




Abstract
HBI-8000 (tucidinostat) is a novel, oral histone deacetylase inhibitor that selectivity inhibits Class I (histone deacetylase 1, 2, 3) and Class II (histone deacetylase 10) with direct anti-tumor activity through various mechanisms of action, including epigenetic reprogramming and immunomodulation. It has been approved in China for the treatment of relapsed or refractory peripheral T-cell lymphoma.
This multicenter, prospective phase I dose-escalation trial evaluating the safety of twice weekly HBI-8000 was conducted in Japan. Eligible patients had non-Hodgkin’s lymphoma and no available standard therapy. The primary endpoint was maximum tolerated dose; secondary endpoints included anti-tumor activity, safety and pharmacokinetics.
Fourteen patients were enrolled in the study. Twelve patients were assessed for dose-limiting toxicity: six patients in the 30 mg BIW cohort had no dose-limiting toxicitys; two of six patients in the 40 mg BIW cohort had asymptomatic dose-limiting toxicitys. Treatment was well tolerated; adverse events were predominantly mild to moderate hematologic toxicities and were managed with dose modification and supportive care. Thirteen patients were included in the efficacy analysis. Objective response was seen in five of seven patients in the 40 mg BIW cohort; three partial responders had adult T-cell leukemia-lymphoma. In the 30 mg BIW cohort, three of six patients had stable disease after the first cycle.
Treatment with HBI-8000 30 and 40 mg BIW were well-tolerated and safe, with hematological toxicities as expected from other studies of histone deacetylase inhibitor. The maximum tolerated dose and recommended dosage for phase II studies of HBI-8000 is 40 mg BIW. Preliminary efficacy results are encouraging.
Introduction
Histone acetylation and deacetylation play important roles in the modulation of chromatin topology and the regulation of gene transcription and have been shown to directly inhibit the proliferation of tumor cells by inducing cell-cycle arrest, differentiation and/or apoptosis in culture. They are believed to do so by triggering the re-expression of silenced genes important to these pathways or by repressing genes relevant to tumorigenesis and progression (1).
The activity of Class I histone deacetylases (HDACs) (HDAC isoenzymes 1, 2, 3 and 8) is elevated in acute myeloid leukemia, acute promyelocytic leukemia, non-Hodgkin’s lymphoma (NHL), as well as numerous solid tumors (2–6), generalizing their role in malignancies. Small interfering ribonucleic acid (siRNA)-mediated inhibition of HDAC1 or HDAC3 resulted in anti-proliferative effects, and HDAC2 inhibition using siRNA sensitized tumor cells to apoptosis (7,8). HDAC inhibitors (HDACi’s) also synergize with p53 by increasing acetylation in the DNA binding domain of p53 to regulate PD-L1 expression in cancer cells (9).
Several HDACi’s have been investigated as anti-cancer agents (10). HDACi’s have shown single-agent activity against T cell lymphomas, cutaneous T cell lymphomas, mantle cell lymphomas and Hodgkin’s disease (11), as well as the potential for diffuse large B-cell lymphomas and follicular lymphoma (12,13). Several HDACi have been approved for the treatment of lymphomas by the United States Food and Drug Administration, including vorinostat for cutaneous T-cell lymphoma (CTCL), romidepsin for CTCL and peripheral T-cell lymphoma (PTCL), belinostat for relapsed or refractory (R/R) PTCL as monotherapy and panobinostat as combination therapy for multiple myeloma.
HBI-8000 (tucidinostat; chidamide in China) is an orally bioavailable benzamide HDACi and is targeting cancer-associated HDAC isoenzymes. In vitro, HBI-8000 inhibits the growth of a wide variety of tumor cell lines. In vivo, HBI-8000 has produced 60–84% inhibition of tumor growth against a panel of mouse and human xenograft models without weight loss or overt signs of toxicity (14). HBI-8000 selectively inhibits the activity of HDAC1, 2, 3 and 10 and induces growth arrest and apoptosis in blood and lymphoid-derived tumor cells, reverses drug resistance of tumor cells and enhances NK-cell and antigen-specific CD8+ cytotoxic T-lymphocyte-mediated cellular antitumor immunity (15). Using cells obtained from patients newly diagnosed with CLL, Kong et al. showed that HBI-8000 has cytostatic and cytotoxic effects on CLL cells at least partially by regulating autophagy activity at the post-transcriptional level. In addition, HBI-8000 in combination with ibrutinib was synergistic and downregulated ibrutinib-induced autophagy (16).
The first clinical studies of HBI-8000 (as chidamide) were conducted in Chinese patients with T-cell lymphomas. Chidamide (HBI-8000) has been authorized in China as Epidaza® at 30 mg twice weekly (BIW) for treatment of R/R PTCL and of locally advanced/metastatic breast cancer in combination with an aromatase inhibitor.
The present phase I study was planned to investigate the tolerability, safety, pharmacokinetics (PK) and efficacy of HBI-8000 in Japanese patients with R/R NHL. The starting dose of 30 mg BIW was based on results of the Chinese clinical studies.
Patients and methods
Patients
The study population included Japanese patients with NHL and no available standard therapy. Patients must have been age 20 years or older, with histologically or cytologically diagnosed NHL and at least one measurable lesion, ECOG performance status of 0–2, estimated life expectancy of >3 months and adequate major organ functions. Patients were excluded if they had organ transplantation or allogeneic stem cell transplantation, extensive radiotherapy, active infection, prolonged corrected QT Interval (QTc), history of seizures or central nervous system involvement, cardiac arrhythmia requiring anti-arrhythmic medication, active clinically significant bleeding or recent thrombotic disorders.
Study design
The study (ClinicalTrials.gov Identifier: NCT02697552) was conducted in accordance with Good Clinical Practice, International Council for Harmonisation (ICH) guidelines, Japan country-specific regulations and the Declaration of Helsinki. It was approved by the Ethics Committees at each study site and informed consent was obtained from each study patient.
This was a multi-center prospective, open-label, non-randomized, dose-escalation study in Japanese patients with R/R NHL. HBI-8000 was administered orally BIW continuously in 4-week cycles. The primary endpoint was to determine the maximum tolerated dose (MTD) of HBI-8000, based on the frequency of dose-limiting toxicities (DLTs). Secondary endpoints included pharmacokinetics (PK), anti-tumor activity (objective response rate, ORR) and safety.
Patients were enrolled and treated using a standard 3 + 3 design and dose escalation was determined by the occurrence of DLTs within 28 days of receiving the first dose. DLTs were defined as any grade 4 hematologic toxicity, grade 3 or higher febrile neutropenia, grade 3 thrombocytopenia with clinically significant hemorrhage, any other grade 3 or higher non-hematologic toxicity, and grade 1 or higher cardiac troponin I (or troponin T) increases that consistently rise on each of 3 consecutive days. All safety data were reviewed to determine whether recruitment to the next dose level could proceed. The starting dose of HBI-8000 was 30 mg BIW and dose escalation was planned up to 50 mg BIW. HBI-8000 5 and 10 mg tablets were administered BIW approximately 30 min after breakfast. Patients were allowed to continue on the study drug until disease progression, unacceptable toxicity or withdrawal of informed consent.
Assessments
The safety data were reviewed by the Data Monitoring Committee (DMC) and the Safety Monitoring Committee (SMC). The MTD was determined by the independent safety experts, including the DMC/SMC. Dose adjustments and interruptions were based on the presence of DLTs and adverse events (AEs).
Anti-cancer activity was determined by the investigator based on evaluation of nodal/extranodal lesions, skin lesions and peripheral blood lesions. Response was evaluated through radiological imaging (computed tomography), hematology, inspection of skin lesions and bone-marrow aspiration or biopsy. In case of suspected lymphomatous or gastrointestinal lesions, endoscopy or biopsy was performed. Tumor assessments were based on Cheson’s response criteria for non-Hodgkin’s lymphoma (17), JCOG response criteria for adult T-cell leukemia-lymphoma (ATL) (18,19) and dates of progression. Skin lesions were evaluated according to a modification of the Severity-Weighted Assessment Tool (mSWAT) (20).
The PK sampling schedule included plasma collected on Days 1 and 25, before dose and up to 72 hours post-dose. Urine was collected in three 24-hour intervals following HBI-8000 dosing on Days 1 and Day 25 of Cycle 1. Plasma and urine HBI-8000 concentrations and metabolites were analyzed using Liquid Chromatography/Mass Spectrometry (LC/MS–MS), and PK parameters were calculated.
Statistical analyses
MTD was based on the number and incidence of DLTs using all patients who received at least one dose of HBI-8000, excluding any patients who did not complete Cycle 1 at the assigned initial dose level for reasons other than the occurrence of a DLT. AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA) (21) version 19.1. Severity of AEs was graded using criteria of the National Cancer Institute- Common Terminology Criteria for Adverse Events CTCAE version 4.03 (22). PK analysis was performed on all patients who received at least one dose of HBI-8000 in Cycle 1 and had at least one measured HBI-8000 concentration at a scheduled time post-dose. Efficacy analysis was performed on all patients who received two or more doses of HBI-8000 and had valid efficacy assessments at screen and post-dose.
Results
The study was conducted from June 2014 to October 2016
Patients
After providing informed consent, 14 patients were enrolled and treated; seven patients received 30 mg of HBI-8000 and seven patients received 40 mg of HBI-8000. The dose was not escalated to 50 mg BIW. Nine patients were ECOG performance status of 0 and five patients were ECOG performance status of 1 at baseline. Six patients had B cell lymphoma and eight had T cell lymphoma, five of whom had ATL. All patients had received prior chemotherapy, and several patients had received other treatments. Baseline characteristics and disease history are shown in Table 1.
Demographic data, baseline characteristics and disease history
| Characteristic | Category | Statistic | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) |
|---|---|---|---|---|---|
| Sex | Male | n (%) | 4 (57.1) | 3 (42.9) | 7 (50.0) |
| Female | n (%) | 3 (42.9) | 4 (57.1) | 7 (50.0) | |
| Age (years) | Median (min, max) | 72.0 (53, 81) | 63.0 (57, 76) | 69.0 (53, 81) | |
| Ethnicity | Japanese | n (%) | 7 (100) | 7 (100) | 14 (100) |
| ECOG performance status | 0 | n (%) | 4 (57.1) | 5 (71.4) | 9 (64.3) |
| 1 | n (%) | 3 (42.9) | 2 (28.6) | 5 (35.7) | |
| Diagnoses | Diffuse large B-cell lymphoma NOS | 2 (28.6) | 1 (14.3) | 3 (21.4) | |
| Follicular lymphoma | 0 (0) | 2 (28.6) | 2 (14.3) | ||
| MALT lymphoma | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Adult T-cell Leukemia/lymphoma | 1 (14.3) | 4 (57.1) | 5 (35.7) | ||
| Peripheral T-cell lymphoma | 2 (28.6) | 0 (0.0) | 2 (14.3) | ||
| Other (Cutaneous gamma delta T-cell lymphoma) | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Prior treatments | Chemotherapy | 7 (100) | 7 (100) | 14 (100) | |
| Radiation therapy | 2 (28.6) | 1 (14.3) | 3 (21.4) | ||
| Immunotherapy | 0 | 1 (14.3) | 1 (7.1) | ||
| UV light therapy | 0 | 1 (14.3) | 1 (7.1) | ||
| Total number of regimens | Median (Min, Max) | 2.0 (1,9) | 3.0 (1,8) | 2.5 (1,9) | |
| Characteristic | Category | Statistic | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) |
|---|---|---|---|---|---|
| Sex | Male | n (%) | 4 (57.1) | 3 (42.9) | 7 (50.0) |
| Female | n (%) | 3 (42.9) | 4 (57.1) | 7 (50.0) | |
| Age (years) | Median (min, max) | 72.0 (53, 81) | 63.0 (57, 76) | 69.0 (53, 81) | |
| Ethnicity | Japanese | n (%) | 7 (100) | 7 (100) | 14 (100) |
| ECOG performance status | 0 | n (%) | 4 (57.1) | 5 (71.4) | 9 (64.3) |
| 1 | n (%) | 3 (42.9) | 2 (28.6) | 5 (35.7) | |
| Diagnoses | Diffuse large B-cell lymphoma NOS | 2 (28.6) | 1 (14.3) | 3 (21.4) | |
| Follicular lymphoma | 0 (0) | 2 (28.6) | 2 (14.3) | ||
| MALT lymphoma | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Adult T-cell Leukemia/lymphoma | 1 (14.3) | 4 (57.1) | 5 (35.7) | ||
| Peripheral T-cell lymphoma | 2 (28.6) | 0 (0.0) | 2 (14.3) | ||
| Other (Cutaneous gamma delta T-cell lymphoma) | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Prior treatments | Chemotherapy | 7 (100) | 7 (100) | 14 (100) | |
| Radiation therapy | 2 (28.6) | 1 (14.3) | 3 (21.4) | ||
| Immunotherapy | 0 | 1 (14.3) | 1 (7.1) | ||
| UV light therapy | 0 | 1 (14.3) | 1 (7.1) | ||
| Total number of regimens | Median (Min, Max) | 2.0 (1,9) | 3.0 (1,8) | 2.5 (1,9) | |
Abbreviations: MALT, mucosa-associated lymphoid tissue; Min, minimum; Max, maximum; NOS, not otherwise specified.
Demographic data, baseline characteristics and disease history
| Characteristic | Category | Statistic | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) |
|---|---|---|---|---|---|
| Sex | Male | n (%) | 4 (57.1) | 3 (42.9) | 7 (50.0) |
| Female | n (%) | 3 (42.9) | 4 (57.1) | 7 (50.0) | |
| Age (years) | Median (min, max) | 72.0 (53, 81) | 63.0 (57, 76) | 69.0 (53, 81) | |
| Ethnicity | Japanese | n (%) | 7 (100) | 7 (100) | 14 (100) |
| ECOG performance status | 0 | n (%) | 4 (57.1) | 5 (71.4) | 9 (64.3) |
| 1 | n (%) | 3 (42.9) | 2 (28.6) | 5 (35.7) | |
| Diagnoses | Diffuse large B-cell lymphoma NOS | 2 (28.6) | 1 (14.3) | 3 (21.4) | |
| Follicular lymphoma | 0 (0) | 2 (28.6) | 2 (14.3) | ||
| MALT lymphoma | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Adult T-cell Leukemia/lymphoma | 1 (14.3) | 4 (57.1) | 5 (35.7) | ||
| Peripheral T-cell lymphoma | 2 (28.6) | 0 (0.0) | 2 (14.3) | ||
| Other (Cutaneous gamma delta T-cell lymphoma) | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Prior treatments | Chemotherapy | 7 (100) | 7 (100) | 14 (100) | |
| Radiation therapy | 2 (28.6) | 1 (14.3) | 3 (21.4) | ||
| Immunotherapy | 0 | 1 (14.3) | 1 (7.1) | ||
| UV light therapy | 0 | 1 (14.3) | 1 (7.1) | ||
| Total number of regimens | Median (Min, Max) | 2.0 (1,9) | 3.0 (1,8) | 2.5 (1,9) | |
| Characteristic | Category | Statistic | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) |
|---|---|---|---|---|---|
| Sex | Male | n (%) | 4 (57.1) | 3 (42.9) | 7 (50.0) |
| Female | n (%) | 3 (42.9) | 4 (57.1) | 7 (50.0) | |
| Age (years) | Median (min, max) | 72.0 (53, 81) | 63.0 (57, 76) | 69.0 (53, 81) | |
| Ethnicity | Japanese | n (%) | 7 (100) | 7 (100) | 14 (100) |
| ECOG performance status | 0 | n (%) | 4 (57.1) | 5 (71.4) | 9 (64.3) |
| 1 | n (%) | 3 (42.9) | 2 (28.6) | 5 (35.7) | |
| Diagnoses | Diffuse large B-cell lymphoma NOS | 2 (28.6) | 1 (14.3) | 3 (21.4) | |
| Follicular lymphoma | 0 (0) | 2 (28.6) | 2 (14.3) | ||
| MALT lymphoma | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Adult T-cell Leukemia/lymphoma | 1 (14.3) | 4 (57.1) | 5 (35.7) | ||
| Peripheral T-cell lymphoma | 2 (28.6) | 0 (0.0) | 2 (14.3) | ||
| Other (Cutaneous gamma delta T-cell lymphoma) | 1 (14.3) | 0 (0) | 1 (7.1) | ||
| Prior treatments | Chemotherapy | 7 (100) | 7 (100) | 14 (100) | |
| Radiation therapy | 2 (28.6) | 1 (14.3) | 3 (21.4) | ||
| Immunotherapy | 0 | 1 (14.3) | 1 (7.1) | ||
| UV light therapy | 0 | 1 (14.3) | 1 (7.1) | ||
| Total number of regimens | Median (Min, Max) | 2.0 (1,9) | 3.0 (1,8) | 2.5 (1,9) | |
Abbreviations: MALT, mucosa-associated lymphoid tissue; Min, minimum; Max, maximum; NOS, not otherwise specified.
Safety
Twelve of 14 patients completed the first cycle of treatment and were included in the assessment of DLTs. Two patients (one in each cohort) were excluded from the assessment as those patients had not completed dosing in Cycle 1. One patient in the 30 mg BIW cohort was excluded because the patient received only 1 dose of HBI-8000, and the other patient in the 40 mg BIW cohort was also excluded because the patient did not complete Cycle 1 at the assigned initial dose due to AE other than an occurrence of a DLT. No DLT was reported in the first three patients at 30 mg BIW and the dosage was escalated to 40 mg BIW. As a DLT was observed in one patient among the first three patients at 40 mg BIW, the cohort was expanded and an additional three patients were enrolled. There were two patients with DLTs among six patients in the 40 mg cohort, and an additional cohort of three patients was recruited at 30 mg BIW according to the protocol. Having two DLTs in the 40 mg BIW cohort, the dose was not escalated to 50 mg BIW.
The first patient in the 40 mg BIW cohort experienced grade 4 neutropenia on day 15 of cycle 1. It was grade 1 on day 18 with the administration of G-CSF and dose interruption. The other patient had grade 3 alanine aminotransferase increased on day 18 of cycle 1. It was improved to grade 2 on day 22 with dose interruption. Both patients resumed taking HBI-8000 at a reduced dose. Given a short time of recovery and nature of the DLTs, and assessment of nature/frequent of the other AEs, the DMC/SMC declared that 40 mg BIW was the MTD.
Thirteen of 14 patients (92.9%) experienced at least one treatment-emergent AE (TEAE) and most of which were considered related to HBI-8000. In both cohorts, the most frequently reported TEAEs were hematologic toxicities including thrombocytopenia/platelet count decreased, anaemia, neutropenia/neutrophil count decreased and white blood cell count decreased (Table 2). Other TEAEs were diarrhoea and fatigue. Among the hematologic TEAEs, incidence of thrombocytopenia was higher in the 40 mg BIW cohort (57.1%).
TEAEs regardless of relationship to study drug (≥10% in total)
| Adverse events | 30 mg (N = 7) n (%) | 40 mg (N = 7) n (%) | Total (N = 14) n (%) |
|---|---|---|---|
| Number of patients with at least one related TEAE | 6 (85.7) | 7 (100) | 13 (92.9) |
| Thrombocytopenia | 2 (28.6) | 4 (57.1) | 6 (42.9) |
| Platelet count decreased | 3 (42.9) | 3 (42.9) | 6 (42.9) |
| Anaemia | 5 (71.4) | 0 (0) | 5 (35.7) |
| Diarrhoea | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Fatigue | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| White blood cell count decreased | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Hypocalcaemia | 0 (0) | 3 (42.9) | 3 (21.4) |
| Dysgeusia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Nausea | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Neutropenia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Pyrexia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Weight decreased | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Decreased appetite | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Hypokalaemia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Back pain | 0 (0) | 2 (28.6) | 2 (14.3) |
| Nasopharyngitis | 0 (0) | 2 (28.6) | 2 (14.3) |
| Abdominal pain | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Insomnia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| C-reactive protein increased | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Headache | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hypertriglyceridaemia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hyperuricaemia | 2 (28.6) | 0 (0) | 2 (14.3) |
| Neutrophil count decreased | 2 (28.6) | 0 (0) | 2 (14.3) |
| Adverse events | 30 mg (N = 7) n (%) | 40 mg (N = 7) n (%) | Total (N = 14) n (%) |
|---|---|---|---|
| Number of patients with at least one related TEAE | 6 (85.7) | 7 (100) | 13 (92.9) |
| Thrombocytopenia | 2 (28.6) | 4 (57.1) | 6 (42.9) |
| Platelet count decreased | 3 (42.9) | 3 (42.9) | 6 (42.9) |
| Anaemia | 5 (71.4) | 0 (0) | 5 (35.7) |
| Diarrhoea | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Fatigue | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| White blood cell count decreased | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Hypocalcaemia | 0 (0) | 3 (42.9) | 3 (21.4) |
| Dysgeusia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Nausea | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Neutropenia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Pyrexia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Weight decreased | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Decreased appetite | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Hypokalaemia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Back pain | 0 (0) | 2 (28.6) | 2 (14.3) |
| Nasopharyngitis | 0 (0) | 2 (28.6) | 2 (14.3) |
| Abdominal pain | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Insomnia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| C-reactive protein increased | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Headache | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hypertriglyceridaemia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hyperuricaemia | 2 (28.6) | 0 (0) | 2 (14.3) |
| Neutrophil count decreased | 2 (28.6) | 0 (0) | 2 (14.3) |
Abbreviations: TEAE, treatment-emergent adverse event.
TEAEs regardless of relationship to study drug (≥10% in total)
| Adverse events | 30 mg (N = 7) n (%) | 40 mg (N = 7) n (%) | Total (N = 14) n (%) |
|---|---|---|---|
| Number of patients with at least one related TEAE | 6 (85.7) | 7 (100) | 13 (92.9) |
| Thrombocytopenia | 2 (28.6) | 4 (57.1) | 6 (42.9) |
| Platelet count decreased | 3 (42.9) | 3 (42.9) | 6 (42.9) |
| Anaemia | 5 (71.4) | 0 (0) | 5 (35.7) |
| Diarrhoea | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Fatigue | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| White blood cell count decreased | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Hypocalcaemia | 0 (0) | 3 (42.9) | 3 (21.4) |
| Dysgeusia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Nausea | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Neutropenia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Pyrexia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Weight decreased | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Decreased appetite | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Hypokalaemia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Back pain | 0 (0) | 2 (28.6) | 2 (14.3) |
| Nasopharyngitis | 0 (0) | 2 (28.6) | 2 (14.3) |
| Abdominal pain | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Insomnia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| C-reactive protein increased | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Headache | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hypertriglyceridaemia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hyperuricaemia | 2 (28.6) | 0 (0) | 2 (14.3) |
| Neutrophil count decreased | 2 (28.6) | 0 (0) | 2 (14.3) |
| Adverse events | 30 mg (N = 7) n (%) | 40 mg (N = 7) n (%) | Total (N = 14) n (%) |
|---|---|---|---|
| Number of patients with at least one related TEAE | 6 (85.7) | 7 (100) | 13 (92.9) |
| Thrombocytopenia | 2 (28.6) | 4 (57.1) | 6 (42.9) |
| Platelet count decreased | 3 (42.9) | 3 (42.9) | 6 (42.9) |
| Anaemia | 5 (71.4) | 0 (0) | 5 (35.7) |
| Diarrhoea | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Fatigue | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| White blood cell count decreased | 3 (42.9) | 1 (14.3) | 4 (28.6) |
| Hypocalcaemia | 0 (0) | 3 (42.9) | 3 (21.4) |
| Dysgeusia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Nausea | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Neutropenia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Pyrexia | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Weight decreased | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Decreased appetite | 1 (14.3) | 2 (28.6) | 3 (21.4) |
| Hypokalaemia | 2 (28.6) | 1 (14.3) | 3 (21.4) |
| Back pain | 0 (0) | 2 (28.6) | 2 (14.3) |
| Nasopharyngitis | 0 (0) | 2 (28.6) | 2 (14.3) |
| Abdominal pain | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Insomnia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| C-reactive protein increased | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Headache | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hypertriglyceridaemia | 1 (14.3) | 1 (14.3) | 2 (14.3) |
| Hyperuricaemia | 2 (28.6) | 0 (0) | 2 (14.3) |
| Neutrophil count decreased | 2 (28.6) | 0 (0) | 2 (14.3) |
Abbreviations: TEAE, treatment-emergent adverse event.
Eleven of 14 treated patients (78.6%) experienced at least one Grade ≥ 3 TEAEs, five in the 30 mg BIW cohort and six in the 40 mg BIW cohort. Grade 4 neutropenia was observed in two patients in the 40 mg cohort, while Grade ≥ 3 neutropenia was reported in one patient in the 30 mg BIW cohort and two patients in the 40 mg BIW cohort (Table 3). There were no patient deaths on study. One patient in each cohort (14.3%) experienced a serious TEAE. Only the one SAE in the 40 mg BIW cohort (Grade 3 abdominal pain) was considered to be possibly related to the study drug.
TEAEs Grades 3 and 4 regardless of relationship to study drug
| AE | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) | ||||
|---|---|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | ||
| Number of patients with at least one TEAE | 5 (71.4) | 0 (0) | 4 (57.1) | 2 (28.6) | 9 (64.3) | 2 (14.3) | |
| Neutropenia | 1 (14.3) | 0 (0) | 0 (0) | 2 (28.6) | 1 (7.1) | 2 (14.3) | |
| Thrombocytopenia | 0 (0) | 0 (0) | 2 (28.6) | 0 (0) | 2 (14.3) | 0 (0) | |
| Neutrophil count decreased | 2 (8.6) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | |
| White blood cell count decreased | 1 (4.3) | 0 (0) | 1 (14.3) | 0 (0) | 2 (14.3) | 0 (0) | |
| Anaemia | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Lymphocyte count decreased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Enterocolitis | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Abdominal pain | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Pyrexia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Blood creatine phosphokinase increased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hyperglycaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hypertriglyceridaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Parkinsonism | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema multiforme | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema nodosum | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Rash generalized | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| AE | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) | ||||
|---|---|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | ||
| Number of patients with at least one TEAE | 5 (71.4) | 0 (0) | 4 (57.1) | 2 (28.6) | 9 (64.3) | 2 (14.3) | |
| Neutropenia | 1 (14.3) | 0 (0) | 0 (0) | 2 (28.6) | 1 (7.1) | 2 (14.3) | |
| Thrombocytopenia | 0 (0) | 0 (0) | 2 (28.6) | 0 (0) | 2 (14.3) | 0 (0) | |
| Neutrophil count decreased | 2 (8.6) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | |
| White blood cell count decreased | 1 (4.3) | 0 (0) | 1 (14.3) | 0 (0) | 2 (14.3) | 0 (0) | |
| Anaemia | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Lymphocyte count decreased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Enterocolitis | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Abdominal pain | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Pyrexia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Blood creatine phosphokinase increased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hyperglycaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hypertriglyceridaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Parkinsonism | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema multiforme | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema nodosum | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Rash generalized | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
TEAEs Grades 3 and 4 regardless of relationship to study drug
| AE | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) | ||||
|---|---|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | ||
| Number of patients with at least one TEAE | 5 (71.4) | 0 (0) | 4 (57.1) | 2 (28.6) | 9 (64.3) | 2 (14.3) | |
| Neutropenia | 1 (14.3) | 0 (0) | 0 (0) | 2 (28.6) | 1 (7.1) | 2 (14.3) | |
| Thrombocytopenia | 0 (0) | 0 (0) | 2 (28.6) | 0 (0) | 2 (14.3) | 0 (0) | |
| Neutrophil count decreased | 2 (8.6) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | |
| White blood cell count decreased | 1 (4.3) | 0 (0) | 1 (14.3) | 0 (0) | 2 (14.3) | 0 (0) | |
| Anaemia | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Lymphocyte count decreased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Enterocolitis | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Abdominal pain | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Pyrexia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Blood creatine phosphokinase increased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hyperglycaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hypertriglyceridaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Parkinsonism | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema multiforme | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema nodosum | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Rash generalized | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| AE | 30 mg (N = 7) | 40 mg (N = 7) | Total (N = 14) | ||||
|---|---|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | ||
| Number of patients with at least one TEAE | 5 (71.4) | 0 (0) | 4 (57.1) | 2 (28.6) | 9 (64.3) | 2 (14.3) | |
| Neutropenia | 1 (14.3) | 0 (0) | 0 (0) | 2 (28.6) | 1 (7.1) | 2 (14.3) | |
| Thrombocytopenia | 0 (0) | 0 (0) | 2 (28.6) | 0 (0) | 2 (14.3) | 0 (0) | |
| Neutrophil count decreased | 2 (8.6) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | |
| White blood cell count decreased | 1 (4.3) | 0 (0) | 1 (14.3) | 0 (0) | 2 (14.3) | 0 (0) | |
| Anaemia | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Lymphocyte count decreased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Enterocolitis | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Abdominal pain | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Pyrexia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Blood creatine phosphokinase increased | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hyperglycaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Hypertriglyceridaemia | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Parkinsonism | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema multiforme | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Erythema nodosum | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 0 (0) | |
| Rash generalized | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 0 (0) | |
Six patients (42.9%) experienced TEAEs that led to treatment discontinuation. These included two patients in the 30 mg BIW cohort (blood creatine phosphokinase increased and perivascular dermatitis) and four patients in the 40 mg BIW cohort (abdominal pain, neutropenia, computerized tomogram thorax abnormal and erythema multiforme). Eight patients (57.1%) (four patients in each cohort) experienced TEAEs that led to dose reduction or dose delay, most commonly reported were platelet count decreased [four of 14 patients (28.6%)] and neutrophil count decreased [two of 14 patients (14.3%)].
Cardiovascular assessments including serial ECGs, transthoracic echocardiogram findings and troponin assessments did not reveal clinically relevant findings. Absolute QTcF >450 ms was observed in three patients in the 30 mg BIW cohort and two patients in the 40 mg BIW cohort; absolute QTcF >480 ms was observed in one patient in the 40 mg BIW cohort; none were prolonged to >500 ms. One patient in the 30 mg BIW cohort and three patients in the 40 mg BIW cohort had QTcF increase of >30 ms from baseline; none were prolonged >60 ms.
Pharmacokinetics
Following single and repeat oral administration of HBI-8000, median tmax occurred at 2.42–5.00 hours followed by a gradual biexponential decline. Mean t1/2 appeared to be dose- and time-independent, ranging from 17.1 to 21.6 hours. Accumulation was minimal, with a mean 23–24% increase in AUC(0-tau) and a mean 16–27% increase in Cmax following BIW dosing up to Day 25. Steady state is expected to have been achieved by Day 25 of BIW dosing. An average of 25.3 and 21.9% of the administered dose of HBI-8000 in the 30 and 40 mg cohort, respectively, was excreted unchanged in urine over the dosing interval (Table 4).
HBI-8000 plasma and urine PK parameters for Cycle 1
| Parameter (unit) | Arithmetic mean (CV%) | |||
|---|---|---|---|---|
| 30 mg (N = 7) | 40 mg (N = 7) | |||
| Day 1 (N = 7) | Day 25 (N = 6) | Day 1 (N = 7) | Day 25 (N = 4) | |
| AUC(0-inf) (ng⋅h/ml) | 4000 (33.6) | NA | 7160 (51.5) | NA |
| AUC(0-tau) (ng⋅h/ml) | 3740 (32.5) | 4870 (27.1) | 6760 (54.0) | 6010 (58.3) |
| Cmax (ng/ml) | 199 (52.8) | 240 (33.2) | 590 (78.7) | 385 (56.7) |
| tmax (h) median (min, max) | 3.98 (2.50, 11.93) | 5.00 (2.47, 12.02) | 2.42 (1.52, 5.95) | 4.19 (0.78, 12.00) |
| t1/2 (h) | 17.1 (18.4) | 21.6 (24.4) | 19.4 (33.5) | 18.7 (11.0) |
| CLr (l/h) | 1.88 (49.9) | 1.71 (70.8)a | 1.75 (42.1) | 1.69 (37.1) |
| Parameter (unit) | Arithmetic mean (CV%) | |||
|---|---|---|---|---|
| 30 mg (N = 7) | 40 mg (N = 7) | |||
| Day 1 (N = 7) | Day 25 (N = 6) | Day 1 (N = 7) | Day 25 (N = 4) | |
| AUC(0-inf) (ng⋅h/ml) | 4000 (33.6) | NA | 7160 (51.5) | NA |
| AUC(0-tau) (ng⋅h/ml) | 3740 (32.5) | 4870 (27.1) | 6760 (54.0) | 6010 (58.3) |
| Cmax (ng/ml) | 199 (52.8) | 240 (33.2) | 590 (78.7) | 385 (56.7) |
| tmax (h) median (min, max) | 3.98 (2.50, 11.93) | 5.00 (2.47, 12.02) | 2.42 (1.52, 5.95) | 4.19 (0.78, 12.00) |
| t1/2 (h) | 17.1 (18.4) | 21.6 (24.4) | 19.4 (33.5) | 18.7 (11.0) |
| CLr (l/h) | 1.88 (49.9) | 1.71 (70.8)a | 1.75 (42.1) | 1.69 (37.1) |
Abbreviations: AUC, Area Under the Curve; AUC(0-inf), AUC from zero (pre-dose) extrapolated to infinity; AUC(0-tau), AUC from zero (pre-dose) to the end of the dosing interval, tau; CLr, renal clearance; Cmax, maximum observed plasma concentration; CV%, coefficient of variation (in percent); Max, maximum; Min, minimum; NA = not applicable; tmax, time of Cmax; t1/2, apparent terminal half-life.
an = 5.
HBI-8000 plasma and urine PK parameters for Cycle 1
| Parameter (unit) | Arithmetic mean (CV%) | |||
|---|---|---|---|---|
| 30 mg (N = 7) | 40 mg (N = 7) | |||
| Day 1 (N = 7) | Day 25 (N = 6) | Day 1 (N = 7) | Day 25 (N = 4) | |
| AUC(0-inf) (ng⋅h/ml) | 4000 (33.6) | NA | 7160 (51.5) | NA |
| AUC(0-tau) (ng⋅h/ml) | 3740 (32.5) | 4870 (27.1) | 6760 (54.0) | 6010 (58.3) |
| Cmax (ng/ml) | 199 (52.8) | 240 (33.2) | 590 (78.7) | 385 (56.7) |
| tmax (h) median (min, max) | 3.98 (2.50, 11.93) | 5.00 (2.47, 12.02) | 2.42 (1.52, 5.95) | 4.19 (0.78, 12.00) |
| t1/2 (h) | 17.1 (18.4) | 21.6 (24.4) | 19.4 (33.5) | 18.7 (11.0) |
| CLr (l/h) | 1.88 (49.9) | 1.71 (70.8)a | 1.75 (42.1) | 1.69 (37.1) |
| Parameter (unit) | Arithmetic mean (CV%) | |||
|---|---|---|---|---|
| 30 mg (N = 7) | 40 mg (N = 7) | |||
| Day 1 (N = 7) | Day 25 (N = 6) | Day 1 (N = 7) | Day 25 (N = 4) | |
| AUC(0-inf) (ng⋅h/ml) | 4000 (33.6) | NA | 7160 (51.5) | NA |
| AUC(0-tau) (ng⋅h/ml) | 3740 (32.5) | 4870 (27.1) | 6760 (54.0) | 6010 (58.3) |
| Cmax (ng/ml) | 199 (52.8) | 240 (33.2) | 590 (78.7) | 385 (56.7) |
| tmax (h) median (min, max) | 3.98 (2.50, 11.93) | 5.00 (2.47, 12.02) | 2.42 (1.52, 5.95) | 4.19 (0.78, 12.00) |
| t1/2 (h) | 17.1 (18.4) | 21.6 (24.4) | 19.4 (33.5) | 18.7 (11.0) |
| CLr (l/h) | 1.88 (49.9) | 1.71 (70.8)a | 1.75 (42.1) | 1.69 (37.1) |
Abbreviations: AUC, Area Under the Curve; AUC(0-inf), AUC from zero (pre-dose) extrapolated to infinity; AUC(0-tau), AUC from zero (pre-dose) to the end of the dosing interval, tau; CLr, renal clearance; Cmax, maximum observed plasma concentration; CV%, coefficient of variation (in percent); Max, maximum; Min, minimum; NA = not applicable; tmax, time of Cmax; t1/2, apparent terminal half-life.
an = 5.
HBI-8000 plasma concentrations were measured and quantifiable up to 72 hours after dose administration on Days 1 and 25 of Cycle 1 for both the 30 and 40 mg cohorts (Figure 1).
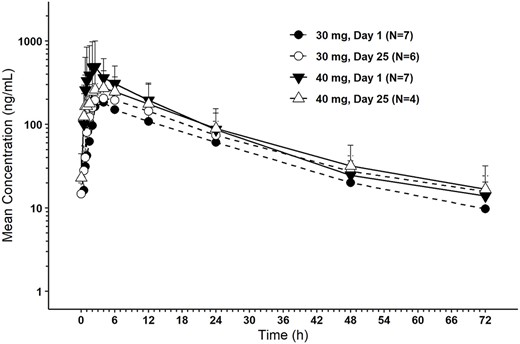
HBI-8000 plasma concentration-time profiles for Cycle 1.
Efficacy
Efficacy analysis included 13 patients (six patients in the 30 mg BIW cohort and seven patients in the 40 mg BIW cohort). One patient in the 30 mg BIW cohort received only one dose of HBI-8000 and was excluded from the efficacy analysis.
In the 30 mg BIW cohort, complete response unconfirmed (CRu) was reported for one patient. ORR was 16.7%, and disease control (complete response [CR] + CRu + partial response [PR] + stable disease [SD]) was observed in four patients (66.7%). In the 40 mg BIW cohort, CR was reported for one patient, CRu for one patient and PR for three patients. ORR was 71.4%, and disease control was observed in all patients (100%).
Objective response in patients with NHL other than ATL was observed in one of five patients in the 30 mg BIW cohort (20%) and two of three patients in the 40 mg BIW cohort (66.7%) (Table 5). Disease control was observed in three of five patients in the 30 mg BIW cohort (60%) and all three patients in the 40 mg BIW cohort (100%). Objective response in ATL was observed in three of four patients in the 40 mg BIW cohort (75%). Disease control was observed in one patient in the 30 mg BIW cohort (100%) and all four patients in the 40 mg BIW cohort (100%) (Table 5).
Best overall response by tumor type (efficacy analysis set)
| Lesion | Response | 30 mg (N = 6) n (%) | 40 mg (N = 7) n (%) |
|---|---|---|---|
| NHL other than ATL | Number of patients | 5 | 3 |
| CR | 0 (0) | 1 (33.3) | |
| CRu | 1 (20.0) | 1 (33.3) | |
| SD | 2 (40.0) | 1 (33.3) | |
| PD | 2 (40.0) | 0 (0) | |
| Objective response | CR, CRu or PR | 1 (20.0) | 2 (66.7) |
| Disease control | CR, CRu, PR or SD | 3 (60.0) | 3 (100) |
| ATL | Number of patients | 1 | 4 |
| PR | 0 (0) | 3 (75.0) | |
| SD | 1 (100) | 1 (25.0) | |
| Objective response | CR or PR | 0 (0.0) | 3 (75.0) |
| Disease control | CR, PR or SD | 1 (100) | 4 (100) |
| Lesion | Response | 30 mg (N = 6) n (%) | 40 mg (N = 7) n (%) |
|---|---|---|---|
| NHL other than ATL | Number of patients | 5 | 3 |
| CR | 0 (0) | 1 (33.3) | |
| CRu | 1 (20.0) | 1 (33.3) | |
| SD | 2 (40.0) | 1 (33.3) | |
| PD | 2 (40.0) | 0 (0) | |
| Objective response | CR, CRu or PR | 1 (20.0) | 2 (66.7) |
| Disease control | CR, CRu, PR or SD | 3 (60.0) | 3 (100) |
| ATL | Number of patients | 1 | 4 |
| PR | 0 (0) | 3 (75.0) | |
| SD | 1 (100) | 1 (25.0) | |
| Objective response | CR or PR | 0 (0.0) | 3 (75.0) |
| Disease control | CR, PR or SD | 1 (100) | 4 (100) |
Notes: Patients with no tumor assessment evaluation are considered non-responders. Abbreviations: CR, complete response; CRu, unconfirmed complete response; NHL, non-Hodgkin’s lymphoma; PD, progressive disease; PR, partial response; SD, stable disease.
Best overall response by tumor type (efficacy analysis set)
| Lesion | Response | 30 mg (N = 6) n (%) | 40 mg (N = 7) n (%) |
|---|---|---|---|
| NHL other than ATL | Number of patients | 5 | 3 |
| CR | 0 (0) | 1 (33.3) | |
| CRu | 1 (20.0) | 1 (33.3) | |
| SD | 2 (40.0) | 1 (33.3) | |
| PD | 2 (40.0) | 0 (0) | |
| Objective response | CR, CRu or PR | 1 (20.0) | 2 (66.7) |
| Disease control | CR, CRu, PR or SD | 3 (60.0) | 3 (100) |
| ATL | Number of patients | 1 | 4 |
| PR | 0 (0) | 3 (75.0) | |
| SD | 1 (100) | 1 (25.0) | |
| Objective response | CR or PR | 0 (0.0) | 3 (75.0) |
| Disease control | CR, PR or SD | 1 (100) | 4 (100) |
| Lesion | Response | 30 mg (N = 6) n (%) | 40 mg (N = 7) n (%) |
|---|---|---|---|
| NHL other than ATL | Number of patients | 5 | 3 |
| CR | 0 (0) | 1 (33.3) | |
| CRu | 1 (20.0) | 1 (33.3) | |
| SD | 2 (40.0) | 1 (33.3) | |
| PD | 2 (40.0) | 0 (0) | |
| Objective response | CR, CRu or PR | 1 (20.0) | 2 (66.7) |
| Disease control | CR, CRu, PR or SD | 3 (60.0) | 3 (100) |
| ATL | Number of patients | 1 | 4 |
| PR | 0 (0) | 3 (75.0) | |
| SD | 1 (100) | 1 (25.0) | |
| Objective response | CR or PR | 0 (0.0) | 3 (75.0) |
| Disease control | CR, PR or SD | 1 (100) | 4 (100) |
Notes: Patients with no tumor assessment evaluation are considered non-responders. Abbreviations: CR, complete response; CRu, unconfirmed complete response; NHL, non-Hodgkin’s lymphoma; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
The data from this phase I study demonstrated that treatment with HBI-8000 30 and 40 mg BIW was well tolerated and safe in Japanese patients with previously treated advanced NHL. The DLTs were grade 4 neutropenia and grade 3 alanine aminotransferase increased in the 40 mg BIW cohort. MTD and recommended phase II dose (RP2D) was determined as 40 mg BIW because the DLTs were asymptomatic laboratory results and were resolved with supportive care or dose interruption.
TEAEs reported in the present study were generally mild to moderate in severity and easily manageable. The most frequently reported TEAEs were hematological toxicities such as thrombocytopenia/platelet count decreased, anaemia and neutropenia/neutrophil count decreased that could be managed with dose interruptions or reductions. Grade ≥ 3 TEAEs observed in two or more patients were thrombocytopenia, neutropenia/neutrophil count decreased and white blood cell count decreased. Hematological toxicities were commonly observed in other studies of HDACi (23,24). The reduction in platelets may be explained by a class effect of HDACi, delay in megakaryocyte differentiation (25), which is non-myeloablative. This suggests that closed monitoring and management of hematological parameters are necessary during the treatment.
While slight QTc elevations were documented, no clinically meaningful prolonged QTc, absolute QTcF >500 ms, was noted in our study. QTc prolongation is reported to be considered as a class effect for HDACi (26,27), but there were no concurrent cardiac symptoms in our study. In pre-clinical study, HBI-8000 did not induce any effect on cardiovascular system including changes in QTc prolongation. However, the number of patients in our study was limited and more data on the QTc prolongation were planned to be assessed in further studies.
The TEAEs reported in the present study were consistent with those observed in Chinese phase II studies; e.g. thrombocytopenia, neutropenia and fatigue were the frequently reported TEAEs in those studies (28,29). Therefore, the safety profile was similar to the Chinese studies although the dosage in those studies was 30 mg BIW, lower than the RP2D determined in our study. A major reason for the difference of RP2D is considered to be differences in study design between the Japanese and Chinese studies.
Preliminary efficacy results of our study showed a sign of effectiveness of HBI-8000 in this patient population with R/R NHL, including three out of five responses in patients with ATL. Moreover, the ORR in the 40 mg BIW cohort was 71.4% (five of seven patients), which is higher than the ORR in the 30 mg BIW cohort. A more desirable clinical outcome is expected by selecting 40 mg BIW as RP2D.
The present study demonstrates that HBI-8000 was well-tolerated at the dose levels tested in Japanese patients with NHL. The preliminary efficacy results are favorable, especially in the 40 mg BIW cohort and in patients with ATL. Data support phase II development at 40 mg BIW as a starting dose. Phase II trials to evaluate efficacy and safety in R/R ATL patients (Japan) and R/R PTCL patients (Japan and Korea) have been conducted and will be reported separately. HBI-8000 was approved in Japan for treatment of R/R ATL in June 2021 and was also approved for the treatment of R/R PTCL in November 2021.
Funding
This work was supported by HUYA Bioscience International, LLC (HUYABIO International), San Diego, USA.
Conflict of interest statement
Yoshida reports grants from Bayer Pharmaceuticals. Takamatsu reports grants from Chugai Pharmaceutical, Takeda Pharmaceutical and Astellas Pharmaceutical, and honoraria from Takeda Pharmaceutical, Janssen Pharmaceutical and Celgene. Gillings reports CEO of HUYABIO International, LLC, indirectly owns the shares of the HUYABIO International, LLC., GHO Capital Management Limited, and QHP LLC. Onogi reports employment from Huya Japan G.K., Lee reports employment from HUYABIO International, LLC, Tobinai reports consulting fees from HUYABIO International, LLC., Zenyaku Kogyo, Mundipharma and Daiichisankyo. The other authors have no conflict of interest.
Acknowledgements
The authors would like to thank the patients, families and all staff in each hospital for their support in performing the present study. We would also like to thank Dr Noriko Usui (Jikei University School of Medicine), Dr Norio Komatsu (Juntendo University School of Medicine) and Dr Kazunori Onishi (Hamamatsu University School of Medicine) for their review of the clinical data as members of the Data Monitoring Committee and Safety Monitoring Committee. Karen Rittweger provided medical writing assistance, funded by HUYA Bioscience International. All authors had reviewed and approved the manuscript and submission.
References
JCOG [Internet].
Author notes
Present address: Department of Immunology, Nagoya University Graduate School of Medicine, Nagoya, Japan

{kind=link}