




Abstract
Patient-derived xenograft model is a powerful and promising tool for drug discovery and cancer biology studies. The application of previous metastatic colorectal cancer models has been greatly limited by its low success rate and long time to develop metastasis. Therefore, in this study, we aim to describe an optimized protocol for faster establishment of colorectal cancer metastatic patient-derived xenograft mouse models.
Smaller micro tissues (˂150 μm in diameter) mixed with Matrigel were engrafted subcutaneously into NSG mice to generate the passage 1 (P1) patient-derived xenograft. The micro tumours from P1 patient-derived xenograft were then excised and orthotopically xenografted into another batch of NSG mice to generate a metastatic colorectal cancer patient-derived xenograft, P2. Haematoxylin and eosin and immunohistochemistry staining were performed to compare the characters between patient-derived xenograft tumours and primary tumours.
About 16 out of 18 P1 xenograft models successfully grew a tumour for 50.8 ± 5.1 days (success rate 89.9%). Six out of eight P1 xenograft models originating from metastatic patients successfully grew tumours in the colon and metastasized to liver or lung in the NSG recipients for 60.9 ± 4.5 days (success rate 75%). Histological examination of both P1 and P2 xenografts closely resembled the histological architecture of the original patients’ tumours. Immunohistochemical analysis revealed similar biomarker expression levels, including CDH17, Ki-67, active β-catenin, Ki-67 and α smooth muscle actin when compared with the original patients’ tumours. The stromal components that support the growth of patient-derived xenograft tumours were of murine origin.
Metastatic patient-derived xenograft mouse model could be established with shorter time and higher success rate. Although the patient-derived xenograft tumours were supported by the stromal cells of murine origin, they retained the dominant characters of the original patient tumours.
Introduction
Globally, colorectal cancer (CRC) is ranked as the third most common cancer worldwide, contributing 10.6% of the total number of new cases diagnosed in 2018 (1). Generally, CRC patients have an overall good 5-year survival rate of 65%. However, once the CRC metastasizes to distant organs, the 5-year survival rate drops to <14% (2). Thus, newer and more effective therapies are still in need to improve the survival chances, especially for the advanced stages of the disease. To the best of our knowledge, most tumour masses, including those of CRCs, are composed of highly heterogeneous populations (3,4). The cancer heterogeneity is highly correlated with drug resistance and cancer relapse, hence resulting in the difficulty to completely eradicate them with the currently available therapies (3). With the increasing focus on precision medicine and personalized therapy for cancer patients, conventional mouse models and immortalized cancer cell lines are no longer appropriate for effective drugs screening. This is because most established genetic mouse models harbour a limited repertoire of genetic mutations although immortalized cancer cell lines could not reflect the full heterogeneity of patients’ tumours (5). Hence, patient-derived xenograft (PDX) mouse model was currently thought of as a promising tool for translational research and drug screening, since they could mimic the phenotypes and genotypes of the original tumour (6). With the use of the PDX mouse model, screening for effective therapeutic approaches could be done more reliably.
So far, most researchers have used heterotopic transplantation method through subcutaneous inoculation to generate their CRC PDX mouse models, even when it involves the tumours of metastatic patients (7–11). This is because it is an easier method and the monitoring of tumour sizes can be done more accurately for any type of tumours. Unfortunately, subcutaneous PDX mouse model could not mimic the original tumour niche that allows it to progress into the metastatic stage. In contrary, orthotopic xenograft models are able to recapitulate the natural microenvironment and demonstrate better pharmacological correlation with the human cancers, which makes it an ideal model to study the metastatic process for development of novel therapeutic approaches to treat malignant metastatic CRCs (6,12).
In this study, we have optimized the diameter of primary tumour for xenograft in order to shorten the time as well as to improve the success rate of tumour growth in the CRC PDX mouse model. A metastatic PDX mouse model was also established via an orthotopic xenograft and the tumour characteristics between the primary tumours and their generations of PDX mouse model were compared.
Patients and methods
Patients and samples
Patient colorectal specimens and patients’ information shown in Table 1 were collected with informed consent from the Department of General Surgery, Chinese PLA General Hospital, Beijing. All specimens were kept in an antibiotic-containing Dulbecco’s modified eagle medium solution on ice and were processed within 2 h after the surgery. Each fresh tumour specimen was cut into three portions, whereby one portion was used for tumour xenograft, another portion was fixed in 4% formalin solution for haematoxylin and eosin (H&E) or immunohistochemistry (IHC) staining and the remaining tumour tissue was frozen in liquid nitrogen for future studies. All the experimental procedures carried out in the study were approved by the Institutional Ethics Board of the Hospitals and Tsinghua University (Identification number: F16-00228; A5061-0).
Clinical pathological features of the colorectal cancer patients
| Patient ID | Sex | Age | Tumour origin | TNM stage | Metastasis | Differentiation | Heterotopic xenograft | Orthotopic xenograft |
|---|---|---|---|---|---|---|---|---|
| hCC001 | M | 68 | Primary | T3N1M0 | No | Moderate–poor | Yes | Yes (failed) |
| hCC002 | M | 66 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC003 | M | 82 | Primary | T3N0M0 | No | Moderate | Yes | No |
| hCC004 | F | 83 | Primary | T4N1M0 | No | Moderate–poor | Yes | No |
| hCC005 | M | 53 | Primary | T2N0M0 | No | Moderate | Yes (failed) | No |
| hCC006 | M | 55 | Primary | T2N1M0 | No | Moderate–poor | Yes | No |
| hCC007 | M | 59 | Primary | T4N2M0 | No | Poor | Yes | No |
| hCC008 | M | 53 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC009 | F | 32 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC010 | F | 59 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC011 | F | 54 | Primary | T4aN0M1 | Liver | Poor | Yes | Yes |
| hCC012 | M | 61 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC013 | F | 67 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC014 | F | 60 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes (failed) |
| hCC015 | F | 48 | Primary, metastasis | T4N1M1 | Peritoneal | Moderate–poor | Yes (failed) | No |
| hCC016 | M | 63 | Primary | T4N1M1 | Peritoneal | Poor | Yes | Yes |
| hCC017 | M | 70 | Primary | T3N1M1 | Liver | Poor | Yes | Yes |
| hCC018 | F | 65 | Primary | T3N2M1 | Lung/liver | Moderate–poor | Yes | Yes |
| Patient ID | Sex | Age | Tumour origin | TNM stage | Metastasis | Differentiation | Heterotopic xenograft | Orthotopic xenograft |
|---|---|---|---|---|---|---|---|---|
| hCC001 | M | 68 | Primary | T3N1M0 | No | Moderate–poor | Yes | Yes (failed) |
| hCC002 | M | 66 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC003 | M | 82 | Primary | T3N0M0 | No | Moderate | Yes | No |
| hCC004 | F | 83 | Primary | T4N1M0 | No | Moderate–poor | Yes | No |
| hCC005 | M | 53 | Primary | T2N0M0 | No | Moderate | Yes (failed) | No |
| hCC006 | M | 55 | Primary | T2N1M0 | No | Moderate–poor | Yes | No |
| hCC007 | M | 59 | Primary | T4N2M0 | No | Poor | Yes | No |
| hCC008 | M | 53 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC009 | F | 32 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC010 | F | 59 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC011 | F | 54 | Primary | T4aN0M1 | Liver | Poor | Yes | Yes |
| hCC012 | M | 61 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC013 | F | 67 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC014 | F | 60 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes (failed) |
| hCC015 | F | 48 | Primary, metastasis | T4N1M1 | Peritoneal | Moderate–poor | Yes (failed) | No |
| hCC016 | M | 63 | Primary | T4N1M1 | Peritoneal | Poor | Yes | Yes |
| hCC017 | M | 70 | Primary | T3N1M1 | Liver | Poor | Yes | Yes |
| hCC018 | F | 65 | Primary | T3N2M1 | Lung/liver | Moderate–poor | Yes | Yes |
hCC, human colorectal cancer; M, male; F, female.
Clinical pathological features of the colorectal cancer patients
| Patient ID | Sex | Age | Tumour origin | TNM stage | Metastasis | Differentiation | Heterotopic xenograft | Orthotopic xenograft |
|---|---|---|---|---|---|---|---|---|
| hCC001 | M | 68 | Primary | T3N1M0 | No | Moderate–poor | Yes | Yes (failed) |
| hCC002 | M | 66 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC003 | M | 82 | Primary | T3N0M0 | No | Moderate | Yes | No |
| hCC004 | F | 83 | Primary | T4N1M0 | No | Moderate–poor | Yes | No |
| hCC005 | M | 53 | Primary | T2N0M0 | No | Moderate | Yes (failed) | No |
| hCC006 | M | 55 | Primary | T2N1M0 | No | Moderate–poor | Yes | No |
| hCC007 | M | 59 | Primary | T4N2M0 | No | Poor | Yes | No |
| hCC008 | M | 53 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC009 | F | 32 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC010 | F | 59 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC011 | F | 54 | Primary | T4aN0M1 | Liver | Poor | Yes | Yes |
| hCC012 | M | 61 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC013 | F | 67 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC014 | F | 60 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes (failed) |
| hCC015 | F | 48 | Primary, metastasis | T4N1M1 | Peritoneal | Moderate–poor | Yes (failed) | No |
| hCC016 | M | 63 | Primary | T4N1M1 | Peritoneal | Poor | Yes | Yes |
| hCC017 | M | 70 | Primary | T3N1M1 | Liver | Poor | Yes | Yes |
| hCC018 | F | 65 | Primary | T3N2M1 | Lung/liver | Moderate–poor | Yes | Yes |
| Patient ID | Sex | Age | Tumour origin | TNM stage | Metastasis | Differentiation | Heterotopic xenograft | Orthotopic xenograft |
|---|---|---|---|---|---|---|---|---|
| hCC001 | M | 68 | Primary | T3N1M0 | No | Moderate–poor | Yes | Yes (failed) |
| hCC002 | M | 66 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC003 | M | 82 | Primary | T3N0M0 | No | Moderate | Yes | No |
| hCC004 | F | 83 | Primary | T4N1M0 | No | Moderate–poor | Yes | No |
| hCC005 | M | 53 | Primary | T2N0M0 | No | Moderate | Yes (failed) | No |
| hCC006 | M | 55 | Primary | T2N1M0 | No | Moderate–poor | Yes | No |
| hCC007 | M | 59 | Primary | T4N2M0 | No | Poor | Yes | No |
| hCC008 | M | 53 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC009 | F | 32 | Primary | T3N0M0 | No | Moderate–poor | Yes | No |
| hCC010 | F | 59 | Primary | T3N1M0 | No | Moderate | Yes | No |
| hCC011 | F | 54 | Primary | T4aN0M1 | Liver | Poor | Yes | Yes |
| hCC012 | M | 61 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC013 | F | 67 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes |
| hCC014 | F | 60 | Primary | T3N1M1 | Liver | Moderate | Yes | Yes (failed) |
| hCC015 | F | 48 | Primary, metastasis | T4N1M1 | Peritoneal | Moderate–poor | Yes (failed) | No |
| hCC016 | M | 63 | Primary | T4N1M1 | Peritoneal | Poor | Yes | Yes |
| hCC017 | M | 70 | Primary | T3N1M1 | Liver | Poor | Yes | Yes |
| hCC018 | F | 65 | Primary | T3N2M1 | Lung/liver | Moderate–poor | Yes | Yes |
hCC, human colorectal cancer; M, male; F, female.
Establishment of PDX mouse models
Tumour samples were collected immediately after surgery, washed with antibiotic-containing phosphate buffered saline (PBS), cut into smaller tissues at micro sizes and filtered using the 150-μm filter mesh. Tumour fragments <150 μm were collected and mixed with concentrated Matrigel at a ratio of 1:1. Subsequently, the tissues were subcutaneously implanted into a group of 6- to 8-week-old NSG mice. When the tumours grew to ~500 mm3 in the mice, those samples were considered as ‘tumorigenic’ and labelled as ‘P1’. Tumour size was measured with a calliper-like instrument. Tumour volume (V) was calculated by the formula: V = (length + [width]2)/2. To develop a metastatic PDX mouse model, this tumorigenic P1 samples originating from the tumour of metastatic patients were then prepared into ˂150-μm tumour micro tissues and injected into the cecal wall of anesthetized NSG mice. The orthotopically engrafted recipient mice were fed with acidified water supplemented with 2 mg/ml neomycin sulphate and 200 μg/ml polymyxin B sulphate (Inalco, San Luis Obispo, CA). Recipient mice were sacrificed to confirm the presence of metastasis when the mice became inactive with sparse and dry hair. The harvested tumour tissues were fixed for histological and IHC analyses. Those tumours that were successfully grown in colon, liver or lung were labelled as ‘P2’.
H&E and IHC staining
Patient tumour specimens or xenografts were fixed with 4% neutral formaldehyde and embedded in paraffin for H&E as well as IHC staining. For H&E analysis, the 5 μm thick sections were stained with the H&E solution (ZSGB-BIO, Beijing, China) in accordance to the manufacturer’s instruction. For IHC staining, antigen retrieval was performed with the microwave heat-induced epitope retrieval method. All specimens were incubated with rabbit polyclonal anti-CDH17 antibody (Proteintech), rabbit monoclonal Ki-67 antibody (SP6, Thermo Scientific), anti-active β-catenin (ABC) antibody (D13A1, Cell signalling technology), mouse monoclonal anti-α-smooth muscle actin (a-SMA) antibody (1A4, Boster) and mouse monoclonal anti-human mitochondria antibody (113-1, Abcam) at 4°C overnight, followed by incubation with secondary antibodies at 37°C for 1 h. The histology slides stained with HRP-conjugated secondary antibodies were incubated with the horseradish peroxidase (HRP) substrate, DAB for 2–5 min, counterstained with haematoxylin and mounted for imaging using ECLIPSE 90i microscope (Nikon, Tokyo, Japan) and Axio Scan.Z1 slide scanner (Zeiss, Oberkochen, Germany).
Results
Clinical characteristics of the patients and generation of metastatic PDX mouse models
A total of 18 colorectal patients’ samples were collected to establish PDX mouse models. There are 10 non-metastatic samples and 8 metastatic samples. The clinical pathological features of the cancer patients were listed in Table 1.
Heterotopic injection (involving subcutaneous inoculation in most cases) is often chosen to establish PDX mouse models. In the published methods to generate CRC PDX mouse models via subcutaneous xenograft, tumour specimens were usually cut into fragments of about 1–10 mm in diameter (13–16) or digested into single cells with collagenase (9,17). However, in this study, we have optimized the method by cutting the tissues into small pieces and then filtering tumour pieces by 100-mesh filter, which could collect the micro tumour tissues of<150 μm in diameter (Fig. 1A). On the other hand, an equal volume of concentrated matrigel was mixed with the micro tumours in PBS, which could maintain proper nutrient supply for the micro tumours. However, the matrigel was used only in the single-cell xenograft but not in the fragment xenograft in the previous methods. The average time for a tumour growing up to 500 mm3 in the first engraftment was 50.8 ± 5.1 days, which was shorter compared with 60–120 days in the previous methods. Besides, the success rate of the tumour formation in our PDX mouse models is much higher (88.9%) compared with the published reports which ranged from 20 to 74% (9,15,18–20) (Table 2).
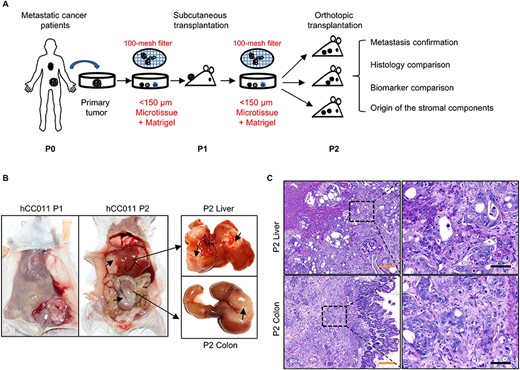
Establishment of a metastatic CRC PDX mouse model. (A) Flow chart of the development of metastatic CRC PDX mouse model. The differences compared with previous methods were labelled in red colour. (B) Representative PDX images from heterotopic and orthotopic xenograft models of hCC011. (C) Representative H&E data for metastasis confirmation of the orthotopic models. CRC, colorectal cancer; PDX, Patient-derived xenograft model; H&E, haematoxylin and eosin. Scale bars, 50 μm in black, 200 μm in yellow.
Additionally, most PDX mouse models of CRC so far have been established via subcutaneous injection, of which an appropriate metastatic model could not be developed due to the lack of a suitable microenvironment for tumour growth. Therefore, these heterotopic grafts are not considered as appropriate models to study cancer metastasis progression as well as for screening of drugs that target metastasis. Hence in our study, we have also established a stable metastatic PDX mouse model for CRC. For the tumour tissues derived from the eight clinical patients who are metastatic, the established P1 tumours were then orthotopically transplanted into the colon cecal wall. Six samples successfully formed tumours in situ and further metastasized to liver and lung after 60.9 ± 4.5 days (Table 2). Metastatic tumours in colon and liver can be clearly seen, as indicated by the black arrows (Fig. 1B). The tumour histological morphology was further investigated using H&E staining. Poorly differentiated human tumour cells were significantly much larger than the original mouse colon and liver cells that surrounded the tumour cells (Fig. 1C).
Histology comparison between the original tumour and xenograft tumour
To investigate whether there are any differences in the histological morphology and tissue characteristics as the tumour tissue was propagated in the mice, xenografted tumours derived from three clinical patient cases were randomly selected to compare their histological changes. These included the original samples, P1 xenografted tumours, P2 orthotopically xenografted tumours and the metastatic tumours. The data revealed a comparable histological morphology among these samples (Fig. 2). For hCC013, the P2 orthotopic xenografts recapitulated the moderately differentiated adenocarcinoma of the original tumour. As for hCC018, the ring cell carcinoma was better reflected in P2 as compared with the one in P1 xenografts. There were also some enriched poorly differentiated carcinomas in P1 heterotopic xenografts in both hCC013 and hCC018. These findings indicated that orthotopic xenograft had a better resemblance to the original tumour histology of the primary tumour.
Tumour formation rate and latency period of xenografts
| Current system | Published system (9,15,18–20) | ||
|---|---|---|---|
| P1 | P2 | P1 | |
| Mouse | NSG | NSG | NSG, NOD-severe combined immuno deficient (SCID), nu/nu |
| Injected tumour size (in diameter) | ˂150 μm | ˂150 μm | 1–10 mm or stable single cells |
| Injection position | Subcutaneous site | Colon | Subcutaneous site |
| Tumour formation rate | 88.9% (16/18) | 75% (6/8) | 14–74% |
| Tumour latency (days) | 50.8 ± 5.1 | 60.9 ± 4.5 | 60–120 |
| Metastasis | No | 100% (6/6) | NA |
| Current system | Published system (9,15,18–20) | ||
|---|---|---|---|
| P1 | P2 | P1 | |
| Mouse | NSG | NSG | NSG, NOD-severe combined immuno deficient (SCID), nu/nu |
| Injected tumour size (in diameter) | ˂150 μm | ˂150 μm | 1–10 mm or stable single cells |
| Injection position | Subcutaneous site | Colon | Subcutaneous site |
| Tumour formation rate | 88.9% (16/18) | 75% (6/8) | 14–74% |
| Tumour latency (days) | 50.8 ± 5.1 | 60.9 ± 4.5 | 60–120 |
| Metastasis | No | 100% (6/6) | NA |
P1, first Passage PDX; P2, second Passage PDX; NA, no analysis.
Tumour formation rate and latency period of xenografts
| Current system | Published system (9,15,18–20) | ||
|---|---|---|---|
| P1 | P2 | P1 | |
| Mouse | NSG | NSG | NSG, NOD-severe combined immuno deficient (SCID), nu/nu |
| Injected tumour size (in diameter) | ˂150 μm | ˂150 μm | 1–10 mm or stable single cells |
| Injection position | Subcutaneous site | Colon | Subcutaneous site |
| Tumour formation rate | 88.9% (16/18) | 75% (6/8) | 14–74% |
| Tumour latency (days) | 50.8 ± 5.1 | 60.9 ± 4.5 | 60–120 |
| Metastasis | No | 100% (6/6) | NA |
| Current system | Published system (9,15,18–20) | ||
|---|---|---|---|
| P1 | P2 | P1 | |
| Mouse | NSG | NSG | NSG, NOD-severe combined immuno deficient (SCID), nu/nu |
| Injected tumour size (in diameter) | ˂150 μm | ˂150 μm | 1–10 mm or stable single cells |
| Injection position | Subcutaneous site | Colon | Subcutaneous site |
| Tumour formation rate | 88.9% (16/18) | 75% (6/8) | 14–74% |
| Tumour latency (days) | 50.8 ± 5.1 | 60.9 ± 4.5 | 60–120 |
| Metastasis | No | 100% (6/6) | NA |
P1, first Passage PDX; P2, second Passage PDX; NA, no analysis.
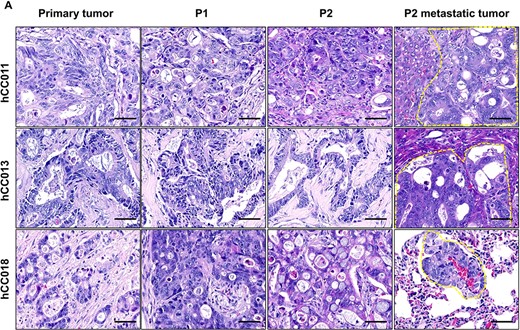
Histological comparison between primary tumours and PDX tumours. (A) Representative H&E images for three cases of metastatic CRC and their PDX models. Scale bars, 50 μm in black.
Biomarker comparison between the original tumours and xenograft tumours
To further assess whether these xenografts have retained the biomarker profile of its original tumour, IHC analysis was performed in these tumour sections probed with antibodies against CDH17, Ki-67, ABC and α-SMA. Figure 3 showed representative pictures of the two cases (hCC0011 and hCC018) and its corresponding xenografts. In both cases, similar levels of CDH17 and Ki-67 were detected in the primary tumour, P1 and P2 xenografts, as well as P2 metastatic tumour. Meanwhile, the ABC level was seen to be increased in the xenografts. However, the ABC expression pattern was different in both the cases. In hCC011, the ABC was mainly accumulated in the nucleus, but for hCC018, ABC was found to be present in the cytoplasm and this expression pattern was also reflected in their xenografts. As for α-SMA, it is a marker to investigate whether the cancer cells have caused any damages to the basement membrane. From our findings, we observed that the membrane barriers were disrupted significantly by the invasive tumour cells in both the cases. This indicated that the tumour cells in both the two cases exhibited strong invasive and metastatic capabilities. These results demonstrated that our xenografts faithfully reproduced the original human CRCs’ characteristics.
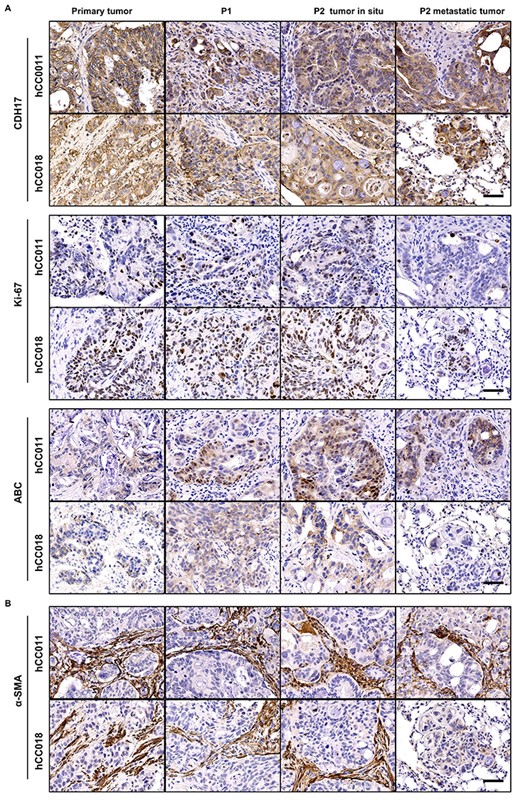
Biomarker expression comparison between primary and PDX tumours. (A) IHC analysis of the normal biomarker (CDH17, ki-67, ABC) expression levels in the hCC0011 and hCC018 metastatic CRC and their PDX models. (B) IHC analysis of α-SMA expression levels of the 2 cases of metastatic CRC and their PDX models. IHC, immunohistochemistry; ABC, Active β-catenin; α-SMA, α-smooth muscle actin. Scale bars, 50 μm in black.
Origin of the stromal cells supporting growth of PDX tumours
To specifically determine the origin of stromal components that support the growth of successfully engrafted patient xenograft. We have done IHC with anti-human mitochondria antibody, which could specifically react with human cells but not mouse cells. As seen in Fig. 4, the antibody specific for a human mitochondrial antigen recognized both tumour cells and stromal cells in the patient specimens of CRC, but only labelled the tumour cells in both the P1 and P2 PDX tumours. This indicated that the stromal cells supporting the growth of these PDX tumours were of murine origin. The human stromal cells which were engrafted as part of the original tumour did not survive and were no longer detectable in the PDX tumours. This is consistent with previous reports (18,21–23).
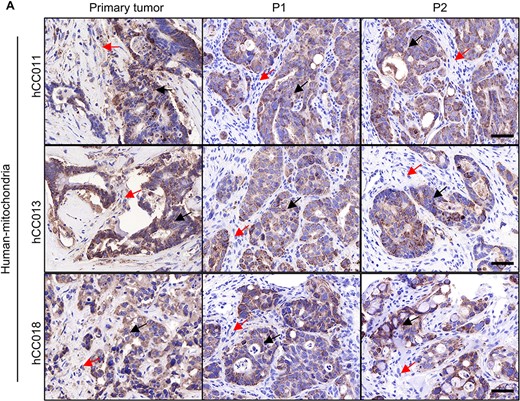
Origin of the stromal cells in PDX tumours. (A) IHC staining with a human-specific mitochondrial antibody in both the primary tumours and PDX tumours. Human tumour cells were indicated by black arrows, and stromal cells were indicated by red arrows. Scale bars, 50 μm in black.
Discussion
Aggressiveness of the advanced CRC has been a major clinical challenge for decades. The low 5-year survival rate for metastatic CRC patients indicates that better therapeutic agent and preclinical model is necessary. CRC PDX mouse models are able to retain clinically relevant features and hence, are important for the development of a more personalized therapeutic strategy. However, generation of PDX mouse models is highly limited by its lower success rate and longer time required (5,6,24,25). Another issue is that implantation method of most CRC PDX is subcutaneous injection. However, the PDX mouse model via subcutaneous injection is not an ideal model for metastasis related study and anti-metastasis drug screening, since the tumour microenvironment and metastasis pathway differ between an orthotopic and a heterotopic xenograft. Although Sylvia Julien et al. generated the metastatic CRC PDX mouse model via tumour engraftment into the cecum of SCID mice, the success rate was only 32% (18).
To solve these issues, we have developed an improved protocol to generate CRC PDX mouse models, whereby the duration for xenograft tumours formation was significantly shortened with a higher success rate. This could be attributed to two factors, as micro tumour tissues with sizes ˂150 μm in diameter were more fitted to maintain the tumour microenvironment and these smaller tumour tissues were also ensured receipt of sufficient nutrients from the body of the recipient mice as compared with using larger tumour tissues reported in other papers. On top of that, we also successfully developed the CRC metastatic PDX mouse models via orthotopic xenograft method using the established P1 xenograft tumours. P1 xenograft tumour was used because it is difficult to generate PDX mouse model with primary tumours via orthotopic transplantation (26). From our previous attempts, only one out of four orthotopic xenograft samples derived from the patients was generated successfully (data not shown). Even then, the duration for tumour growth and metastasis development had taken up to 6 months (27). In order to shorten the tumour formation time and to increase the success rate, it is necessary to first engraft the patient tumours into NSG mice through subcutaneous xenograft method, followed by using the P1 PDX tumours to develop the metastatic PDX mouse models. The metastatic PDX tumours generated have also maintained the histological characteristics of the original patient tumours. This indicated that our orthotopic xenograft model could be a better preclinical model to study the CRC metastasis, as well as for the development of new therapeutic strategies.
Nevertheless, although we had successfully developed the metastatic xenograft models originating from the metastatic patients, there are still several limitations in our study. This is because the in situ tumours and metastatic tumours were difficult to be monitored throughout the formation and growth process in orthotopic xenograft. Besides that, the use of our PDX mouse models to study human tumour metastasis and drug screening is still limited by several factors. This includes the lack of a functional immune system and stromal components of human origin, as the interaction between human stromal cells and the immune system confers the biggest challenge in the PDX mouse models for all type of cancers (6,28).
In conclusion, the xenograft success rate has been improved by using our xenograft protocol. Besides that, the tumorigenesis duration has been shortened remarkably. More importantly, the metastatic CRC PDX mouse models were able to be developed and they have maintained the histological characteristics of the original patient tumours and metastatic tumours. Our research findings are expected to be able to provide a comprehensive basis for future—PDX study. These improved PDX generation methods were also expected to be useful in the developmental study of therapeutic strategies and drug discovery for personalized treatment.
Acknowledgements
The authors would like to thank Dr Zai Chang in the Animal Facility, Tsinghua University for his animal care support. Besides that, we would also like to show our appreciation to Dr Wei Guo for his support and experimental trouble shooting in the study.
Conflicts of interest
The authors declare that they have no conflict of interest.
Funding statement
None.

{kind=link}
{kind=link}
{kind=link}
{kind=link}