




Abstract
The development of PD-1 pathway inhibitors has dramatically altered the treatment of advanced/recurrent non-small-cell lung cancer patients. However, the prognostic significance of their ongoing usage is controversial, especially for patients who have not progressed for a period of time. If discontinuation has no negative impact on survival, suspension may reduce side effects from toxicity and help alleviate the economic burdens on health insurance systems and patients. This randomized controlled trial enrolls patients who have responded well to PD-1 pathway inhibitors for >12 months. The aim is to confirm the non-inferiority of discontinuation of PD-1 pathway inhibitors, relative to continuation, in terms of overall survival. A total of 216 patients will be enrolled over 3 years. This trial has been registered in the Japan Registry for Clinical Trials as jRCT1031190032 (https://jrct.niph.go.jp/). An ancillary study examining the prognostic and predictive role of circulating tumor DNA using Guardant360® is planned.
Introduction
The development of immune checkpoint inhibitors (ICIs) that block the PD-1 pathway has dramatically changed treatment practices for advanced non-small-cell (squamous or non-squamous) lung cancer patients. Treatment with nivolumab, the first drug of this kind approved for lung cancer in Japan, prolonged progression-free survival (PFS) and overall survival (OS) in two pivotal phase III trials (termed the CheckMate 017 (1) and 057 (2) studies) in the second-line setting. Based on similar results, the approvals of pembrolizumab and atezolizumab soon followed (KEYNOTE-010 study (3), POLAR (4) and OAK (5) studies). In the first-line setting, a total of eight randomized controlled trials (pembrolizumab monotherapy in the KEYNOTE-024 (6) and 042 (7) studies; combination therapy of pembrolizumab with platinum-based chemotherapy in the KEYNOTE-021 (8), 189 (9), and 407 (10) studies; combination therapy of atezolizumab with platinum-based chemotherapy in the Impower-130 (11) and 150 (12) studies; and nivolumab and ipilimumab in the CheckMate-227 study (13)) showed statistically significant increases in PFS and/or OS in either or both of the entire population and the PD-L1-positive subsets, leading to approval of label expansions.
Excluding patients who have driver gene mutations (EGFR, ALK, ROS1, RET and BRAF), the current standard treatments for treatment-naïve advanced non-small-cell lung cancer patients with a PD-L1 tumor proportion score (TPS) ≥50% are pembrolizumab monotherapy or pembrolizumab in combination with platinum-based chemotherapy (cisplatin or carboplatin plus pemetrexed for non-squamous and carboplatin plus paclitaxel for squamous patients). For the rest of the treatment-naïve population, one must select from the following regimens, based on the patient’s background, PD-L1 status and so on: (i) pembrolizumab monotherapy or the abovementioned combined regimen; (ii) a combined regimen of cisplatin or carboplatin together with paclitaxel and bevacizumab; or (iii) a combined regimen of carboplatin plus pemetrexed followed by pemetrexed maintenance. Note that combination therapy of atezolizumab with chemotherapy (carboplatin, paclitaxel and bevacizumab followed by bevacizumab and atezolizumab maintenance; just approved in Japan) or combined use of ipilimumab and nivolumab (not approved in Japan) will become an option in the near future. In the second-line setting, standard treatments are ICI monotherapy or combined use of docetaxel and ramucirumab.
Current guidelines require that effective ICIs be continuously administered until treatment termination (e.g. due to disease progression or unacceptable toxicity). Even though their physical toxicity is much lower than those of standard chemotherapies, their ‘financial toxicity’ (i.e. economical burdens) is a serious concern, not only for patients and their families but also for society. Due to the rising costs of novel ICIs, many countries do not always reimburse patients for treatment with these agents, resulting in fewer patients receiving these treatments. In addition, ICIs are not effective for every patient, and exploratory findings have shown that testing for existing biomarkers is not sufficient to predict responsiveness. Further, patients sometimes experience non-negligible immune-related toxicities. Considering these issues, determining the optimal usage of ICIs is a critical need for lung cancer communities. Interestingly, long-lasting response to ICIs, even after discontinuation, has been reported for non-small-cell lung cancer (14,15) and melanoma (16) patients. In a randomized post-marketing trial in the second-line setting (termed the CheckMate-153 study (17)), a worsening of PFS and OS was observed in patients after discontinuation of nivolumab. However, determining the impact of discontinuation cannot rely on this trial alone because it was not planned to address a specific statistical hypothesis; patients who showed exacerbation were included, the number of responding patients was much larger in the continuation arm, important patient characteristics (e.g. sex, histology, PD-L1 status) were imbalanced between treatment arms, and most patients were enrolled in the USA.
Based on these backgrounds, we have designed a multicenter, randomized, phase III trial to confirm the non-inferiority of ICI discontinuation compared to continuation, focusing on patients who have had no response to ICIs for 12 months or longer (up to 62 weeks), both in the first- and second-line settings. The landmark time of 12 months was selected due to the following considerations. In the second-line setting, a previous study showed that 25% and 18% patients who responded to nivolumab monotherapy at 24 and 48 weeks relapsed within the next 24 weeks, respectively (18). A discussion with study investigators concluded that 12 months (up to 62 weeks) is the appropriate landmark to select homogeneous population who might show long-lasting effect of PD-1 pathway inhibitors. Furthermore, the study investigators considered the factors regarding feasibility such as informed consent of randomization and enrollment completion within an admissible interval. The trial is conducted by the Lung Cancer Study Group of Japan Clinical Oncology Group (JCOG).
The study protocol was approved by the JCOG Protocol Review Committee in March 2017 and by the institutional review board of each participating institution prior to initiating patient enrollment. Patient enrollment was initiated in July 2019. This trial has been registered in the Japan Registry for Clinical Trials as jRCT1031190032 (https://rctportal.niph.go.jp/en/detail?trial_id=jRCT1031190032).
Protocol digest of JCOG1701
Objectives
The aim of this study is to confirm the non-inferiority of discontinuation of ICIs versus their continuation in patients with advanced/recurrent non-small-cell lung cancer who have had no response to ICIs for 12 months or longer.
Study design
This study is a multi-institutional, two-arm, open-label, randomized phase III trial. The study scheme is shown in Fig. 1.
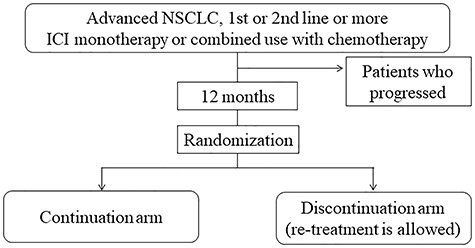
Study scheme.
Endpoints
The primary endpoint is OS. OS is defined as the period from randomization to death due to any cause, using the last contact date as the end date for surviving patients. The secondary endpoints are PFS, time to treatment failure of strategy (TFS), response rate and PFS after resuming ICIs and safety. PFS is defined as the period from randomization to disease progression or death due to any cause, censored at the last confirmed date with no progression. For the experimental discontinuation group, TFS is defined as the duration from the date of randomization to the date of observing the following events: (i) progression after resuming the same agents used before enrollment; (ii) resuming due to the patients’ request and/or doctors’ decision; (iii) initiating other treatments during drug holidays; and (4) deaths. TFS is censored at the last confirmed date at which none of the above-listed events had occurred. Note that the events (i)–(iii) represent a failure of strategy that breaks immune checkpoint inhibiting agents, which cannot be assessed by PFS alone. For the control continuation group, TFS is identical to PFS. Response rate is defined as the proportion of complete or partial response among the eligible patients according to the RECIST ver1.1.
Eligibility criteria
Inclusion criteria
- (1)
Histologically confirmed non-small-cell lung cancer (adenocarcinoma, squamous carcinoma, large cell neuroendocrine carcinoma [LCNEC], carcinoid tumor, large cell carcinoma, adenosquamous carcinoma, sarcomatoid cancer, unclassifiable cancer and salivary gland tumor) or cytologically (including biopsy) confirmed non-small-cell lung cancer (adenocarcinoma, squamous carcinoma, adenocarcinoma-suggested, squamous-suggested, LCNEC-suggested and not otherwise specified).
- (2)
Treated with either of the following for 52 weeks or more and not over 62 weeks, with the latest ICI treatment occurring within the 28 days before registration: (a) ICI monotherapy (pembrolizumab in the first/second line, atezolizumab or nivolumab in the second line); (b) combined therapy of ICI with chemotherapy (carboplatin or cisplatin together with pemetrexed or carboplatin together with paclitaxel and bevacizumab for non-squamous cell carcinoma, paclitaxel or nab-paclitaxel together with carboplatin for squamous cell carcinoma) in the first line.
- (3)
Response to ICI monotherapy or combination therapy with platinum-based chemotherapy, as measured by comparing CT imaging obtained before the treatment and within 4 weeks before registration.
- (4)
20 years of age or older.
- (5)
ECOG performance status, 0 or 1.
- (6)
None of the treatments were performed in the 8 weeks prior to registration:
Radiotherapy (for metastatic sites)
Chest drainage for more than 24 hours
Pleurodesis
Surgery with general anesthesia
- (1)
No adverse reactions with grades 3 or 4, related to anti-PD-1 or PD-L1 inhibitors.
- (2)
No pneumonitis with grade 3 or 4, even not related to the pretreated anti-PD-1 or PD-L1 inhibitors.
- (3)
No active, chronic or recurrent autoimmune disease.
- (4)
No symptomatic brain metastasis, meningeal metastasis or spinal metastasis that requires radiotherapy or surgery.
- (5)
No superior vena cava syndrome, pericardial effusion, pleural effusion, or ascites with grades 3 or 4.
- (6)
Adequate function of major organs.
- (7)
Written informed consents.
Exclusion criteria
- (1)
Synchronous or metachronous (within 2 years) malignancies.
- (2)
Infectious disease requiring systemic treatment.
- (3)
Pyrexia of 38 degrees centigrade or higher.
- (4)
Female during pregnancy, within 28 days of post-parturition, during lactation, or a male expecting partner’s pregnancy.
- (5)
Severe psychological disorders.
- (6)
Patients requiring systemic steroid medication or immunosuppressants for non-autoimmune disease.
- (7)
Poorly controlled diabetes mellitus.
- (8)
Poorly controlled hypertension.
- (9)
History of unstable angina pectoris with new onset or exacerbation within 3 weeks before registration or myocardial infarction within 6 months before registration.
- (10)
Positive serum HBs antigen.
Randomization
Patient registration is performed through a web-based system of the JCOG Data Center, Tokyo, Japan. Patients are randomized to the continuation or discontinuation group in a 1:1 ratio using the minimization method with random component, balancing with the institution, histology (squamous versus non-squamous) and drugs/timings (first-line pembrolizumab versus second-line atezolizumab versus second-line nivolumab versus combined therapy with chemotherapy).
Treatments
In the continuation group, the same ICI as used before registration is continued. For non-squamous patients treated with combined therapy with chemotherapy, only the maintenance part (pemetrexed and pembrolizumab) is continued. Widely accepted doses per one course are used: atezolizumab/nivolumab/pembrolizumab monotherapy 1200/240/200 mg/body for 2/3/3 weeks, pembrolizumab 200 mg/body and pemetrexed 500 mg/m (2) for 3 weeks.
In the discontinuation group, the ICI treatment (including combined therapies) administered before registration is suspended. When any of the following criteria is met, the same ICI treatment as was administered before registration can be resumed: (i) progression detected by imaging studies (e.g. appearance of new lesions, a 10% or greater increase in the diameter or sum of diameters); (ii) appearance or exacerbation of symptoms associated with the tumor; (iii) clear clinical exacerbation.
Follow-up
All randomized patients will be followed up for at least 3 years. Radiographic tumor evaluations are performed and assessed, according to RECIST (version 1.1), by each investigator at least every 6 weeks for the first 24 weeks, every 8 weeks for the next 24 weeks and every 12 weeks for the remaining follow-up period. In the discontinuation group, the proportion of patients that resume their treatments, and their reasons for doing so, will be monitored semiannually so that the difference between two randomization groups can be maintained.
Statistical analysis
This randomized phase III trial is designed to confirm the non-inferiority of discontinuation of ICIs versus continuation in terms of OS for patients who have shown no exacerbation for 12 months or longer with ICI treatment in both the first- and second-line settings. Based on Schoenfeld and Richter’s method (19), the estimated sample size is 216 patients to observe 106 events required, assuming the 2-year survival of both groups is 70% with the non-inferiority margin of 12% (1.53 in hazard ratio), power of 70%, a one-sided alpha-level of 5%, a planned accrual period of 3 years and a follow-up period of 3 years after enrolling the last patient. The primary analysis will be the stratified Cox regression analysis with histology and drugs/timings as stratification factors. If the non-inferiority null hypothesis is rejected, a more stringent testing against margin of 1.46 (corresponding to 10% decrease in 2-year survival) will be performed. No multiplicity adjustment is required for this two-step procedure due to the closed testing principle. All statistical analyses will be performed at the JCOG Data Center.
Interim analysis and monitoring
Two interim analyses are planned. The first one will be performed after half of the planned patients are enrolled. The second interim analysis will be performed at the time considered appropriate based on a regular monitoring about half a year after the end of registration in consultation with JCOG Data Center and Study Coordinator. The Lan-DeMets method with an O’Brien and Fleming-type alpha spending function will be used to adjust the multiplicity of the two interim analyses and the primary analysis (20). Only in the first interim analysis, a strict stopping rule for TFS and OS is incorporated because there is no preliminary evidence showing a non-inferiority trend of discontinuation. The Data and Safety Monitoring Committee of the JCOG will independently review the interim analysis reports and decide the early termination of the trial, if necessary. The JCOG Data Center and Study Coordinator will conduct central monitoring and issue a monitoring report every 6 months to evaluate study progress, improve data integrity and record patient safety. For quality assurance, on-site audits will be performed by the JCOG Audit Committee.
Participating institutions
- (1)
Aichi Cancer Center Hospital
- (2)
Asahikawa Medical Center
- (3)
Cancer Institute Hospital of Japanese Foundation for Cancer Research
- (4)
Fujita Health University
- (5)
Gunma Prefectural Cancer Center
- (6)
Hiroshima University Hospital
- (7)
Hokkaido University Hospital
- (8)
Hyogo College of Medicine
- (9)
Itami City Hospital
- (10)
Japanese Red Cross Okayama Hospital
- (11)
Juntendo University Hospital
- (12)
Kanagawa Cancer Center
- (13)
Kansai Medical University Hospital
- (14)
Kitasato University School of Medicine
- (15)
Kurashiki Central Hospital
- (16)
Kurume University School of Medicine
- (17)
Kyushu University Hospital
- (18)
Miyagi Cancer Center
- (19)
Nagasaki University Hospital
- (20)
Nagoya University School of Medicine
- (21)
National Cancer Center Hospital
- (22)
National Cancer Center Hospital East
- (23)
National Center for Global Health and Medicine (NCGM)
- (24)
National Hospital Organization Hokkaido Cancer Center
- (25)
National Hospital Organization Kinki-Chuo Chest Medical Center
- (26)
National Hospital Organization Shikoku Cancer Center
- (27)
National Hospital Organization Yamaguchi-Ube Medical Center
- (28)
Niigata Cancer Center Hospital
- (29)
Nippon Medical School Hospital
- (30)
Osaka City General Hospital
- (31)
Osaka City University Hospital
- (32)
Osaka General Medical Center
- (33)
Saitama Cancer Center
- (34)
Sendai Kousei Hospital
- (35)
Shizuoka Cancer Center
- (36)
Shinshu University School of Medicine
- (37)
Tochigi Cancer Center
- (38)
Tokushima University Hospital
- (39)
Tokyo Metropolitan Cancer and Infectious diseases Center Komagome Hospital
- (40)
Toranomon Hospital
- (41)
Wakayama Medical University, School of Medicine
- (42)
Yokohama Municipal Citizen’s Hospital
Biomarker research
In an ancillary study, blood plasma will be collected at five time points (at enrollment, 12/24/40 weeks after enrollment and disease progression), and the circulating tumor DNA will be analyzed using Guardant360®. The primary aim is to assess whether the baseline or longitudinal change of variant allele frequency, which reasonably reflects the tumor burden, can help identify prognostic or predictive patient subsets. This ancillary study includes enrollment at institutions where a specific approval from the institutional review board was obtained.
Conflict of interest statement
T.M., T.K. and S.K. have nothing to disclose. S.N. reports personal fees from AstraZeneca, Taiho Pharmaceutical, Chugai and Pfizer, outside the submitted work. YG reports grants and personal fees from Eli Lilly, Chugai, Taiho Pharmaceutical, Pfizer, Novartis, MSD, Guardant Health and Ono Pharmaceutical; personal fees from Boehringer Ingelheim, AstraZeneca and Illumina; and grants from Kyorin and Daiichi Sankyo, outside the submitted work. Y.O. reports personal fees from Chugai, Bristol-Myers Squibb and MSD, outside the submitted work. H.M. reports grants and personal fees from AstraZeneca, Chugai, Lilly Japan, Taiho Pharmaceutical and Takeda; grants from AbbVie, Daiichi Sankyo and IQVIA; and personal fees from Ono Pharmaceutical, Bristol-Myers Squibb Japan and MSD, outside the submitted work. K.T. reports grants and personal fees from Ono, Boehringer Ingelheim and Chugai and personal fees from AstraZeneca, Eli Lilly, Bristol-Myers Squibb, AbbVie, Novartis, Pfizer, Kyowa Kirin, MSD and Taiho, outside the submitted work. Y.O. reports grants and personal fees from Ono Pharmaceutical, Chugai, MSD and Bristol-Myers Squibb during the conduct of the study; grants and personal fees from AstraZeneca, Eli Lilly, Taiho, Kyorin, Novartis and Janssen; personal fees from Boehringer Ingelheim, Pfizer, Kyowa Hakko Kirin, Celltrion, Amgen and Nippon Kayaku; and grants from Sumitomo Dainippon, Ignyta and Kissei, outside the submitted work.
Funding
This study has been supported in part by the National Cancer Center Research and Development Fund (29-A-3) and by the Japan Agency for Medical Research and Development (AMED) under Grant Number JP19ck0106457h0002. The ancillary biomarker study is partly supported by Guardant Health, Inc.
Yuichiro Ohe on behalf of the Lung Cancer Study Group/Japan Clinical Oncology Group.

{kind=link}