitis. Under the same conditions, intrastromal injection of sFasL DNA completely failed to induce keratitis. Importantly, when comparable levels of sFasL and mFasL plasmid DNA were injected together, the extent of inflammation was dramatically reduced compared to mFasL alone.126 Similar results were obtained in a model of LPS-induced keratitis, where administration of an AV vector encoding mFasL exacerbated neutrophil-mediated inflammation, corneal opacity, and neovascularization, whereas administration of an AV vector encoding sFasL prevented LPS-induced keratitis, resulting in a clear cornea.126 Together, these data support the notion that sFasL antagonizes the proinflammatory effects of mFasL and may actively suppress innate immune activation.
Retinal detachment
Retinal detachment, the separation of photoreceptors from the underlying retinal pigment epithelium (RPE), results in the apoptotic death of photoreceptors.127,128 In addition, following retinal detachment, activated microglia and/or infiltrating macrophages accumulate in the subretinal space and are thought to further contribute to the death of photoreceptors.129 In experimental models, retinal detachment leads to transcriptional up-regulation and activation of the Fas-mediated apoptosis pathway.130 Subsequent studies in rat and mouse models confirmed an essential role of Fas activation in photoreceptor death following retinal detachment by showing that photoreceptor death was significantly reduced in Fas-deficient lpr mice, as well as in animals treated with either Fas receptor neutralizing antibodies, siRNA against the Fas receptor, or the small peptide inhibitor Met12 that binds Fas and prevents FasL engagement.131,132
In patient populations, detection of increased levels of sFasL in the subretinal fluid at the time of retinal detachment has been associated with the future development of proliferative vitreoretinopathy, suggesting a proinflammatory role for sFasL.133 However, cultured RPEs have been shown to release both sFasL, as well FasL-V,106 and the immunoassay used to detect FasL in subretinal fluid could not distinguish sFasL from FasL-V. To further study the relative contribution of mFasL and sFasL in the photoreceptor death following retinal detachment, we evaluated two strains of mice previously demonstrated as susceptible (BALB/c) and resistant (B6129) to retinal detachment-induced photoreceptor death. When FasL function was eliminated genetically in BALB/c FasL-knockout mice, there was a significant reduction in photoreceptor death.134 In contrast, in B6/129 ΔCS mice, genetically modified to express only mFasL, there was a significant increase in photoreceptor death, and photoreceptor loss in ΔCS mice could be prevented by a subretinal injection of recombinant sFasL.134 Together, these data demonstrate the extent of vision loss following retinal detachment depends on the form of FasL that is actually expressed at the site of injury; mFasL triggers increased photoreceptor cell death whereas sFasL prevents photoreceptor cell death.
Age-related macular degeneration (AMD)
Choroidal neovascularization (CNV) is the hallmark of “wet” AMD where abnormal choroidal vessels infiltrate into the outer retina of the macula. Activated microglia and infiltrating macrophages also contribute to local inflammation at the site of CNV and have been shown to play a prominent role in the pathogenesis of AMD.135,136 These new blood vessels often bleed and leak fluid, causing retinal edema and detachment with subsequent loss of vision. FasL has been implicated in the inhibition of neovascularization in several in vivo models of both corneal and retinal neovascularization (suture induced, hyperoxia induced, and laser induced), where increased neovascularization was observed in FasL-deficient gld mice.137–139 In all cases, it was concluded that FasL controlled neovascularization by inducing apoptosis of infiltrating Fas+ endothelial cells. This idea was further supported by studies using RPE65-FasL transgenic mice that overexpressed full-length WT FasL on RPE cells. Expression of this transgene prevented laser-induced CNV,140 supporting the idea that FasL expressed on the RPE induced apoptosis of infiltrating Fas+ endothelial cells. However, another study suggests that FasL+ macrophages also induce apoptosis of Fas+ vascular endothelial cells, further contributing to the inhibition of CNV.141
The relevance of FasL cleavage in CNV associated with AMD was subsequently investigated using systemic administration of doxycycline, an MMP inhibitor, to inhibit cleavage of mFasL.142 Doxycycline-treated mice developed reduced CNV lesions in a laser-induced CNV model. Reduced angiogenesis correlated with increased FasL expression on choroidal tissue and decreased sFasL in the serum and eye as measured by ELISA, consistent with the idea that inhibition of mFasL cleavage led to increased expression of proapoptotic mFasL, and more effective elimination of Fas+ infiltrating vascular endothelial cells.137 However, examination of surgically excised choroidal neovascular membranes from patients diagnosed with “wet” AMD did not find a correlation between the expression of mFasL on RPE cells and either the size of neovascular membranes, or apoptosis of choroidal endothelial cells.143,144 One important problem in the in vivo analysis of sFasL and mFasL function is the difficulty of specifically regulating FasL cleavage. Although MMP inhibitors such as doxycycline can block proteolytic FasL cleavage, as mentioned earlier, they also have a variety of other anti-inflammatory and anti-angiogenic effects that are independent of the Fas/FasL pathway.145
Glaucoma
Glaucoma, the leading cause of irreversible blindness worldwide, is characterized by the progressive apoptotic death of RGCs.146–149 FasL was first linked to RGC apoptosis in an experimental rat model of chronic ocular hypertension, where Western blot analysis of the neural retina revealed a significant up-regulation of FasL expression following elevated intraocular pressure (IOP) and immunohistochemistry localized the increased FasL expression to the retinal microglia.12,150 Importantly, studies with both a total FasL-knockout mouse line and Fas-deficient lpr mice subsequently demonstrated that apoptosis of RGCs in both inducible and chronic mouse models of glaucoma was dependent upon the FasL-Fas signaling pathway.13,151 Moreover, RGCs were shown to up-regulate expression of the Fas receptor and FADD, a key component of the FasL-mediated apoptosis pathway, coincident with RGC apoptosis.150 Together the data pointed to microglial cells as a key effector of glaucoma. However, microglial cells and Mueller glial cells (nonneuronal cells that provide support and protection for neurons) are also Fas+ and astrocytes and infiltrating macrophages express both Fas and FasL (Fig. 2).12,13,150Such expression of FasL and Fas by multiple cell types in the retina suggest a more complicated crosstalk between additional FasL+ and Fas+ cell types as glaucoma progresses from the early signs of glial activation and inflammation to RCG death.
The proinflammatory cytokine TNFα has also been implicated as a critical link between elevated IOP and glaucoma; intravitreal injection of TNFα can lead to RCG death, even in the absence of elevated IOP.152–154 However, TNFα does not directly kill RGCs152 and FasL is required for TNFα-triggered RGC death.13 This synergy between FasL and TNFα in the development of glaucoma parallels the role for both cytokines in EAE, as described earlier.81
The FasL-induced death of RGCs by microglial cells and potentially other FasL+ nonlymphoid cells was initially hard to reconcile with the fact that FasL is constitutively expressed in the eye and responsible for immune privilege. Studies from our own group have now pointed to mFasL as the key effector of RGC apoptosis in both chronic (DBA/J) and inducible (TNFα and microbead) models of glaucoma, as we found that mFasL-only ΔCS mice developed accelerated and much more severe glaucoma than mice that express WT FasL.13,107 As mentioned earlier, FasL primarily exists as nonapoptotic, noninflammatory sFasL in the healthy, nonglaucomatous eye. However, as glaucoma develops, there is a shift from sFasL to the dominant expression of mFasL, presumably reflecting a loss of metalloproteinase cleavage.107 Moreover, intravitreal AAV2-mediated gene delivery of sFasL provided complete and long-term protection of RGCs and their axons in both the chronic DBA/2J mouse model and the inducible microbead mouse model of glaucoma.107 sFasL-mediated neuroprotection also correlated with reduced retinal microglia and muller glia activation and reduced production of proinflammatory mediators, highlighting the importance of FasL-Fas signaling in both apoptosis and inflammation. In addition, directly targeting the Fas receptor with same small peptide antagonist used in the retinal detachment studies described earlier132 also inhibited microglia activation and inflammation and provided significant neuroprotection in the microbead-induced mouse model of glaucoma.151 Overall, these studies further illustrate the neurodestructive effect of mFasL in the pathogenesis of glaucoma and reveals how blocking mFasL-mediated activation of the Fas receptor through the use of sFasL or a small peptide Fas inhibitor prevents microglia activation and the development of neuroinflammation and provides significant neuroprotection to both the RGCs and their axons (summarized in Fig. 3).
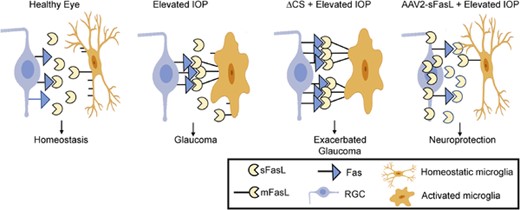
Membrane-associated FasL (mFasL) and soluble FasL fragment (sFasL) exhibit opposing effects on the development of glaucoma. In the healthy eye, Fas ligand (FasL) is constitutively cleaved and sFasL maintains a noninflammatory homeostatic environment. Following elevated intraocular pressure (IOP), microglia become activated and up-regulate expression of FasL, with a shift from sFasL to mFasL contributing to the apoptosis of retinal ganglion cells (RGCs). In ΔCS mice, the inability to cleave FasL results in accelerated and much more severe glaucoma. Intravitreal injection of AAV2-sFasL results in constitutive sFasL production by long-lived neurons and provides significant neuroprotection of RGCs and axons
SUMMARY AND FUTURE DIRECTIONS
Numerous studies have linked constitutive FasL expression in ocular tissues to the immunoprivileged status of the eye with evidence that FasL can induce the apoptotic death of infiltrating Fas+ effector cells following allogeneic corneal transplants. In addition to inducing apoptosis, mFasL is also a potent inducer of inflammation and contributes to a range of ocular pathologies associated with neurodestruction or death of nonhematopoietic target tissues in the eye. Because many ocular tissues also express Fas, FasL activity needs to be tightly regulated. We propose that FasL in the eye is regulated by constitutive cleavage of membrane-bound FasL and the release of a soluble fragment, sFasL, that blocks both apoptosis and inflammation. Based on cleavage-site specificity, we believe that ADAM10 is responsible for FasL cleavage in the eye although other metalloproteinases cannot be ruled out at this time. It follows that inflammation or the onset of other ocular pathologies interfere with FasL cleavage and results in the accumulation of the more pathogenic mFasL isoform. Exactly how FasL cleavage is regulated remains to be determined but first we need to confirm the role of ADAM10 and better understand when and where it is expressed.
Another unanswered question is exactly how sFasL attenuates mFasL activity. One possible mechanism is steric hindrance. sFasL could simply compete with mFasL for binding to Fas, but it is hard to envision how sFasL monomers could effectively block interactions between Fas multimers and FasL multimers that form in the lipid rafts associated with DISC assembly. Alternatively, sFasL may bind Fas outside of lipid rafts and thereby block the formation of the Fas/FasL multimers and activation of the apoptotic/inflammatory pathways. The feasibility of a steric hindrance model is supported by data on the small molecule inhibitor Met 12. Met12 binds Fas directly and prevents Fas from interacting with FasL. Met12 is a peptide derived from the Met protein, a heterodimer tyrosine kinase receptor known for its ability to bind hepatocyte growth factor and also associate with Fas.132 Intriguingly, Met is highly expressed in macrophages, RPE, and the cornea, but not the retina,155 raising the possibility that it may also regulate Fas signaling in certain ocular tissues.
Alternatively, it is also possible that sFasL plays a more active role in immunomodulation. Studies from Legembre and colleagues have shown that naturally cleaved sFasL can engage Fas and activate a PLCg1→PI3K→Akt pathway associated with cell survival, induction of T cell motility, and tumor invasion.80,92,156,157 To what extent sFasL actively regulates the inflammatory process in glaucoma and other ocular disorders remains an area of active investigation that has important implications for therapeutic applications of sFasL. If sFasL can be shown to play an active role limiting ocular inflammation, it may be possible to use these pathways to regulate pathologies beyond those mediated by mFasL.
AUTHORSHIP
M.G-K. and A.M-R. contributed equally to the conceptualization and writing of this review.
ACKNOWLEDGMENTS
The authors acknowledge support from NIH R01EY01645, R21EY022433, R01EY021543, and R21EY030276. Figures 2 and 3 were created with Biorender.
DISCLOSURE
M.G-K. serves on the Scientific Advisory Board of ONL Therapeutics.
REFERENCES
Abbreviations
- AMD
age-related macular degeneration
- ARDS
Acute respiratory distress syndrome
- CKI
casein kinase I
- CNV
Choroidal neovascularization
- FasL
Fas ligand
- FasL-V
FasL microvesicles
- IOP
Intraocular pressure
- mFasL
membrane-associated FasL
- PRR
Proline-rich region
- RGC
retinal ganglion cell
- RPE
retinal pigment epithelium
- sFasL
naturally cleaved soluble FasL fragment
- sFasL-extra
soluble FasL extracellular domain
- ΔCS
mouse line with mutation in cleavage site that render it uncleavable

{kind=link}
{kind=link}
{kind=link}
{kind=link}