






Abstract
Two transplant recipients (1 kidney and 1 hematopoietic stem cell) received maribavir (MBV) after cytomegalovirus (CMV) infection clinically resistant to standard therapy. Both patients achieved CMV DNA clearance within 30 and 18 days; however, the UL97 C480F variant emerged, causing recurrent CMV infection after a cumulative 2 months of MBV and 15 or 4 weeks of ganciclovir treatment, respectively. C480F was not detected under ganciclovir before MBV treatment. Recombinant phenotyping showed that C480F conferred the highest level of MBV resistance and ganciclovir cross-resistance, with impaired viral growth. Clinical follow-up and genotypic and phenotypic studies are essential for the assessment and optimization of patients with suspected MBV resistance.
Antiviral therapy for cytomegalovirus (CMV) plays an important role in the clinical management of solid organ transplant (SOT) and hematopoietic stem cell transplant (HSCT) recipients. However, CMV antiviral therapy can be complicated by drug resistance–associated mutations in the phosphotransferase UL97 and the DNA polymerase UL54 [1]. New therapeutic molecules (letermovir, maribavir [MBV], and brincidofovir) have been developed against different pharmacologic targets; however, resistance mutations have already been described [2].
MBV is a phase 3 antiviral inhibitor of CMV DNA synthesis, viral gene expression, encapsidation, and viral capsid egress by inhibition of pUL97 [3]. Phase 2 trials using high MBV doses for the treatment of CMV infection refractory or resistant to conventional therapy have shown plasma CMV DNA clearance after 6 weeks in 67%–77% of patients. However, about 30% presented CMV recurrent infection with UL97 mutations conferring MBV resistance [4].
MBV resistance mutations have primarily been mapped to the UL97 gene and also show compensatory mutations in UL27 [5]. Only a few mutations in UL97, such as the recently described C480F, cause cross-resistance to ganciclovir (GCV) [6].
Our study emphasizes that clinical follow-up, in combination with genotypic and phenotypic studies, is essential for the assessment and optimization of patients with suspected maribavir resistance. In this study we report MBV-GCV resistance mutations and their associated phenotype in the context of 2 transplant recipients.
MATERIALS AND METHODS
Sample Collection
Clinical isolates were obtained from 1 HSCT recipient from the Hospital Clinic of Barcelona (Spain), where MBV was approved for compassionate use, and 1 kidney transplant recipient from the Hospital of Bellvitge (Barcelona, Spain), who was enrolled in the MBV study (Shire, SHP620-303), both with suspected resistance to antiviral agents [7]. Plasma samples and biopsies were sent to the Hospital Clinic of Barcelona to undergo genotypic assays. Plasma CMV monitoring, treatment follow-up, and sample collection and storage were performed as described previously [8].
CMV Viral Load Measurement
Extraction of DNA was performed in MagNA Pure Compact (Roche, Switzerland). The CMV viral load was measured by quantitative real-time polymerase chain reaction (PCR) Cobas CMV (Roche, Switzerland) according to the manufacturer’s instructions.
Genotypic Antiviral Resistance Testing
Genotypic testing was based on PCR amplification of CMV UL97 in the 2 fragments associated with resistance mutations to GCV (residues 400–670) and MBV (270–482) and in 4 fragments of UL54 (300–1000), followed by Sanger sequencing as described elsewhere [8]. UL27 was genotyped (284–602) by Sanger sequencing using the primers described by Chou et al [6].
Phenotypic Assay by Recombinant Bacterial Artificial Chromosome Technology
UL97 C480F mutation, with an unknown phenotype at the time of genotypic detection, was individually tested at the French National Reference Center for Herpesviruses (Limoges) using a phenotypic assay with recombinant bacterial artificial chromosomes (BAC) technology as described previously [9]. C480F single mutation was introduced by “en passant” mutagenesis into a human cytomegalovirus (HCMV) BAC [10] containing an enhanced green fluorescent protein (EGFP) gene in the unique short region derived from the AD169 laboratory strain (provided by M. Messerle). The recombinant BAC was transfected into MRC-5 cells (bioMérieux, Lyon, France) using the liposomal reagent Transfast (Promega, Madison, Wisconsin) following the manufacturer’s instructions. The presence of the mutation was confirmed by Sanger sequencing.
A focus reduction assay in 48-well MRC-5 culture plate with a multiplicity of infection (MOI) of 0.01 was used to assess antiviral susceptibility in triplicate to MBV, GCV, and foscarnet (FOS), and 50% inhibitory concentrations (IC50) of the mutant were compared to those of the wild-type control HCMV BAC.
To estimate the impact of the C480F mutation on viral fitness, the recombinant strain and the AD169-EGFP wild-type control were inoculated into 48-well MRC-5 culture with an MOI of 0.01. The number of fluorescent cytopathic foci was counted from days 1 to 8 postinoculation to establish viral growth curves for each recombinant.
Ethical Approval
This study was approved by the Ethical Committee of Hospital Clinic of Barcelona (reference number HCB/2018/0634) as the reference committee for the participating hospitals endorsed by the Grupo de Estudio de la Infección en el Trasplante (GESITRA) according to the Guidance on Good Clinical Practice (CPMP) and the International Conference on Harmonisation (ICH) CPMP/ICH/135/95 regulations. Patient consent was obtained for inclusion in the study.
RESULTS
Clinical Follow-up of Cases With the Novel C480F Mutation
Patient 1 was a 38-year-old CMV-seropositive (R+) woman who received a myeloablative allogenic HSCT from a CMV-seronegative (D–) sibling donor due to acute myeloid leukemia (Figure 1A). Prophylaxis for graft-vs-host disease (GVHD) consisted of tacrolimus. At day 41 after transplant, the patient experienced diarrhea and abdominal pain. Lower gastrointestinal (GI) endoscopy revealed diffuse edema and multiple small ulcers in the colon. Gland apoptosis showed histological evidence of acute GI-GVHD. The patient was treated with prednisone 1 mg/kg every 24 hours and beclomethasone 2 mg every 6 hours showing clinical improvement but on day 45, CMV replication was detected in plasma, and valganciclovir (VGCV) 900 mg every 12 hours was started. After 1 week, VGCV was discontinued because of severe cytopenia, and FOS 60 mg/kg every 12 hours was administered for 1 week, resulting in CMV DNA clearance. Two months later, the patient was diagnosed with GI-CMV disease and treated with GCV 5 mg/kg every 12 hours. After 3 weeks, GCV was discontinued due to the development of cytopenia. Since viral loads were not reduced, MBV 400 mg every 12 hours was started and followed for 6 weeks with adequate CMV clearance. One week after stopping MBV, CMV reactivation was detected and MBV was restarted. After 3 weeks of MBV, there was a progressive viral load increase, and resistance mutation genotyping showed the emergence of the UL97 C480F variant, with a phenotype unknown at that time, in addition to the UL54 F669L sensitive polymorphism. The patient was then treated with GCV 2.5 mg/kg every 12 hours and again switched to FOS 60 mg/kg every 24 hours for cytopenia until death caused by multiorgan failure related to uncontrolled GI-CMV disease on day 258.
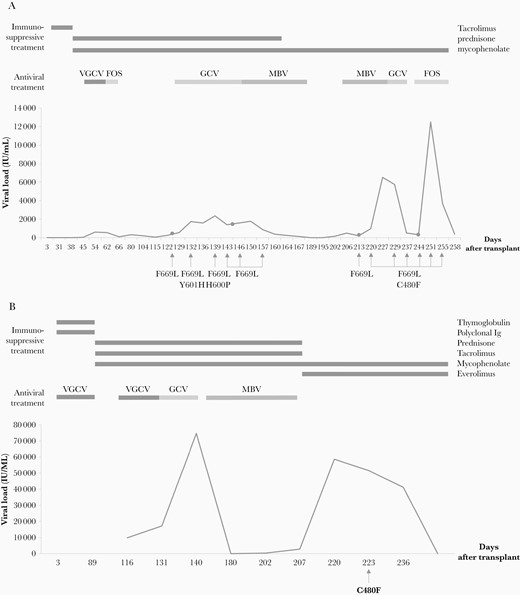
Clinical follow-up of patient 1 (A) and patient 2 (B). Viral loads were tracked against days after transplantation. Immunosuppressive and antiviral treatment outset and end are indicated. Genotypic assays were performed in the clinical isolates indicated with an arrow. Gastrointestinal biopsies are indicated with a dot; substitutions found in the UL54 and UL97 (bold) genes are shown below. Abbreviations: FOS, foscarnet; GCV, ganciclovir; Ig, immunoglobulin; MBV, maribavir; VGCV, valganciclovir.
All of the plasma and biopsy samples obtained from the initial CMV replication until patient death were retrospectively genotyped. The naturally occurring polymorphism UL54 F669L was detected in all of the isolates. Two plasma samples showed previously uncharacterized UL54 mutations (Y601H, H600P) under GCV treatment on days 136 and 139. Neither mutation was detected in the subsequent esophageal or duodenum biopsies or in plasma samples.
UL97 C480F was first found in a plasma sample (962 IU/mL) 72 days after the first MBV treatment, which is 13 days after the second treatment with MBV. This variant emerged and spread under MBV, GCV, and FOS treatments, being detected in every plasma sample and colon and gastric biopsy from that time until death.
No mutation was detected in UL27, which is also related to MBV resistance. No resistance mutation to VGCV-GCV, FOS, or MBV was found at baseline. All fragments were verified with bidirectional sequencing of the PCR product for all target genes with technical and biological repetitions.
Patient 2 was a 44-year-old woman who received a third D+R+ kidney transplant due to chronic kidney failure (Figure 1B). Prophylactic treatment during the first 3 months included VGCV (450 mg every 72 hours increased to 900 mg every 24 hours after normalization of kidney function), thymoglobulin, polyclonal antibodies, and corticoids. Maintenance therapy consisted of mycophenolate, tacrolimus, and prednisone. One month after the end of primary prophylaxis, the first CMV viral load (9885 IU/mL) was detected with no associated symptoms, and VGCV 900 mg every 12 hours was started. Clinical symptoms (diarrhea) and an increase in viremia appeared 9 days posttherapy and was treated with GCV 5 mg/kg every 12 hours. Viremia continued to increase (74 687 IU/mL) after 2 weeks on GCV treatment, and GCV was replaced by MBV 400 mg every 12 hours for 2 months until clearance.
Sixteen days after MBV discontinuation, asymptomatic viremia was determined (58652 IU/mL) and a resistance mutation study was requested. C480F was detected in UL97. No amino acid changes were found in UL54 or UL27. However, no specific anti-CMV drug was prescribed owing to the lack of response to previous episodes, and immunosuppression treatment was modified to everolimus and a reduction in mycophenolate until the loss of viremia without CMV-associated clinical outcomes. Unfortunately, clinical isolates from this patient could not be recovered for retrospective genotypic studies.
Phenotypic Analysis of the C480F Mutation
Recombinant virus with the novel C480F mutation was constructed from the standard AD169-BAC with the EGFP reporter gene. C480F was verified by sequencing of the UL97 fragment. The IC50 values for the C480F mutant were compared with the AD169 control strain for GCV, FOS, and MBV (Supplementary Table 1). C480F IC50 demonstrated resistance to GCV, but for MBV could not be calculated as MBV concentrations did not reduce the foci to 50%, clearly demonstrating a pattern of high-level resistance (Figure 2A). This assay confirmed that C480F conferred high resistance to MBV and GCV cross-resistance.
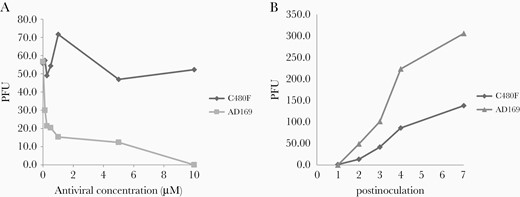
Phenotypic assay of the C480F mutation. The C480F recombinant cytomegalovirus (CMV) strain was compared with an AD169 CMV control strain. A, Maribavir plaque reduction curves. Plaque-forming units (PFUs) were counted at day 5 postinoculation. Viral susceptibility was assessed in triplicate. B, Comparative growth curves of viral strains. Both strains were inoculated at an equal multiplicity of infection of 0.01. PFUs were counted from days 1 to 7 postinoculation. Data shown are the mean of 3 replicates set up simultaneously.
Growth assay showed a slower replication of the C480F variant compared with AD169, demonstrating deteriorated viral fitness (Figure 2B).
Discussion
This study describes 2 clinical cases in which the recently reported MBV-GCV cross-resistance mutation C480F was found under MBV therapy. Both patients achieved CMV DNA clearance within 18 days (patient 1) and 30 days (patient 2) under MBV treatment; however, the C480F variant emerged, causing recurrent CMV infection detected 13 days after the initiation of a second MBV therapy and 16 days after 2 months of MBV, respectively. Both patients received 400 mg every 12 hours of MBV, a dosage that presented similar efficacy to that of VGCV for clearance CMV viremia. However, a higher dosage related to resistance development has been reported [4, 11].
Previous trials inquired whether C480F was selected under GCV or MBV [6]. In the present study, C480F was not detected under GCV treatment before the initiation of MBV, and it was not present at baseline before GCV or any anti-CMV drug in patient 1. The cumulative duration of treatment before clinical resistance associated with the emergence of this mutant was 2 months of MBV for both patients, 1 month of VGCV/GCV for patient 1, and nearly 4 months of VGCV/GCV for patient 2.
The C480F substitution is located in the catalytic loop of UL97, outside the codon range of the ATP-binding site, specified for GCV resistance mutation studies in most laboratories. GCV resistance mutations cluster in the UL97 domains involved in substrate recognition (residues 460, 520, and 590–607), whereas the majority of mutations located in the vicinity of the ATP-binding site (residues 353–397 and 409–411) confer resistance to MBV but not to GCV [4]. Most commonly selected after exposure, MBV or GCV resistance mutations do not confer cross-resistance to the other [12]. However, only a few studies have reported GCV-MBV cross-resistance mutations in clinical specimens, such as V466G, P521L, or the novel C480F, which were found at distant sites from the ATP-binding loop [13, 14].
The C480F mutation has previously been detected in a patient treated with MBV [14] but was not phenotyped. The same mutation was found in the 2 transplant recipients in this study, and phenotyping of the uncharacterized C480F mutation demonstrated that it confers the highest level of resistance to MBV ever found in addition to GCV cross-resistance, similar to what Chou et al recently described [6].
Genotyping artifacts must be considered when interpreting single-nucleotide mutations. We sequenced the 2 known resistant regions for MBV and GCV of UL97 to have a widely overviewed genotype. Using bidirectional sequencing, we verified all of the variants found in technical replicates and in different biological samples during the follow-up of the 2 study subjects. Clinical follow-up is highly recommended for routine diagnosis due to the possible sudden emergence of mutants and the limited sensitivity of Sanger sequencing for detecting small populations.
The C480R substitution has also previously been reported in AD169 exposed to the methylenecyclopropane analogue conferring GCV-MBV cross-resistance and severe growth defects [15], such as those observed with C480F. These data are consistent with the fact that the residue 480 is essential for UL97 phosphotransferase function and HCMV replication.
Mapping of the UL97 mutation that confers resistance to MBV and GCV may be useful for designing alternative UL97 inhibitor antivirals to avoid cross-resistance. Expanding the scope of standard diagnostic genotyping (residues 355–680) can provide better insight into the frequency of genetically induced cases of drug resistance with poor response to antiviral treatment and the incidence of each resistance mutation. Estimating the phenotypes of new UL97 mutations from similar or adjacent mutations is inadvisable, and the phenotyping of uncharacterized mutations is essential not only for clinical significance but also for determining levels of resistance.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Financial support. This work was supported by Fondo de Investigación en Salud (FIS) PI 17/0150 of the Instituto de Salud Carlos III; Ministerio de Ciencia e Inovacion del Gobierno de España; the Agency for Health Technology Assessment and Ministerio de Economía y Competitividad.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}