





Abstract
Inflammasomes are multimeric protein complexes that induce the cleavage and release of bioactive IL-1β and cause a lytic form of cell death, termed pyroptosis. Due to its diverse triggers, ranging from infectious pathogens and host danger molecules to environmental irritants, the NOD-like receptor protein 3 (NLRP3) inflammasome remains the most widely studied inflammasome to date. Despite intense scrutiny, a universal mechanism for its activation remains elusive, although, recent research has focused on mitochondrial dysfunction or potassium (K+) efflux as key events. In this review, we give a general overview of NLRP3 inflammasome activation and explore the recently emerging noncanonical and alternative pathways to NLRP3 activation. We highlight the role of the NLRP3 inflammasome in the pathogenesis of metabolic disease that is associated with mitochondrial and oxidative stress. Finally, we interrogate the mechanisms proposed to trigger NLRP3 inflammasome assembly and activation. A greater understanding of how NLRP3 inflammasome activation is triggered may reveal new therapeutic targets for the treatment of inflammatory disease.

1 INTRODUCTION
Inflammasomes are cytosolic multimeric protein complexes that assemble upon exposure to pathogen and host-derived danger signals to modulate the host immune response. Assembly of the inflammasome triggers autoproteolytic cleavage and activation of the inflammatory cysteine aspartic protease, caspase-1. Active caspase-1 subsequently induces the cleavage and maturation of the proinflammatory cytokines, pro-IL-1β and pro-IL-18, and cleaves gasdermin D (GSDMD) to cause pore formation and induce cellular lysis, or pyroptotic cell death.1,2,3 Since the discovery of the nucleotide-binding oligomerization domain (NOD)-like receptor 1 (NLRP1) inflammasome by the laboratory of Jürg Tschopp in 2002,4 several cytosolic pattern recognition receptors (PRRs) have now been shown to assemble functional inflammasomes, or trigger caspase-1 activation, upon sensing pathogen- or host damage-associated molecular patterns (PAMPS and DAMPS).5 These include members of the NLR family, such as NLRP3, NLRC4 (IPAF), and the less well studied human NLRP2, NLRP7 and mouse NLRP6, NLRP9b, and NLRP12 sensor proteins,6,7,8,9,10,11 the HIN-200 absent in melanoma-like receptors, AIM2 and IFI-16,12,13 and the tripartite motif (TRIM) family receptor, Pyrin.14 To date, the majority of inflammasome sensors exhibit selectivity for physiological triggers. NLRP1 is activated by anthrax lethal toxin secreted by Bacillus anthracis,15,16 while AIM2 senses cytosolic bacterial, viral or host dsDNA,12,17,18 and Pyrin is triggered by Rho GTPase inactivating bacterial toxins/effectors, such as Clostridium difficile toxin A/B.14 In the case of NLRC4, it does not directly sense Gram-negative bacteria (e.g., Salmonella Typhimurium) but instead utilizes NLR family of apoptosis inhibitory protein (NAIP) co-receptors that specifically sense flagellin and components of the type III secretion system (T3SS).19,20,21,22 It is important to note that in humans there is a sole NAIP co-receptor, while in mice NAIP5 and NAIP6 sense flagellin, and NAIP1 and NAIP2 recognize needle and inner rod proteins of the T3SS, respectively.19,20,21,22 In contrast to the selective nature of most inflammasome sensors, NLRP3 recognizes a diverse range of pathogen and danger molecules, including bacterial toxins (e.g., pore-forming toxin nigericin), host metabolites (e.g., uric acid, cholesterol crystals) and environmental irritants (e.g., asbestos, silica), which has implicated the NLRP3 inflammasome in the pathogenesis of numerous infectious and inflammatory diseases.23
Upon sensing a danger signal, inflammasome sensors recruit the bipartite adaptor, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), via common pyrin domain (PYD) interactions, or in the case of CARD containing receptors (i.e., NLRC4, NLRP1) associate via CARD-CARD interactions. This facilitates ASC polymerization through homotypic PYD-PYD interactions to form long helical prion-like filaments, which are condensed into a discrete ASC speck, in a CARD-dependent manner.24,25,26,27 In turn, ASC recruits pro-caspase-1 via its N-terminal CARD domain (CARD-CARD interaction) to promote proximity-induced caspase-1 dimerization and autoproteolysis to release the large (p20) and small (p10) subunits. Until recently, it was widely assumed that the labile caspase-1 tetramer comprising two p20 and two p10 subunits (p20/10) was the active form. Undeniably, superphysiological levels of recombinant p20/10 caspase-1 is catalytically active.28,29,30 However, recent work by the laboratory of Kate Schroder has indicated that, in fact, the inflammasome-associated full-length caspase-1 (p46), as well as the transient p33/p10 species, are the bioactive forms.30 Instead, the p20/10 tetramers are the result of p33/10 self-cleavage to terminate caspase-1 signaling.30 In support of this notion, the CARD-containing NLRC4 and NLRP1b inflammasome sensors can interact directly with caspase-1 to drive pyroptosis (and limited IL-1β activation) independent of caspase-1 proteolysis.31,32 Moreover, dimerized caspase-1 has been shown to induce the activation and secretion of IL-1β in the absence of proteolysis and cell death.33 Hence, it remains of interest to reassess the cell-specific kinetics of activation/inactivation of caspase-1 by different inflammasome complexes.
The primary functions of active caspase-1 is to induce the cleavage of pro-IL-1β and -IL-18 to their bioactive forms, and cleave GSDMD (at Asp 276 in mice and Asp275 in humans) to release an active effector N-terminal GSDMD (GSDMD-NT) p30 fragment that causes pyroptotic cell death.2,3,34 GSDMD-NT binds and inserts into lipid membranes, and oligomerizes to form transmembrane pores (∼10–20 nm) that causes a loss of cell membrane integrity and the lytic release of cytoplasmic contents (e.g., HMGB1, IL-1α).35,36,37,38 GSDMD pores also facilitate the release of mature IL-1β and IL-18, and this has recently been suggested to occur immediately prior to pyroptotic cell death.39,40 It is worth mentioning that caspase-1 and GSDMD are not essential for cell death to all inflammasome triggers.3,34,41,42,43 In fact, in certain cases NLRP3 itself is dispensable, such as upon exposure to lysosomal-disrupting agents, crystalline and particulate matter.44,45,46 Likewise, inflammasome-associated IL-1β secretion can occur in a GSDMD-independent manner, via caspase-8-mediated apoptosis or secondary necrosis/pyroptosis, and can even occur largely in the absence of cell death.41,47,48 Furthermore, caspase-8 is recruited to several inflammasomes to drive caspase-1-independent IL-1β activation and/or apoptotic cell death.43,49,50 Remarkably, it has now also become apparent that there is bidirectional crosstalk between apoptotic and pyroptotic cell death pathways. For example, under certain conditions, caspase-1 can cleave and activate caspase-3 and -7 to drive apoptosis.51,52 Conversely, caspase-3 and -7 cleave and inactivate GSDMD (at Asp87) to limit pyroptosis.52
Despite our improved knowledge of inflammasome assembly and downstream signaling, the upstream events that trigger activation of the most widely studied inflammasome, the NLRP3 inflammasome, remain contentious. Mitochondrial damage and oxidative stress are widely touted to be the universal mechanism to trigger NLRP3, yet, there is considerable evidence that potassium ion (K+) efflux is essential in most cases. In this review, we give an overview of NLRP3 inflammasome activity in health and disease, particularly highlighting the recent link with mitochondrial damage and oxidative stress in metabolic disease. We discuss the ambiguities surrounding mechanisms of NLRP3 activation, such as ion flux, mitochondrial damage, and reactive oxygen species (ROS) generation.
2 MODES OF NLRP3 INFLAMMASOME ACTIVATION
2.1 Canonical activation of the NLRP3 inflammasome
The NLRP3 sensor (also known as NALP3 and Cryopyrin, encoded by the NLRP3 or CIAS gene) has been extensively studied due to its involvement in many disease pathologies and the diverse nature of its triggers, which include host DAMPS, such as extracellular ATP, uric acid, and cholesterol crystals, PAMPs (e.g., bacterial mRNA and DNA, DNA/RNA viruses, fungi, and protozoa) and environmental irritants (e.g., silica, asbestos, aluminum hydroxide).23 NLRP3 comprises a C-terminal leucine-rich repeat (LRR) that may act as the sensor domain, a NACHT (or NOD) domain that enables ATP-dependent oligomerization and activation, and a PYD domain at the N-terminus to recruit the adaptor protein ASC.
Activation of the NLRP3 inflammasome in innate immune cells is generally thought of as a 2-step process (Fig. 1). In the first step (signal 1), microbial engagement of TLRs, or sterile priming (i.e., TNF receptor 1 [TNFR1] signaling), activates NF-κB-mediated transcription of NLRP3 and other proinflammatory genes, including pro-IL-1β. The second signal, such as a DAMP or PAMP, then triggers assembly and activation of the NLRP3 inflammasome via an ill-defined manner that may involve one or more events, including K+ efflux, mitochondrial damage and ROS generation, calcium (Ca2+) influx, and lysosomal rupture (see Section 4, Figs. 1 and 4). Notable exceptions to this 2-step model, include the spontaneous NLRP3 activation observed upon LPS stimulation of murine bone marrow dendritic cells (BMDCs) and human monocytes in vitro, as well as the P2X7 receptor (P2X7R)-independent activation of NLRP3 in mice upon LPS challenge (discussed further in Sections 2.2.1 and 2.2.2).53,54,55
![Modes of canonical NLRP3 inflammasome activation. Activation of the canonical NLRP3 inflammasome requires a priming signal, signal 1, via engagement of pattern recognition receptors (PRRs) that induce NF-κβ-mediated transcriptional up-regulation of inflammasome machinery, namely NLRP3, pro-IL-1β and pro-IL-18. Signal 2, an NLRP3 activation signal, is triggered by a variety of DAMPS (e.g., cholesterol crystals, ATP) and PAMPS (e.g., pore-forming toxins, viral RNA). PAMPS and DAMPS trigger formation of the multimeric inflammasome complex comprising NLRP3, ASC and pro-caspase-1 via an ill-defined mechanism that may involve K+ efflux and/or Ca2+ influx, lysosomal rupture, ROS production and mitochondrial dysfunction (see Fig.). NEK7-NLRP3 interactions are essential to promote NLRP3 oligomerization and the subsequent recruitment of ASC via homotypic pyrin domain interactions. ASC oligomerization leads to amyloid-like fibril structures that can interact with capase-1 via common CARD domains and promote proximity-induced caspase-1 activation. Active caspase-1 (putative p33/10 and p46 caspase-1 subunits [dotted line box]), in turn, cleaves substrates pro-IL-1β and pro-IL-18 to their bioactive forms. Caspase-1 also cleaves GSDMD to release the N-terminal fragment that can translocate to the plasma membrane and form oligomeric pores to induce a lytic form of cell death, termed pyroptosis, and facilitate IL-1β release](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jleukbio/105/2/10.1002_JLB.MR0318-124R/2/m_jlb10292-fig-0001-t.gif?Expires=1750231060&Signature=ZYnnyDSRU1PmysK3BesUa6LLHBJ84x6uc9LLAcZevmdrf2wsSu4QNNMEB8lkB-UGejTT0OqWyoM7M1raK5fDxcIZUZY4OfhD4bNfXBD2bHYYAwnY2XWcIqZUBc7mOF3no7Vk3pEzu7S72udqXJBJi3ckBm1MegQTeUbOwt~B1tmB3WwXIYTCifphllvVyCv2HwHI8THubWdVAK6f4STxI4XHSepFGdXogr3IG~aigQFe0wyUkUQFokusJmT27oQTbSGreBU7pHURmNWAL92NJ4gzHKl6iCQUAgWLnPFxyQNtT6iQTd95TB0NMi4y7djBMFw36FvpeSPViYN-qGTrhg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Modes of canonical NLRP3 inflammasome activation. Activation of the canonical NLRP3 inflammasome requires a priming signal, signal 1, via engagement of pattern recognition receptors (PRRs) that induce NF-κβ-mediated transcriptional up-regulation of inflammasome machinery, namely NLRP3, pro-IL-1β and pro-IL-18. Signal 2, an NLRP3 activation signal, is triggered by a variety of DAMPS (e.g., cholesterol crystals, ATP) and PAMPS (e.g., pore-forming toxins, viral RNA). PAMPS and DAMPS trigger formation of the multimeric inflammasome complex comprising NLRP3, ASC and pro-caspase-1 via an ill-defined mechanism that may involve K+ efflux and/or Ca2+ influx, lysosomal rupture, ROS production and mitochondrial dysfunction (see Fig.). NEK7-NLRP3 interactions are essential to promote NLRP3 oligomerization and the subsequent recruitment of ASC via homotypic pyrin domain interactions. ASC oligomerization leads to amyloid-like fibril structures that can interact with capase-1 via common CARD domains and promote proximity-induced caspase-1 activation. Active caspase-1 (putative p33/10 and p46 caspase-1 subunits [dotted line box]), in turn, cleaves substrates pro-IL-1β and pro-IL-18 to their bioactive forms. Caspase-1 also cleaves GSDMD to release the N-terminal fragment that can translocate to the plasma membrane and form oligomeric pores to induce a lytic form of cell death, termed pyroptosis, and facilitate IL-1β release
In recent work by the laboratories of Bruce Beutler, Gabriel Núñez, and Veit Hornung, the serine threonine kinase NIMA-related kinase 7 (NEK7) was identified as a critical positive regulator of NLRP3 inflammasome assembly downstream of K+ efflux and ROS.56,57,58 NEK7 was shown to interact via its catalytic domain with the LRR and NACHT domain of NLRP3 to facilitate inflammasome activation in a manner independent of its kinase activity.56,57 Notably, this interaction can reportedly be disrupted by intrinsic apoptosis regulator cytochrome c to presumably limit pyroptotic cell death.59 Similarly, the anticancer ent-kaurene diterpenoid, Oridonin, which is the major constituent of Rabdosia rubescens, was recently suggested to covalently bind cysteine 279 of the NACHT domain to block NEK7-NLRP3 interactions.60
It should be acknowledged that in addition to transcriptional regulation (i.e., signal 1), TLR signaling also induces post-translational regulation of NLRP3 activation to expedite an inflammatory response.61,62,63,64,65 Brief exposure of macrophages to TLR agonists that signal via MyD88 (10–20 min), and NLRP3 activators nigericin and ATP, or Listeria monocytogenes, induces rapid IL-1 receptor associated kinase-1 (IRAK1)/IRAK4- and mitochondrial ROS (mtROS)-dependent inflammasome assembly and activation.64,65 Further to this, Juliana et al. earlier showed that both LPS and ATP are capable of inducing the deubiquitylation of NLRP3 to regulate inflammasome activity, although surprisingly, only LPS required mtROS to trigger deubiquitinase (DUB) activity.63 The DUB, BRCC3 (BRCC36 in humans), was recently revealed to deubiquitylate NLRP3 to induce inflammasome activation.66,67 A number of additional ubiquitin modification events have now been proposed to regulate NLRP3 inflammasome assembly and activation. For example, the E3 ubiquitin ligase HOIL-1, a linear ubiquitination chain assembly complex (LUBAC) component, was suggested to ubiquitylate ASC to promote NLRP3 inflammasome assembly; however, related complex member SHARPIN was found to only be required for inflammasome priming.68,69 In contrast, the DUB A20 has been shown to limit NLRP3 activation by deubiquitylating inflammasome-associated IL-1β.70 Several E3 ubiquitin ligases, including F-box L2 (FBXL2) and TRIM31, as well as membrane-associated RING finger protein 7 (MARCH7), have also been reported to promote K48-linked polyubiquitylation of NLRP3 to promote its proteasomal or autophagolysomal degradation.71,72,73 It has now become increasingly clear that multiple post-translational modification events fine-tune NLRP3 inflammasome assembly and activation, including ubiquitylation, phosphorylation, acetylation, and nitrosylation; however, most results require further validation (reviewed in Poudel et al.).74
2.2 Noncanonical and alternative pathways to NLRP3 activation
2.2.1 Noncanonical caspase-11-mediated activation of the NLRP3 inflammasome
In 2011, the laboratory of Vishva Dixit identified that the murine inflammatory caspase, caspase-11, acts as the cytosolic sensor of intracellular LPS derived from Gram-negative bacteria to promote noncanonical activation of NLRP375 (Fig. 2). In this pioneering work, Dixit and colleagues confirmed reports by the laboratory of Junying Yuan that the widely used caspase-1-deficient mouse lines derived in 129 embryonic stem cells lacked caspase-11 expression.76 This effect was caused by a mutation in the 129 caspase-11 locus, which due to its close proximity to caspase-1, could not be segregated upon backcrossing onto the C57BL/6 background.75 Subsequent examination of caspase-11-deficient bone marrow-derived macrophages (BMDMs) revealed that caspase-11 was essential for NLRP3 inflammasome-mediated IL-1β and IL-18 activation in response to cholera toxin B, Escherichia coli, Citrobacter rodentium, and Vibrio cholerae, but not canonical activators, such as ATP and MSU.75 Importantly, while caspase-11 activation triggered the NLRP3-caspase-1 inflammasome to activate IL-1β and IL-18, caspase-11 alone was sufficient to trigger pyroptotic cell death and lethal sepsis.75 Caspase-11 has now been shown to be critical for inducing lethal sepsis in response to LPS,75,77 and protecting mice from Gram-negative bacterial infections78,79,80,81 and acute dextran sulfate sodium-induced colitis.82,83
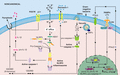
The noncanonical caspase-11-dependent pathway to NLRP3 inflammasome activation. Activation of the noncanonical caspase-11 NLRP3 inflammasome requires TLR- and type I IFN-induced transcription of pro-caspase-11, guanylate binding proteins (GBP), as well as NLRP3 machinery. Caspase-11 is activated within the cytosol by LPS derived from Gram-negative bacteria (e.g., Burkholderia, Legionella) that escape phagocytic vacuoles in a GBP-dependent manner, or accesses the cytosol via outer membrane vesicles (OMVs). The cytosolic hydrophobic hexa-acetylated lipid A moiety of LPS directly binds the CARD domain of caspase-11 to facilitate caspase-11 oligomerization and activation. Active caspase-11 induces the cleavage and release of the N-terminal domain of GSDMD to form pores in the cell membrane and cause pyroptotic cell death. Active caspase-11 also reportedly cleaves the pannexin-1 hemichannel to facilitate the release of ATP to perpetuate pyroptosis via P2X7R signalling, and activates NLRP3 via K+ efflux
Several recent studies have shed light on how murine caspase-11, and human homologs caspase-4 and -5, are activated by the hexa-acetylated lipid A moiety of LPS to trigger NLRP3 inflammasome activity and cause pyroptosis in innate immune cells.75,84,85,86,87 The laboratory of Feng Shao proposed that caspase-11, -4, and -5 specifically bind the lipid A moiety of LPS via their CARD domains to induce caspase oligomerization and activation.84 It is important to note, that caspase-11 can be activated via transfection, electroporation or cholera toxin A delivery of LPS to the intracellular compartment.75,77,87 In activated dendritic cells, and possibly retinal epithelial cells, caspase-11 can also reportedly be triggered by oxidized phospholipids (OxPAPC) released from dying cells.88,89 Moreover, high doses of OxPAPC components have been shown to trigger both noncanonical and canonical NLRP3 activation in macrophages.90,91 Perplexingly, a recent study disputed these findings and concluded that OxPAPC binds caspase-11 to actually limit NLRP3 activation by Gram-negative bacteria.92 More recently, it has become evident that outer membrane vesicles derived from Gram-negative bacteria (e.g., Pseudomonas aeruginosa, Neisseria meningitides) are also sensed by specific inflammatory caspases to direct innate immune cell responses.93,94
Upon activation, caspase-11, akin to caspase-1 in the canonical pathway, induces pyroptotic cell death by directly cleaving and activating GSDMD2,3,34 (Fig. 2). However, it has additionally been shown that active casapase-11 can contribute to pyroptosis, in a GSDMD-independent manner, via the cleavage of the pannexin-1 hemichannel to allow ATP release and engagement of the purinergic P2X7R.95 Similar to most canonical triggers of NLRP3 activation, noncanonical caspase-11-mediated activation of NLRP3 is dependent on K+ efflux,95,96 and it has been suggested that caspase-11 mediated opening of the pannexin-1 channel allows K+ efflux.95 Remarkably, despite efforts at dissecting apart these pathways in myeloid cells in vitro, it now appears that caspase-11-induced GSDMD activity is not required in the hematopoietic compartment for lethal endotoxic shock in vivo.97,98 In fact, in response to E. coli endotoxin and live bacteria, the hematopoietic system initiates inflammatory events via type I IFN and TNF production, which then in turn promotes pro-pyroptotic caspase-11 and pro-apoptotic caspase-8 collaborative activity in target tissue cells (e.g., gut epithelial cell death) to amplify injury.98
Despite improved knowledge on how caspase-11 activates NLRP3 and GSDMD to drive inflammatory events, the upstream events required for activation remain ill defined. A number of studies have identified that type I IFN signaling, induced via TLR3/4 and Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF), can amplify the expression of caspase-11.77,78,87,100 Recently, the complement-related carboxypeptidase B1 was implicated in up-regulation of caspase-4, -5, and -11 downstream of type I IFN.101 Notably, type I IFNs (and type II IFNs) also influence caspase-11 activation at another level, via the induction of IFN-inducible GTPases, including guanylate binding proteins (GBPs) and IFN-inducible protein IRGB10.102,103,104 It has been shown that GBPs and IRGB10 can liberate bacterial ligands from bacteria-containing vacuoles, and possibly outer membrane vesicles, to allow sensing by caspase-11 (Fig. 2).103,104 Interestingly, a vacuole-independent role for GBPs in the rapid noncanonical and canonical activation of NLRP3 has recently been reported upon Chlamydia muridarum infection of macrophages.105 Moreover, IRGB10 has not only been shown to permit caspase-11-mediated NLRP3 inflammasome activation to Francisella novicida, but also lyses the bacterial membrane, releasing sufficient levels of DNA to activate the AIM2 inflammasome.106 Hence, IFN-inducible GTPases appear to play a vital role in coordinating inflammasome responses to Gram-negative bacteria, and it is now emerging that bacteria have evolved effector mechanism to evade these antibacterial defense mechanisms.107
2.2.2 Alternative and cell death-induced pathways to NLRP3 inflammasome activation
Apoptosis is an evolutionarily conserved programmed cell death pathway to remove superfluous or damaged cells, promote pathogen clearance and maintain immune tolerance. Intrinsic “mitochondria-dependent” apoptosis is triggered by a range of cellular stressors, including growth factor withdrawal and ultra violet irradiation. Regulated by the balance of pro- and anti-apoptotic BCL-2 family members, the irreversible step culminating in cell death is activation of BAX and BAK oligomeric pores in the mitochondrial membrane that allows the release of mitochondrial contents, including cytochrome c. Subsequently, cytochrome c orchestrates formation of an apoptosome complex (comprising cytochrome c, apoptosis protease activating factor-1 [APAF-1] and pro-caspase-9) that induces energy-dependent autoactivation of caspase-9 to cleave effector caspases, such as caspase-3 and -7, to induce apoptosis (Fig. 3B).108 In contrast, death receptor signaling, via members of the TNFR superfamily, such as TNFR1, and members of the TLR family (i.e. TLR3 and TLR4), activates a death or “ripoptosome” complex comprising Fas-associated protein with death domain (FADD), receptor interacting protein kinase 1 (RIPK1) and pro-caspase-8, which leads to a caspase cascade and cellular demise (Fig. 3B). If caspase-8 activity is compromised, TLR and TNFR1 signaling can also induce a lytic form of cell death, called necroptosis. Necroptosis requires the kinase activity of RIPK1 and RIPK3, and the pseudokinase, Mixed lineage kinase domain-like (MLKL). Phosphorylation of MLKL by RIPK3 induces a conformational change that exposes the N-terminal four-helix bundle (4HB) killing domain and promotes its oligomerization and migration to the plasma membrane, where it inserts into the membrane to cause lytic cell death109 (Fig. 3B).
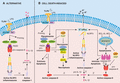
Alternative and cell death-induced pathways to NLRP3 inflammasome activation. A. The alternative NLRP3 inflammasome is activated in human monocytes by LPS. LPS signaling via TRIF recruits RIPK1, FADD and caspase-8 to trigger NLRP3 activation in a K+ efflux-independent manner. Unique to this pathway is the lack of requirement for ASC speck formation and pyroptosis. B. Multiple programmed cell death pathways trigger NLRP3 activation via K+ efflux. In the presence of LPS priming: (i) IAP loss induces formation of a RIPK1-RIPK3-FADD-caspase-8 complex, whereby activation of extrinsic apoptotic caspase-8 can activate IL-1β directly or trigger NLRP3 inflammasome activation via K+ efflux. (ii) When IAPs are absent and/or caspase-8 levels are compromised, a RIPK1/3 kinase- and MLKL-dependent necroptotic cell death pathway triggers NLRP3 activation in a K+ efflux-dependent manner. (iii) Specific activation of intrinsic effector molecules BAX and BAK, upon loss of pro-survival proteins MCL-1 and BCL-XL, triggers IAP loss to promote caspase-8 activity. BAX and BAK signaling activates effector caspases, caspase-3 and caspase-7, which cause K+ efflux to activate NLRP3 and are also required for activation of caspase-8 to cleave and activate IL-1β directly. Of note, pyroptotic effector GSDMD is dispensable for cell death and IL-1β secretion in all modes of cell-death mediated NLRP3 inflammasome activation
There is growing literature that there is significant crosstalk between modes of cell death and inflammatory signaling pathways, particularly the NLRP3 inflammasome.110 Caspase-8 has long been acknowledged to exhibit some overlap in substrate specificity with caspase-1. In 2008, the laboratory of Rudi Beyaert demonstrated that recombinant caspase-8, but not caspase-3, could cleave IL-1β at the same site as caspase-1.111 TLR3 and TLR4 ligation and protein synthesis inhibition were also shown to promote caspase-8 mediated cleavage of IL-1β.29,111 In fact, TLR-TRIF signaling alone was sufficient to trigger the formation of a signaling complex comprising FADD, RIPK1, RIPK3, and caspase-8 to induce both caspase-8 and caspase-1-mediated activation of IL-1β in BMDCs.54 Many subsequent studies have demonstrated that in TLR- and TNF-primed myeloid cells, a diverse range of cell stressors can activate caspase-8 to trigger NLRP3, or directly cleave pro-IL-1β and pro-IL-18 in myeloid cells (reviewed in Feltham et al.).110 For example, ligation of the death receptor Fas in macrophages induces a FADD-caspase-8 complex to activate IL-1β directly, in a RIPK3-independent manner.112 Whilst, treatment of LPS-primed BMDCs with pro-apoptotic chemotherapeutics, such as doxorubicin or staurosporine, induces RIPK3-dependent IL-1β production, dominantly via direct caspase-8 proteolysis.113 How diverse stressors trigger capsase-8-mediated IL-1β activity remains unclear; however, it is likely to involve changes in the level of cell death inhibitors.113,114 In line with this notion, chemical or genetic loss of all 3 E3 ubiquitin ligase inhibitor of apoptosis (IAP) proteins, namely cellular IAP1 (cIAP1) and cIAP2 and X-linked IAP (XIAP), in TLR- or TNF-primed BMDMs, BMDCs, and neutrophils induced RIPK1-RIPK3-caspase-8-mediated apoptosis (RIPK1-capase-8 in neutrophils), proteolysis of IL-1β and NLRP3 activation29,115,116,117,118 (Fig. 3B). It is important to note, that XIAP loss is essential for TLR-mediated activation of this inflammatory pathway in innate immune cells.115,116,119 Moreover, it has recently been shown that TLR-MyD88 and TNF-TNFR2-mediated proteasomal degradation of cIAP1, and its adaptor TRAF2, is required to induce caspase-8-driven NLRP3 inflammasome and IL-1β activation in the absence of XIAP.119 Intriguingly, loss of the NF-κB inhibitor and DUB, A20, also promotes TLR4-induced, RIPK3-dependent NLRP3 activation, presumably via caspase-8 activation.70 Whereas, loss of cellular FADD-like IL-1β-converting enzyme-inhibitor protein (c-FLIP), an endogenous capase-8 inhibitor, exaggerates caspase-8-mediated IL-1β secretion in response to a mimetic of the natural IAP inhibitor, second mitochondria-derived activator of caspases (SMAC).120
A number of recent studies have proposed that caspase-8 can associate directly with the NLRP3 inflammasome to drive IL-1β activation and apoptotic cell death.41,50,70,121 Work performed in the laboratory of Thirumala-Devi Kanneganti suggested that the FADD-RIPK3-caspase-8 ripoptosome complex associates with the NLRP3 inflammasome to promote both canonical and noncanonical caspase-11-mediated (e.g., C. rodentium) inflammasome activity.121 An alternative model proposed that upon Yersinia infection a RIPK1-FADD-caspase-8 complex can directly cleave caspase-1, in the absence of NLRP3 and NLRC4.122 Complicating the interpretation of these findings is the fact that caspase-8 plays a fundamental role in the transcription of cytokines (e.g. pro-IL-1β) and NLRP3 itself, as evidenced by defective TLR3/4 and Gram-negative bacterial inflammatory cytokine responses in Ripk3−/−Caspase-8−/−, Ripk3−/−FADD−/−, and Mlkl−/−FADD−/− mice.121,123,124,125 Is it worth noting that there are conflicting reports as to whether defective canonical NF-κB activation is responsible for reduced transcriptional responses in Ripk3−/−Caspase-8−/− macrophages.121,123,124 In addition, other groups have reported relatively normal canonical NLRP3 inflammasome activation upon sufficient TLR-priming in Ripk3−/−Caspase-8−/− myeloid cells (as indicated by ASC oligomerization and caspase-1 cleavage), suggesting that these findings warrant closer examination.116,122,123 Nevertheless, a recent study by Olaf Groß and colleagues established that similar to other inflammasomes, caspase-8 (as well as FADD, RIPK1 and c-FLIP) can indeed be recruited to the canonical NLRP3 inflammasome to activate IL-1β and induce secondary pyroptosis, although, this phenomenon was only pronounced in the absence of caspase-1 protease, or GSDMD, activity.41 Intriguingly, reminiscent of the noncanonical caspase-11 inflammasome pathway, a few studies have now proposed that caspase-8 can act upstream of the NLRP3 inflammasome (Fig. 3A, B).53,116 Chemical loss of the IAP proteins in LPS-primed BMDMs was first reported to induce RIPK1-RIPK3-caspase-8-dependent (and RIPK kinase independent) activation of NLRP3 via ROS and K+ efflux29,116,126 (Fig. 3B). However, divergent from the noncanonical pathway, GSDMD did not play a major role in cell death or IL-1β secretion.119 The requirement for upstream caspase-8 activity was established by James Vince and colleagues upon making the observation that NLRP3-associated caspase-1 cleavage events were blocked in unprimed, NLRP3-sufficient, Ripk3−/−Caspase-8−/− macrophages upon IAP inhibition.116 The laboratory of Veit Hornung later coined the term the “alternative NLRP3 inflammasome” to describe a similar TLR-TRIF-RIPK1-FADD-caspase-8-mediated route to NLRP3 inflammasome activation in human BlaER1 monocytic cells, which was surprisingly not dependent on RIPK3, ASC oligomerization or K+ efflux, and occurred in the absence of cell death53 (Fig. 3A).
Work centralized around unravelling how caspase-8 loss triggers lethal necroptotic signaling also uncovered that the RIPK1-RIPK3-MLKL “necrosome” complex can trigger NLRP3 inflammasome activation. The laboratory of David Wallach discovered that deletion of caspase-8 in dendritic cells rendered mice hypersensitive to LPS-induced lethality associated with RIPK3-driven NLRP3 inflammasome activity.127 In caspase-8-deficient BMDCs, TLR2/4 ligation led to spontaneous RIPK1/3 kinase, MLKL and mitochondrial phosphatase PGAM5-dependent NLRP3 inflammasome assembly, and IL-1β and IL-18 activation, but surprisingly not necrotic or pyroptotic cell death.127 Of note, a recent study revealed that PGAM5 is dispensable for necroptotic signaling, and therefore most likely PGAM5 is redundant in this mode of NLRP3 activation.128 Other groups, however, have reaffirmed that TLR ligation triggers RIPK3 and MLKL-mediated NLRP3 activation in caspase-8-deficient BMDMs and human BlaER1 monocytes, as well as in RIPK1-deficient monocyte/macrophages.53,116,129 It has also become apparent that the RIPK3-MLKL-driven NLRP3 inflammasome pathway can be further amplified in the absence of caspase-8, via the chemical or genetic loss of the IAP proteins, particularly XIAP115,116,126 (Fig. 3B). Of note, these necroptotic cell death signaling pathways to NLRP3 inflammasome activation were dependent on the kinase activities of RIPK1 and/or RIPK3, as well as the common event, K+ efflux.115,116,126
Controversially, mitochondrial dysfunction and apoptosis have been suggested to be the universal trigger for canonical NLRP3 activation (see Section 4.2). Until recently, whether direct activation of mitochondria-dependent apoptosis could drive NLRP3 inflammasome activation in innate immune cells remained unclear. Work by the laboratory of Thomas Naderer recently identified that upon infection of macrophages, Legionella inhibits host cell protein synthesis, and accordingly, reduces pro-survival MCL-1 levels, thereby rendering infected cells exquisitely sensitive to BAX/BAK-dependent apoptotic cell death upon BCL-XL inhibition.130 Remarkably, two subsequent studies have now discovered that in LPS-primed macrophages, activation of BAX and BAK signaling can trigger a novel K+ efflux-dependent pathway to NLRP3 inflammasome and IL-1β activation (Fig. 3B; Fig. #4, 11, 130,131). BAX/BAK activation, upon chemical/genetic BCL-XL and MCL-1 loss, or treatment with myxobacteria-derived cyclic peptides, vioprolides (targets BCL-2 and MCL-1), was shown to cause IAP degradation to promote caspase-8-mediated activation of IL-1β.131,132 However, wholly unexpectedly in the paper by Vince et al., caspase-8 did not act upstream of NLRP3 activation, as posited by previous studies.53,116 Instead, intrinsic apoptotic activation of effector caspases, caspase-3 and -7, triggered NLRP3 activation via K+ efflux, which was presumably caused by plasma membrane damage.131 Remarkably, caspase-3 and caspase-7 activity was also found to act upstream of caspase-8 activation.131 Furthermore, cellular demise and IL-1β secretion proceeded largely in the absence of caspase-1 and caspase-3, and their respective pyroptotic cell death effectors GSDMD and GSDME.2,3,131,133,134 Given the potential for three programmed cell death pathways to converge on NLRP3 activation, one could collectively consider them alternative cell death-induced inflammasomes.
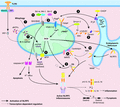
Proposed models for NLRP3 inflammasome activation via mitochondrial dysfunction. Canonical stimuli are proposed to trigger NLRP3 inflammasome assembly and activation by damaging the mitochondria and inducing mitochondrial ROS (mtROS) production, releasing oxidized mitochondrial DNA (mtDNA) and promoting the mitochondrial localization of NLRP3 and ASC for activation. (1) Mitochondrial autophagy, mitophagy, limits NLRP3 activation by eliminating damaged or stressed mitochondria from the cell. (2-6) NLRP3 activation can be mediated by VDAC1/2-mediated mtROS production and (3) BAX/BAK activation, which can both be repressed by pro-survival protein BCL-2. (4, 6) ER stress induces C/EPB homologous protein (CHOP)-mediated transcriptional control of IP3R-dependent Ca2+ efflux from the ER into the cytosol. This induces transport of Ca2+ ions into the mitochondria that generates mtROS. (5) mtROS induces deubiquitylation of NLRP3 to promote inflammasome activation, and (6) may facilitate mtDNA release to activate NLRP3 by inducing mitochondrial pore transition (MPT) pore opening. (7) TLR4-IRF1 mediated transcriptional up-regulation of CMPK2 can generate short strands of mtDNA that exit the mitochondria in an oxidized form to bind and activate the NLRP3 inflammasome. (8) Depletion of mitochondrial NAD+ upon Sirtuin 2 inhibition may lead to hyper-acetylation of α-tubulin and induce Dynein-mediated transport of ASC on the mitochondria to NLRP3 on the ER, and facilitate NLRP3 activation. (9, 10) Cardiolipin and the mitochondrial antiviral signaling protein (MAVS) can reportedly tether NLRP3 to the mitochondria for activation in a mtROS-dependent manner. (11) Specific activation of BAX/BAK triggered by loss of BCL-XL and MCL-1 activity can trigger NLRP3 activation, via activation of downstream effector caspases, caspase-3 and -7, in a K+ efflux-dependent manner (see Fig.)
3 NLRP3 IN HEALTH AND DISEASE
3.1 Cryopyrin associated periodic syndromes and inflammatory disease
Historically, NLRP3 was first linked to the development of Familial cold autoinflammatory syndrome (FCAS) and Muckle-Wells syndrome (MWS), which are both characterized by dysregulated systemic inflammation leading to intermittent episodes of rash, arthralgia, fever, and conjunctivitis.135 Subsequently, a spectrum of typically dominantly inherited autoinflammatory disease syndromes have been described in patients harboring “gain-of-function” mutations in NLRP3 that precipitate in IL-1β activation. These include the mild to moderate features observed in FCAS and MWS, through to the severe and often fatal disease manifestations exhibited in chronic infantile neurologic, cutaneous, articular (CINCA) syndrome, also known as neonatal-onset multisystem inflammatory disease (NOMID).136,137 In 2004, the laboratory of Jürg Tschopp established that these NLRP3 mutations induced spontaneous inflammasome assembly and caused pathogenic IL-1β secretion.6 These syndromes are now collectively known as cryopyrin-associated periodic syndromes (CAPS). Most CAPS patients (∼60-75%) are heterozygous germline carriers of missense mutations in the NLRP3 gene located on chromosome 1 (1q44), with 209 different nucleotide variants listed in the infevers database and ∼90 mutations described to date.137 However, in CINCA/NOMID somatic mutations during fetal development account for ∼40% of the affected paediatric patients. It is also now emerging that somatic NLRP3 mosaicism may account for CAPS symptoms in previously documented “mutation negative” patients.138 Importantly, most of the mutations in NLRP3 disrupt exon 3, which encodes the NACHT domain that is required to control NLRP3 oligomerization. Generation of knock-in mice harboring CAPS patient point mutations has confirmed the requirement for elevated myeloid cell-specific NLRP3 inflammasome-mediated IL-1β activation to drive disease pathologies.139 Furthermore, the rapid remission of CAPS symptoms in 97% of patients following anti-IL-1β monoclonal antibody (i.e., Canakinumab) therapy has confirmed the pathologic role for IL-1β in these syndromes.140
In addition to these autoactivating NLRP3 mutations associated with CAPS, single nucleotide polymorphisms in NLRP3 have been associated with increased caspase-1 activity in common diseases, such as Crohn's disease, rheumatoid arthritis, ankylosing spondylitis, and celiac disease.141 The importance of NLRP3 inflammasome-mediated activation of IL-1β and IL-18 in the pathogenesis of common diseases has largely been validated in preclinical models, including models of Alzheimer's disease,142 acute graft-versus-host-disease,143 multiple sclerosis,144 liver inflammation and fibrosis,145 gout,146 obesity and type 2 diabetes,147 and cardiovascular disease (e.g., atherosclerosis).148 Moreover, the results from the phase I clinical trial of Canakinumab (Canakinumab Anti-inflammatory Thrombosis Outcomes Study [CANTOS] clinical trial) in cardiovascular indications, suggest that inhibiting NLRP3 inflammasome or IL-1β activity holds promise for cardiovascular and metabolic diseases.149,150 This potential has led to a growing interest in developing compounds that target inflammasome machinery. In particular, agents that target NLRP3 itself, such as CP-456,733 (also known as MCC950), have been developed and tested successfully in a range of preclinical disease models (2018). Impeding the progression of NLRP3 antagonists to the clinic is the lack of knowledge surrounding mechanisms of activation, protein structure, and drug targeting.
3.2 Mitochondrial dysfunction-induced NLRP3 activation in metabolic disorders
NLRP3 inflammasome activation is commonly linked with metabolic disorders that exhibit well-defined host-derived triggers. These include uric acid crystals in gout,146 islet amyloid polypeptide (IAPP) in type 2 diabetes,152 crystalline cholesterol generated from oxidized LDL uptake in atherosclerosis,148 and fibrillar peptide amyloid-beta in Alzheimer's disease.153 These crystalline and amyloid NLRP3 agonists activate the canonical NLRP3 pathway largely via “frustrated” phagocytosis and lysosomal rupture (see Section 4.3.2). In this section, we discuss the emerging concept that oxidative stress and mitochondrial dysfunction are key mechanisms triggering NLRP3 activation in aging and obesity-induced metabolic syndrome.
3.2.1 Mitochondrial perturbation in metabolic disease and aging
One common denominator in the development of metabolic disease and degenerative aging is mitochondrial dysfunction. This is not surprising, because mitochondria are the central organelle responsible for many key metabolic processes, of which, the most important is the stepwise conversion of glucose or fats into the cellular energy source ATP. In the case of glucose, pyruvate generated via glycolysis is next converted into acetyl-coenzyme A (acetyl-coA). Acetyl-coA is further metabolized within the mitochondria matrix via the Krebs cycle (also known as the citric acid cycle or tricarboxylic acid [TCA] cycle) to yield NADH and FADH2, which then undergo oxidative phosphorylation via the electron transport chain to generate ATP.154 Mitochondrial fatty acid β-oxidation is another critical pathway to yield energy upon uptake of free fatty acids liberated via lipolysis. Fatty acid oxidation of long chain acyl-CoA esters, which form the backbone for phospholipid, cholesterol, and triacylglycerol synthesis, generates acetyl-coA that is utilized by the Krebs cycle and oxidative phosphorylation to generate ATP.154 Interestingly, a side product of oxidative phosphorylation is the production of mtROS that can cause cellular and mitochondrial DNA (mtDNA) damage.154 Both mtROS and oxidized mtDNA have been proposed to directly activate the NLRP3 inflammasome (see Section 4.2).
The role of mitochondrial dysfunction in aging is well established; by the fact that aging mitochondria lose their integrity, exhibit morphologic abnormalities, and decreased oxidative phosphorylation, as well as a general decrease in mitochondrial mass and mtDNA copy-numbers (2013). Similarly, mitochondria isolated from the skeletal muscle and adipose tissue of obese and/or insulin-resistant patients display decreased oxidative phosphorylation capacity, lipid metabolism, and reduced mitochondrial size that is associated with impaired glycolysis rates.156,157 It is now becoming abundantly clear that these mitochondrial defects associated with metabolic stress may contribute to NLRP3 activation in metabolic disease. For example, reduced activity of key glycolytic enzyme, hexokinase, is not only associated with defective mitochondrial bioenergetics in metabolic disease,158 but can also activate the NLRP3 inflammasome (see Section 4.1).159 Moreover, and as discussed below, defective glucose metabolism is also linked to oxidative stress and thioredoxin-interacting protein (TXNIP)-mediated NLRP3 activation,160,161 albeit controversially (see Section 4.2.2). Accumulation of free fatty acids, particularly saturated fatty acid palmitate, in nonadipose tissues (e.g., muscle, pancreatic β-cells and cardiomyocytes) induces mitochondria-dependent apoptotic cell death and lipotoxicity, which is now associated with the activation of NLRP3 via mtROS production162,163 (see Section 3.2.2). Finally, reduced levels of mitochondrial mitofusin proteins (mediate mitochondrial fusion) and increased levels of dynamin-related protein 1 (DRP1; mediates mitochondrial fission) are associated with metabolic disease progression and NLRP3 activity.164,165 For instance, increased DRP1 activity and mitochondrial fission is linked with NLRP3 activation in high glucose situations (see Section 4.2.4).165
3.2.2 NLRP3 activation in degenerative aging
Loss of metabolic control upon aging is linked to increased canonical NLRP3 inflammasome activity in myeloid cells and chronic low-grade systemic sterile inflammation or “inflammaging.” For example, in aging mice NLRP3 activity promotes a decline in glycemic control, and induces thymic involution and T cell senescence, bone loss, as well as hippocampal astrogliosis that is associated with cognitive decline and dementia.166,167 How NLRP3 is triggered during the aging process remains uncertain, although as discussed above, there is accumulating evidence that defective mitochondrial biogenesis, and an accumulation of DAMPs with age and sedentary lifestyles (e.g., free fatty acids, cholesterol, ceramide) drives NLRP3 activation. With advanced age, elevated TNF levels have also been linked to increased transcriptional up-regulation of NLRP3 for activation. In point of fact, neutralization of TNF in aging mice dampened NLRP3 expression and caspase-1 cleavage in adipose tissues (and liver), improved glucose metabolism, and attenuated insulin resistance.168 Interestingly, one recent study also suggested that the macrophage somatotropic (growth hormone and insulin-like growth factor-1) axis up-regulates NLRP3 expression and activity in aging visceral adipose tissue to drive chronic inflammation and metabolic dysfunction.169 In addition to driving chronic IL-1β activity in aging, NLRP3 activation also maintains macrophage numbers in visceral adipose tissue and blunts lipolysis by increasing the secretion of growth differentiation factor 3 and monamine oxidase A, which lowers the amount of bioavailable lipolysis-inducing norepinephrine.170 This reduced capacity for lipolysis is associated with the common signs of aging, including increased adiposity, lower exercise capacity, reduced thermoregulation during cold stress, and reduced ability to survive starvation. NLRP3 inflammasome activity may also globally contribute to degenerative aging, and most likely tumorigenesis, as downstream IL-1 signaling promotes cell senescence and associated activation of the proinflammatory senescence-associated secretory phenotype.171
3.2.3 Fatty acids and NLRP3 activation in obesity-induced metabolic disease
Nutrient overload through the consumption of saturated fats, cholesterol, and highly processed carbohydrates (i.e., a Western lifestyle diet) induces chronic low-grade inflammation that causes metabolic syndrome. A weight of evidence gathered from obese patients and preclinical models suggests that elevated NLRP3 and IL-1β activity drives this inflammation and contributes to insulin resistance.172 In fact, NLRP3-deficient mice have been reported to exhibit significant protection from high fat diet-induced metabolic syndrome, characterized by reduced sterile inflammation (i.e., a decrease in the frequency of effector T cell infiltrate in visceral adipose tissue), reduced adipocyte hypertrophy and liver steatosis, and preservation of pancreatic β-cell mass that results in improved insulin sensitivity and glucose tolerance.147 A number of studies have now largely confirmed this pathogenic role for NLRP3 and ASC in obesity-related disorders, such as insulin resistance and Type 2 diabetes, nonalcoholic steatohepatitis (NASH) and cardiovascular disease (e.g., atherosclerosis).172,173,174,175,176
Accumulating evidence suggests that gut dysbiosis contributes to NLRP3 inflammasome activation in metabolic syndrome via the release of microbiome-derived LPS to prime the inflammasome.177 In point of fact, morbidly obese people exhibit chronically elevated levels of LPS,178 and mice lacking TLR4 are protected from high fat diet-induced inflammation and disease manifestations associated with metabolic syndrome (e.g., NASH, atherosclerosis).179,180,181,182 Dietary saturated fatty acids can also signal via PRRs, such as TLR4 and the endocytic receptor CD36, to induce transcription of inflammatory cytokines and NLRP3 itself.183,184 Moreover, saturated fatty acids, such as palmitate and its breakdown product ceramide, have been shown to trigger activation of the NLRP3 inflammasome in macrophages and dendritic cells.147,185,186
Exactly how saturated fatty acids trigger NLRP3 remains unclear. Palmitate was shown to inactivate AMP-activated protein kinase (AMPK) and inhibit autophagic clearance of damaged mitochondria, thereby triggering mtROS-induced activation of NLRP3, presumably via oxidized mtDNA release.186 An alternative model proposed that palmitate-induced lipotoxicity induces ER stress responses and the resultant activation of inositol-required enzyme 1 (IRE1) triggers NLRP3 activation. Unexpectedly, this IRE1 pathway induced by saturated fatty acids did not require the unfolded protein response, nor did it require the mobilization of Ca2+ or downstream target TXNIP (see Sections 3.2.3 and 4.2.2).187,188 Instead, IRE1 activation was associated with mtROS-induced NLRP3 activation.188 Remarkably, one recent report suggested that akin to cholesterol crystals and amyloid-β fibrils generated upon CD36-medated phagocytosis,184 saturated fatty acid uptake may induce crystalline structures to cause lysosomal rupture and activate NLRP3 in macrophages.189
Omega-3 polyunsaturated fatty acids are reported to counterbalance saturated fatty acid-induced metabolic dysfunction by altering the gut microbiota and increasing the production of short chain fatty acids (e.g., butyrate). It has been shown that omega-3 fatty acids can block canonical NLRP3 activation and protect from high fat diet-induced metabolic dysfunction, although this effect may be partly due to effects on inflammasome priming.190,191 The laboratory of Vishwa Deep Dixit also recently revealed that the ketone body β-hydroxybutyrate, which is generated by short chain fatty acid metabolism in the liver during periods of energy deficit, can act as a negative metabolic regulator of NLRP3 inflammasome activation to both canonical triggers and lipotoxic fatty acids by blocking K+ efflux.192,193 Moreover, administration of β-hydroxybutyrate, or a ketogenic-diet, have also been shown to suppress inflammasome activity and inflammation in preclinical models of MSU-induced gouty peritonitis and CAPS.192,193 How β-hydroxybutyrate exerts its activities and the levels required to combat chronic inflammation still needs to be resolved.
3.2.4 Hyperglycemia, oxidative stress and TXNIP in NLRP3 activation and metabolic disease
TXNIP is an inhibitor of thioredoxin, a major thiol-reducing and antioxidant protein. Over the last few years TXNIP has garnered attention as an important regulator of glucose and lipid metabolism. Evidence for a connection between TXNIP and metabolic disease was revealed through a genome wide study that reported high expression of TXNIP in pre-diabetic and diabetic patient muscle biopsies.194 Subsequently, specific TXNIP variants have been associated with hyperglycemia and high triglyceride levels in diabetes patients.195 Correlating with this data, high glucose induces TXNIP expression in numerous cell types associated with metabolic disease (e.g., adipocytes, skeletal myocytes, and β-cells), and conversely, insulin can repress TXNIP expression.160,194,196 Moreover, TXNIP promotes glucose uptake by insulin responsive cells,194 and its overexpression promotes β-cell loss (a hallmark of diabetes) via mitochondrial apoptosis upon exposure to high glucose, while its deficiency prevents cell death,197,198 Fitting with a key role in glucose metabolism, mice lacking TXNIP are hypoglycemic, hypoinsulinemic and also exhibit a central defect in glucose release both to a normal diet and upon high fat diet challenge.199 Intriguingly, palmitate has been shown to suppress glucose-mediated TXNIP expression and attenuate ROS generation in β-cells, but not to alter cell death.198,200 Collectively, these results suggest that fatty acids and insulin act to counterbalance TXNIP-dependent β-cell oxidative stress caused by hyperglycemia, and highlights the fact that palmitate utilizes a TXNIP-independent pathway to induce β-cell apoptosis.201
The correlation between oxidative stress, TXNIP activity and NLRP3 was first made following the discovery that TXNIP binds to NLRP3, using a yeast two-hybrid screen.160 Subsequently, ER stressors (e.g., thapsigargin) and/or high glucose have been suggested to trigger NLRP3 inflammasome activation in islets, adipose tissue/adipocytes, macrophage and monocytes in a TXNIP-dependent manner160,196,202 (see Section 4.2.2 for ambiguities surrounding TXNIP involvement in NLRP3 activation). It is worth noting that there are discrepancies between mouse and human as to whether β-cells have a functional NLRP3 inflammasome.203,204 One recent study reported that mouse β-cells only express low levels of NLRP3 machinery, and consequently observed that high glucose failed to activate caspase-1 or IL-1β in WT and NLRP3 autoactivating mutant β-cells.203 Therefore, it will be important to clarify the exact mechanisms and cell-types by which metabolic stressors trigger NLRP3 activity. This will be vital as the ROS-TXNIP-NLRP3 pathway is implicated in the pathology of a growing number of metabolic disease models, such as type 1 and 2 diabetes,205 arterial damage206 and conversion of NASH to fatty liver disease.207
4 MECHANISMS FOR NLRP3 ACTIVATION
Based on the diverse nature of canonical NLRP3 activators, it is widely assumed that there must be a universal mechanism to trigger NLRP3 activation. Currently several common molecular and cellular events have been proposed to induce the activation and assembly of the NLRP3 inflammasome (Figs. 1 and 4), including K+ efflux, Ca2+ mobilization, lysosomal rupture, ROS production, and mitochondrial dysfunction. Of these, K+ efflux is widely believed to be the unifying event for NLRP3 inflammasome activation,45,95 with a few notable exceptions.53,208 Nevertheless, mitochondrial dysfunction has recently emerged as a major, if not controversial, contender. It therefore remains of great debate as to whether there is a single universal event responsible for NLRP3 inflammasome activation. In this section, we will discuss in detail the strengths and weaknesses surrounding the most widely documented mechanisms—K+ efflux and mitochondrial dysfunction—and give a brief overview on the ambiguities surrounding other potential triggers.
4.1 The universal event: potassium (K+) efflux
K+ efflux is the most common upstream event required for NLRP3 inflammasome activation.45 Extracellular ATP binding the purinergic P2X7R, and pore-forming toxins like potassium ionophore nigericin, were the first stimuli demonstrated to lower intracellular K+ levels and activate NLRP3.209,210 Subsequently, the laboratory of Gabriel Núñez has established that almost all known stimuli trigger NLRP3 in a K+ efflux-dependent manner.45 In fact, exposure of LPS-primed macrophages to high extracellular potassium (range 30–60 mM) inhibits NLRP3 activation to ATP, pore-forming toxins, and lysosomal disruptors (i.e., crystalline/particulates and Leu-Leu-O-methyl ester [LL-OMe]). Fittingly, lowering extracellular K+ levels promotes spontaneous NLRP3 activation.211,212 In line with this unifying model for NLRP3 activation, K+ efflux is also required for noncanonical activation of the NLRP3 inflammasome,95 and for most alternative and cell death-induced inflammasome pathways126 (Figs. 2 and 3).
Despite it being clear that perturbations in membrane integrity lead to K+ efflux and activation of NLRP3, how K+ efflux occurs to most NLRP3 triggers remains unresolved. Most studies have focused on pore-forming toxins and H+/K+ ionophores, such as nigericin, and the ATP-triggered P2X7R cation channel. In the case of extracellular ATP binding P2X7R, a larger nonselective pannexin-1 pore was originally implicated in the activation of caspase-1 activation and IL-1β release.213 However, later studies indicated that the pannexin-1 hemichannel was not essential for increased permeability to pore-forming toxins or cell regulatory volume decreases.214,215 In fact, examination of pannexin-1-deficient macrophages suggested that pannexin-1 is dispensable for both DAMP uptake and NLRP3 activity.216 It should be noted, that whilst pannexin-1 is not obligatory for extracellular ATP-induced NLRP3 activation, caspase-11 has also been proposed to cleave and activate pannexin-1 hemichannels to permit the extracellular release of ATP to trigger P2X7R-mediated events (see Section 2.2.1 and Fig.).95 Intriguingly, a K+ channel inhibitor screen recently performed by the laboratory of Asrar Malik, uncovered that a quinine-sensitive two pore TWIK2 potassium efflux channel, may be required for ATP/P2X7R-mediated activation of NLRP3.217 As these studies focused on ATP/P2X7R-mediated events, it remains to be seen how K+ efflux is triggered by an ever-growing number of canonical triggers.
Another conundrum in the K+ efflux model is the mechanism for NLRP3 activation. The work of Núñez and colleagues has clearly demonstrated that K+ efflux acts upstream of NLRP3 activation. In macrophages harboring the autoactivating NLRP3R258W mutation, K+ efflux was not required for NLRP3 activation and high extracellular potassium failed to inhibit inflammasome activity.45 NEK7 has been shown to bind the LRR domain of NLRP3 in a K+ efflux-dependent manner to trigger NLRP3 inflammasome oligomerization and activation.57,58 The fact inflammasome activation in NLRP3R258W mutant macrophages critically required NEK7 but not K+ efflux, may argue for a role for K+ efflux in inducing a conformational change to promote NEK7-NLRP3 interactions.45,57 Despite the clear connect between K+ efflux and NLRP3 activation, it remains to be seen if reduced intracellular K+ levels directly trigger NLRP3 activation, or if other homeostatic changes are required. The laboratory of Rongbin Zhou recently proposed that nigericin and ATP activate chloride intracellular channels (CLICs) downstream of K+ efflux and mtROS, and thereby promote NEK7 interactions with NLRP3.218 However, in direct contradiction, Núñez and colleagues observed K+ efflux and Na+ influx in response to nigericin but no change in intracellular Cl− levels.45 Zhou and colleagues postulated that these discrepancies may be explained by the transient nature of Cl− ion flux in response to activators in their hands. As other groups have also implicated CLICs in the priming and activation of NLRP3,219 it may be worth revisiting the role of ion flux homeostasis in NLRP3 inflammasome activation.
At face value, K+ efflux appears to be the unifying event to trigger NLRP3 inflammasome activation; yet recent reports support the possibility that K+ efflux-independent NLRP3 triggers exist. For example, alternative NLRP3 inflammasome activation via a RIPK1-caspase-8 complex in human monocytes occurs in the absence of cell death (and thus plasma membrane damage) and does not require K+ efflux.53 Likewise, cytosolic Gram-positive bacterial peptidoglycan-N-acetylglucosamine was shown to bind and inhibit the glycolytic enzyme hexokinase resulting in its dissociation from the mitochondrial outer membrane and activation of NLRP3 in a K+ efflux-independent manner. How hexokinase release from the mitochondria activates NLRP3 remains unclear, but Wolf et al. eluded to the possibility that disruption of hexokinase interactions with voltage-dependent anion channels (VDACs) may facilitate mtDNA release.159 Importantly, blockade of glycolysis also reportedly triggered NLRP3 activation by causing mitochondria damage and mtROS production.220 Finally, the laboratory of Olaf Groß recently reported that TLR7 agonist imiquimod, and related molecule CL097, also target the mitochondria, specifically the ROS-producing quinone oxidoreductases NQO2 and mitochondrial Complex I, to trigger NLRP3 activation in a manner independent of K+ efflux but dependent on NEK7.208
4.2 An emerging culprit: Mitochondrial dysfunction
Mitochondria play an essential role in the generation of energy in the form of ATP. However, as discussed above, mitochondria also critically regulate intrinsic “mitochondria-dependent” apoptosis. In addition, mitochondria have also recently been implicated in innate antiviral immune responses. For example, upon sensing cytoplasmic viral RNA, the retinoic acid inducible gene-I-like (RIG-I) receptors, RIG-I and MDA5, signal via the outer mitochondrial membrane adaptor protein mitochondria antiviral signaling (MAVS) to activate NF-κB and IFN regulator factor 3 (IRF3) transcription factors and induce type I IFN production.221 Likewise, mitochondrial apoptotic signaling causes the release of mtDNA, which in the absence of intrinsic apoptotic caspase activity, triggers the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) to drive IFN responses.222,223 Finally, there is an emerging dogma that mitochondrial dysfunction is the universal trigger for NLRP3 activation, via events including ROS and mtDNA release, apoptosis, and mitochondrial localization (Fig. 4).
4.2.1 BCL-2 levels
Mitochondria were first implicated in inflammasome activation in 2007 through work performed in the laboratory of John Reed, which showed that the BCL-2 pro-survival family members, BCL-2 and BCL-XL could bind and suppress NLRP1 inflammasome activation.224 Consistent with this finding, a vaccinia virus homologue of BCL-2, F1L, was later found to also bind and inhibit NLRP1 and promote virulence.225 Possible caveats to the work by Reed and colleagues, is the growing evidence that the bacterial peptidoglycan muramyl dipeptide (MDP) used in this study may not be a ligand of NLRP1.226 This is best exemplified by the fact that macrophages derived form NLRP1b-deficient mice (129 genetic background) respond normally to MDP-conjugated to adjuvant titanium oxide (TiO2) and L18-MDP (that has improved cellular uptake).227 It should be noted that TiO2 is a bona fide NLRP3 trigger.227,228 Key experiments were also performed in the presence of the now recognized NLRP3 trigger, ATP.210 As such, reports of enhanced or diminished inflammasome-associated IL-1β activity in macrophages deficient or overexpressing BCL-2, respectively, is more suggestive of a role for BCL-2 in limiting activation of the NLRP3 inflammasome.161,229 However, more recently, mice overexpressing BCL-2 in the haematopoietic system were shown to exhibit comparable IL-1β responses upon MSU crystal injection, compared to littermate control mice.230 Likewise, BMDMs derived from two independent BCL-2 transgenic mouse lines exhibited normal NLRP3 inflammasome responses to a range of canonical triggers.123
4.2.2 Autophagy, mitophagy, and ROS
Autophagy is a conserved cellular process for the delivery and targeted lysosomal degradation of cytosolic constituents, such as proteins and damaged organelles. In 2008, the laboratory of Shizuo Akira first made the connection between autophagy and NLRP3 inflammasome regulation by demonstrating that loss of autophagy-related protein ATG16L1 in mice rendered them sensitive to spontaneous IL-1β activation upon TLR ligation, and exaggerated canonical NLRP3 responses.231 Subsequently, chemical inhibition of autophagy, and deficiency in a range of autophagy related proteins, including microtubule light chain 3 beta (LC3B), Beclin1, ATG5, and ATG7, have been shown to trigger NLRP3 activation.231,232,233 Moreover, mice deficient in LC3B were highly susceptible to the cecal-ligation and puncture-induced sepsis model and produced excessive levels of NLRP3 inflammasome-associated IL-1β.232
Autophagy has been proposed to limit inflammasome activation and IL-1 family cytokine production and secretion in several ways. One obvious way autophagy could limit NLRP3 activation is by delivering inflammasome machinery into the lysosomes for degradation. Arguing against this theory, is the fact that early studies showed no significant differences in NLRP3, ASC, caspase-1 and pro-IL-1β levels in macrophages lacking autophagy machinery, despite increased NLRP3 activity.231,232 In work performed using chemical inhibitors and activators of autophagy, a role for autophagy in the degradation of pro-IL-1β was counter-proposed.234 Subsequently, TRIM20/MEFV has emerged as an autophagic receptor to promote the degradation of inflammasome machinery, including NLRP3, caspase-1 and NLRP1. In fact, mutations in MEFV, which are associated with Familial Mediterranean Fever, diminish recruitment of autophagy machinery to NLRP3 and promote inflammasome activity.235 Collectively, this work suggests that autophagy acts to remove inflammasome machinery and limit inflammation. Remarkably, it is now emerging that inflammasomes themselves may coordinate their own autophagic destruction via caspase-1 activation.236,237,238
ROS have long been associated with NLRP3 inflammasome activation. Initial studies using RNA interference against NADPH oxidase and ROS scavengers suggested that cellular ROS was the trigger for NLRP3.211,239 However, later studies using peripheral blood mononuclear cells from chronic granulomatous disease patients that exhibit defective NOX1-4 activity, and mice genetically deficient in NADPH oxidase subunits, suggested that NADPH oxidase was not required for NLRP3 activation to canonical activators or upon autophagy inhibition.234,240,241 In 2011, two independent laboratories proposed that stressed mitochondria were the source of ROS essential for NLRP3 activation, and that autophagic clearance of damaged mitochondria (mitophagy) was required to prevent mtROS production and excessive NLRP3 activation161,232 (Fig. 4). Zhou et al. postulated that canonical NLRP3 inflammasome activation was dependent on VDAC1/2 facilitating Ca2+ uptake into the mitochondria to generate mtROS. Increased mtROS subsequently disrupted TXNIP/thioredoxin interactions and allowed TXNIP to bind and recruit NLRP3 and ASC to the mitochondria-associated ER membranes (ER-MAMs) for activation (Fig. 4).161 In contrast, Nakahira et al. suggested that LPS and ATP-induced mtROS triggered mitochondrial permeability transition (MPT) pore opening to cause mtDNA release and inflammasome activation (Fig. 4, 6).232 Notably, in this scenario, NLRP3 was required for mtDNA release, suggesting that mtDNA release acted to amplify inflammasome activity. Subsequently, other groups reported similar models that centralized around mitochondrial damage inducing mtROS and mtDNA release (see Section 4.2.3 and Fig.).229,242 Based on these models, there has been great interest in connecting mitochondria, oxidative stress, and NLRP3 inflammasome activation with disease, as discussed above (see Section 3.2).
Stemming from the work of Zhou and colleagues,161 it is now widely assumed that mtROS is the common event triggering activation of NLRP3. Nevertheless, several groups have now failed to reproduce these findings and thereby weakened the mitochondrial dysfunction model. For instance, the spontaneous NLRP3 activation observed upon inhibition of the mitochondrial respiratory chain complex with rotenone is not reproducible.45,63,243 Similarly, it was inferred that ROS-dependent recruitment of TXNIP to the mitochondria was required for NLRP3 binding and activation; however, a study by the laboratory of Luke O'Neill observed no defect in NLRP3 activation in TXNIP-deficient macrophages exposed to IAPP, MSU, silica, and ATP.152 Intriguingly, a recent study may point to an indirect role for TXNIP in NLRP3 activation, as TXNIP represses LPS-induced generation of nitric oxide that is a putative negative regulator of NLRP3 activation.74,244 Finally, the data supporting the concept that BCL-2 suppresses VDAC1/2 to limit mtROS-induced NLRP3 inflammasome activation has been directly contradicted by another recent study that showed normal canonical NLRP3 activation in BCL-2 transgenic macrophages.123
The laboratory of Tiffany Horng suggested that NLRP3 activators universally trigger Ca2+ mobilization from the ER stores to damage mitochondria, induce ROS, and causes a loss of mitochondrial membrane potential to release mtDNA (Fig. 4, 6). Complicating this model, is the fact that Ca2+ influx has been demonstrated downstream of ROS production induced by crystal and liposome NLRP3 activators.245 It also appears that mtROS is not obligatory for NLRP3 activation to all stimuli, including some pore-forming toxins, influenza, encephalomyocarditis and measles viruses, and even the widely used trigger ATP.45,246,247,248 Moreover, the ROS scavengers and inducers of mtROS utilized to model NLRP3 activation may have off-target effects. For example, Veit Hornung and colleagues documented that ROS scavengers, diphyliodonium (DPI) and N-acetylcysteine (NAC), can blunt LPS-induced NLRP3 inflammasome priming rather than block activation.240 Nevertheless, others have shown that addition of ROS scavengers after TLR priming can still inhibit NLRP3 activity.29,245,249 Intriguingly, mtROS has now been suggested to contribute to nontranscriptional priming of NLRP3 by promoting deubiquitylation of NLRP3 upon LPS stimulation63 (Fig. 4).
The concept that clearance of damaged ROS-producing mitochondria occurs to limit NLRP3 inflammasome activation is largely based on data obtained upon the genetic loss of autophagy machinery or chemical inhibition of autophagy161,231,232 (Fig. 4). Chemicals that induce mitochondrial pore transition (MPT), such as the mitochondrial un-coupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), can activate NLRP3 but also trigger the accumulation of mitochondrial serine/threonine kinase Pink1 to recruit the E3 ubiquitin ligase Parkin (encoded by Park2) and activate mitophagy.161,250 Challenging the belief that mitophagy restricts inflammasome activation, a study by Allam et al. discerned no defect in NLRP3 inflammasome activation in Park2-deficient macrophages to canonical activators ATP, MSU, and alum, as well as apoptotic stimuli, despite the accumulation of mitochondria.123 In contrast, in a more recent study, Parkin deficiency was associated with exaggerated responses to NLRP3 activators.251 The reason for these discrepancies remains unclear, although, Parkin has recently been ascribed nonmitophagic functions that might affect NLRP3 activation, including the K63 ubiquitylation of RIPK1 to promote activation of NF-κB.252
4.2.3 Apoptosis and mtDNA release
Apoptotic cell death is classically viewed as an immunologically silent process; however, as discussed above, targeted activation of apoptotic caspases, caspase-8 and caspase-3/7, can contribute to activation of the NLRP3 inflammasome and IL-1β in innate immune cells115,116,123,131,132 (see Fig. 3B; Fig. #11). In 2012, the laboratory of Moshe Arditi put forward mitochondrial apoptosis as the unifying model for how diverse stimuli trigger NLRP3 (Fig. 4). It was proposed that NLRP3 activators, such as ATP and nigericin, trigger mitochondrial apoptosis to induce the release of oxidized mtDNA that binds and activates NLRP3. Fitting with this idea, apoptosis-inducing drugs, such as the protein translation inhibitor cycloheximide, can trigger NLRP3 activation.229 Discrediting this model, macrophages deficient in essential intrinsic apoptotic effectors BAX and BAK, or alternative effector BOK, or initiator caspase, caspase-9, responded normally to both canonical NLRP3 activators (e.g., alum, Nigericin, and ATP), as well as apoptotic stimuli (e.g., cycloheximide, staurosporine, and ultraviolet irradiation).123,131 Indeed, a growing consensus is that apoptosis-inducing cancer chemotherapeutics promote TLR-induced caspase-8-mediated activation of IL-1β and/or NLRP3 activation.29,50,113
Augustine Choi and colleagues proposed that MPT enables mtDNA release to activate NLRP3232 (Fig. 4). Inconsistent with the notion, NLRP3 activity was an upstream prerequisite for mtDNA release. Evidence for the role of the MPT in NLRP3 activation was also gathered using cyclosporine A, a potent inhibitor of MPT component cyclophilin D.232 A more recent study, however, showed genetically that cyclophilin D is not essential for canonical NLRP3 activation. Furthermore, cyclosporine A still inhibited NLRP3 activation in the absence of cyclophilin D, suggesting that cyclosporine A targets another unknown protein to block NLRP3 activation.123
Recently, the laboratory of Michael Karin reproduced the core finding that mtDNA can activate NLRP3 using transcription factor A, mitochondria (TFAM) knockout macrophages that exhibit reduced mtDNA content.253 Extending these findings, Karin and colleagues suggested that TLR engagement was required to induce the synthesis of new mtDNA, via IRF1-mediated up-regulation of the mitochondrial deoxyribonuleotide kinase, UMP-CMPK2; Fig. #7). Correspondingly, reduced LPS-induced mtDNA synthesis in IRF1- or UMP-CPMK2-deficient BMDMs precipitated in defective oxidized mtDNA release and IL-1β responses to canonical NLRP3 triggers.253 Confusing these matters, however, is a previous report that suggested moderate mtDNA stress caused by TFAM loss in murine embryonic fibroblasts and macrophages leads to reduced total mitochondrial DNA levels and increased cytosolic mtDNA accumulation to trigger cGAS-STING signaling.254 Furthermore, in direct contradiction to this TLR-IRF1-UMP-CMK2 pathway to mtDNA up-regulation, Si Ming Man et al. previously demonstrated that IRF1 is not required for either canonical or noncanonical NLRP3 activation but is obligatory for AIM2 activation.255 A final caveat to the mtDNA model, is that a related study found no defect in ATP-induced NLRP3 responses upon mtDNA depletion with ethidium bromide.256
4.2.4 Mitochondrial localization and homeostasis
Since the original report that mtROS stimulated the recruitment of NLRP3 from the ER to the ER-MAMs to trigger activation,160 several mitochondrial proteins have been linked to this process. For example, mitochondrial damage inflicted by NLRP3 activators is suggested to deplete co-enzyme NAD+ stores. Reduced NAD+ levels, in turn, inactivates the α-tubulin deacetylase sirtuin 2 and promotes acetylated-α-microtubule, dynein-dependent transport of perinuclear mitochondria to the ER-MAMs to bring ASC into the proximity of NLRP3 and drive inflammasome assembly (Fig. 4).257 However, incompatible with this model is the fact that (i) prior to NLRP3 activation ASC is predominantly found in the nucleus and rapidly redistributes to the cytosol upon activation,258 (ii) other reports suggest that NLRP3 is localized to the mitochondria,161 and (iii) the lack of requirement for either mtROS and K+ efflux.257
MAVS was also proposed by the laboratory of Ronald Germain to tether NLRP3 to the mitochondria (Fig. 4) and facilitate activation to soluble (e.g., ATP and nigericin), but not particulate/crystalline matter (e.g., alum, cholesterol crystals, or MSU crystals).259 Challenging this study, several groups have now identified that while MAVS, and interacting protein mitofusin 2, are required for NLRP3 activation to RNA viruses, they do not impact canonical NLRP3 activation.123,247,260,261 Inner mitochondrial membrane phospholipid, cardiolipin, also allegedly binds NLRP3 to localize it to the mitochondria for activation (Fig. 4), as knockdown of cardiolipin synthase attenuated NLRP3 activation.262 Nevertheless, it was recently suggested that mtROS generation at the inflammasome priming stage facilitates both NLRP3 and caspase-1 association with cardiolipin on the outer mitochondria membrane, and thus cardiolipin may act as scaffolding for NLRP3 inflammasome assembly, rather than its activation.263 Notably, if the mitochondria acts as the inflammatory platform and trigger, it remains to be reconciled why in many studies the active inflammasome appears to be cytosolic, as evidenced by a lack of co-localization of ASC specks with organelle markers.264
It is also noteworthy that the mitochondrial GTPase DRP1 has been suggested to be required for NLRP3 activation downstream of RIPK1-RIPK3 upon exposure to dsRNA or RNA viruses, although a latter study failed to reproduce these findings.265,266 It has also recently been proposed that the mitochondrial phosphatase PGAM5, which also maintains mitochondrial homeostasis and triggers mitophagy, is required for mtROS-induced NLRP3 activation in macrophages to not only vesicular stomatosis virus infection but also soluble and particulate triggers.128 It therefore remains plausible that mitochondrial dysfunction may trigger NLRP3 activity under certain circumstances.
4.3 THE MINOR PLAYERS
4.3.1 Calcium mobilization
Intracellular Ca2+ was first implicated in ATP- and Nigericin-induced NLRP3 activation in a study utilizing the Ca2+ chelator BAPTA-AM and ER-Ca2+ ATPase thapsigargin.267 Several studies have now reported that NLRP3 can be triggered by Ca2+ mobilization from the ER stores upon activation of G protein coupled calcium sensing receptor (CaSR) signaling, and Ca2+ influx induced via store operated-Ca2+ entry (SOCE) channels or cationic transient receptor potential (TRP) channels.215,242,245,268,269 Ca2+ mobilization from the ER stores was initially postulated to be a common event to trigger NLRP3 via CaSR, phospholipase C (PLC) and 1,4,5-triphosphate signaling. Indeed, inhibition or knockdown of CaSR, PLC, or the ER Ca2+ release channel, 1,4,5-triphosphate receptor (IP3R), inhibited intracellular Ca2+ levels and ATP-mediated NLRP3 activation. Conversely, high extracellular Ca2+ and direct activation of PLC triggered spontaneous IL-1β activation in the absence of NLRP3 activators.268 A subsequent report suggested that the release of Ca2+ by necrotic cells engaged both the CaSR and related G protein coupled calcium sensing receptor (GPRC6A) to mobilize ER Ca2+ stores and trigger NLRP3.269 A further model proposed that cell swelling followed by regulatory volume decrease triggered TRP vanilloid 2 (TRPV2)-mediated Ca2+ influx and TGF-β-activated kinase 1 (TAK1) activation to activate NLRP3.215 However, this TAK1-dependent pathway has largely been discounted, as genetic deletion of TAK1 has no effect on canonical NLRP3 activation and in fact, promoted spontaneous TLR-induced NLRP3 activation.29,270 In an alternative model, the Transient receptor potential melastatin 2 (TRPM2) channel was implicated in ROS-dependent Ca2+ influx to liposome/crystalline triggers.245 Discordant with the general idea that Ca2+ influx is required for NLRP3 activation, more recent studies have reported that: (i) cytosolic Ca2+ levels were neither essential nor sufficient for NLRP3 activation, compared to the critical effect of K+ efflux, (ii) direct engagement of calcium mobilizing G protein receptors failed to activate NLRP3, (iii) calcium chelator BAPTA-AM and channel blocker 2-APB blocked NLRP3 activation in a manner independent of perturbed Ca2+ levels,271 and (iv) high extracellular Ca2+ (in the presence of high phosphate ion levels) can crystalize and potentially act as a particulate to trigger lysosomal rupture45 (see Section 4.3.2). Therefore, the role of Ca2+ homeostasis in NLRP3 activation remains controversial despite significant genetic evidence for GPRC6A and TRPM2.
4.3.2 Lysosomal rupture
In contrast to soluble NLRP3 activators, particulate and crystalline stimuli, such as silica, alum, β-amyloid, and MSU, as well as bacteria, are phagocytosed and cause lysosomal rupture to promote cathepsin-dependent lysosomal cell death and NLRP3 activation.153,272,273 Recently, cytosolic DNA sensing by cGAS-STING was also shown to induce STING translocation to the lysosome, membrane rupture, and lysosomal cell death-triggered activation of the NLRP3 inflammasome.274 Despite the early belief that this pathway was highly distinct, it has now been established that all particulate/crystalline and lysosomotropic agents induce K+ efflux to activate NLRP3.45
Evidence for this mechanism has largely been gathered using chemical inhibitors that impact phagocytic and lysosomal activities. For example, the potent vacuolar-type H+-ATPase inhibitor bafilomycin A1, which prevents lysosomal acidification, blocked NLRP3 activation to particulate triggers, such as silica and alum.272 Conversely, “low-level” lysosome membrane permeabilization with the soluble lysosomotropic agent, LL-OMe promotes K+ efflux-dependent NLRP3 inflammasome assembly and activation in BMDMs and BMDCs.271 Curiously, doses of LL-OMe that cause extensive lysosomal membrane damage inhibited NLRP3 activation in dendritic cells, via an uncharacterized mechanism that may involve impaired deubiquitylation of NLRP3.271 Several early studies also showed that chemical inhibition of the lysosomal cysteine protease, cathepsin B, using CA-074-Me blocked NLRP3 activation; however, cathepsin B-deficient macrophages did not display major defects in NLRP3 activation to a range of particulates.153,273 This discrepancy now appears to most likely reflect functional redundancy, as CA-074-Me has been shown to target multiple cathepsins.275
5 CONCLUSIONS
There have been considerable attempts made in the last few years to gain a better understanding of the mechanisms behind NLRP3 activation and the downstream events that culminate in IL-1β secretion and cell death. Since the groundbreaking discovery of the noncanonical inflammasome, alternative and programmed cell death signaling pathways have been shown to induce NLRP3 inflammasome assembly and activation. It will therefore be of great interest to mechanistically tease apart these novel pathways, and ascertain their role, particularly specific BAX/BAK activation, in metabolic and common inflammatory diseases. It is worth mentioning that genetic loss or mutation of death receptor and TLR signaling machinery, including XIAP, A20, LUBAC components HOIL and HOIP and most recently RIPK1 and caspase-8, have all been linked to pathogen molecule-associated hyperinflammation linked to NLRP3 and IL-1β activation.110,276,277 A consensus on a universal event required to trigger NLRP3 activation to diverse activators also remains elusive. Based on our current knowledge, it seems clear that K+ efflux remains the most commonly described event precipitating in NLRP3 activation. Mitochondrial dysfunction has emerged as a highly attractive alternative mechanism for canonical NLRP3 activation; however, the large number of conflicting reports and caveats to the proposed models weakens its potential. Despite this controversy, there is considerable evidence in vivo during metabolic diseases that mitochondrial damage and oxidative stress may be involved in NLRP3 inflammasome activation and disease pathogenesis.
AUTHORSHIP
M.Y., D.J.C., D.S.S., and K.E.L. contributed to the writing of this review and generation of figures.
ACKNOWLEDGMENTS
This work was supported by a National Health and Medical Research Council Project grant 1145788 to K.E.L. and operational infrastructure grants through the Australian Government IRISS and the Victorian State Government OIS.
DISCLOSURES
The authors declare no conflicts of interest.
Abbreviations
- AIM2
absent in Melanoma
- AMPK
AMP-activated protein kinase
- APAF-1
apoptosis protease activating factor-1
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- BMDCs
bone marrow dendritic cells
- BMDMs
bone marrow-derived macrophages
- Ca2+
calcium
- CAPS
Cryopyrin-Associated Periodic Syndromes
- CARD
caspase activation and recruitment domain
- CaSR
calcium sensing receptor
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- c-FLIP
cellular FADD-like IL-1β-converting enzyme-inhibitor protein
- cGAS
cyclic GMP-AMP Synthase
- CINCA
chronic infantile neurological, cutaneous, articular
- CLICs
chloride intracellular channels
- DAMPS
damage-associated molecular patterns
- DPI
diphyliodonium
- DRP1
dynamin-related protein 1
- DUB
deubiquitinase
- ER-MAMs
endoplasmic reticulum-mitochondria-associated membranes
- FADD
Fas-associated protein with death domain
- FBXL2
F-box L2
- FCAS
familial cold autoinflammatory syndrome
- GBPs
guanylate binding proteins
- GPRC6A
G protein coupled calcium sensing receptor 6A
- GSDM
gasdermin
- HMGB1
high mobility group box-1
- IAP
Inhibitor of Apoptosis
- IAPP
islet amyloid polypeptide
- IP3R
1,4,5-triphosphate receptor
- IRAK
IL-1 receptor associated kinase
- IRE1
inositol-required enzyme 1
- LC3B
light chain 3 beta
- LL-OMe
Leu-Leu methyl ester
- LRR
Leucine-rich repeat
- LUBAC
linear ubiquitination chain assembly complex
- MARCH7
membrane-associated RING finger protein 7
- MDP
muramyl dipeptide
- MLKL
Mixed lineage kinase domain-like
- MPT
mitochondrial permeability transition pore
- MSU
monosodium urate
- mtDNA
mitochondrial DNA
- mtROS
mitochondrial reactive oxygen species
- MWS
Muckle-Wells syndrome
- NAC
N-acetylcysteine
- NAIP
NLR family of apoptosis inhibitory protein
- NASH
nonalcoholic steatohepatitis
- NEK7
NIMA-related kinase 7
- NLRP3
NOD-like receptor protein 3
- NLRP1
Nucleotide-binding oligomerization domain (NOD)-like receptor 1
- NOMID
neonatal-onset multisystem inflammatory disease
- OxPAPC
oxidized phospholipids
- P2X7R
P2X7 receptor
- PAMPS
pathogen-associated molecular patterns
- PGAM5
PGAM family member 5
- PLC
phospholipase C
- PRR
pattern recognition receptors
- PYD
pyrin domain
- RIPK
receptor interacting protein kinase
- ROS
reactive oxygen species
- SMAC
second mitochondria-derived activator of caspases
- SOCE
store operated-Ca2+ entry
- STING
Stimulator of Interferon Genes
- T3SS
type III secretion system
- TAK1
TGF-β-activated kinase 1
- TCA
tricarboxylic acid
- TFAM
transcription factor A
- TiO2
titanium oxide
- TNFR
tumor necrosis factor receptor
- TRIF
Toll/IL-1R domain-containing adaptor-inducing IFN-β
- TRIM
tripartite motif
- TRP
transient receptor potential
- TRPM2
transient receptor potential melastatin 2
- TRPV2
TRP vanilloid 2
- TXNIP
thioredoxin-interacting protein
- UMP-CMPK2
uridine/cytidine monophosphate kinase-2
- VDAC
voltage-dependent anion channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}