






Abstract
Urinary tract infections are common and costly diseases affecting millions of people. Uropathogenic Escherichia coli (UPEC) is a primary cause of these infections and has developed multiple strategies to avoid the host immune response. Here, we dissected the molecular mechanisms underpinning UPEC inhibition of inflammatory cytokine in vitro and in vivo. We found that UPEC infection simulates nuclear factor-κB activation but does not result in transcription of cytokine genes. Instead, UPEC-mediated suppression of the metabolic enzyme ATP citrate lyase results in decreased acetyl-CoA levels, leading to reduced H3K9 histone acetylation in the promotor region of CXCL8. These effects were dependent on the UPEC virulence factor α-hemolysin and were reversed by exogenous acetate. In a murine cystitis model, prior acetate supplementation rapidly resolved UPEC-elicited immune responses and improved tissue recovery. Thus, upon infection, UPEC rearranges host cell metabolism to induce chromatin remodeling processes that subvert expression of host innate immune response genes.
Uropathogenic Escherichia coli (UPEC) is the most common etiological agent of urinary tract infections (UTI) and male genital tract infections [1, 2]. The effective eradication of UPEC is now challenging because of the increasing rate of resistance to antibiotics [3]. Even following successful treatment of an initial infection, more than 20% of women suffer from recurrent UTI, and 40%–80% of men exhibit impaired semen parameters [4–7]. Hence, an understanding of the molecular mechanisms underlying this complicated disease is crucial for developing novel therapeutic strategies for its treatment.
Activation of the innate immune response is essential for the effective elimination of pathogens, and accordingly many have evolved mechanisms of subverting this process. In UPEC, the virulence factor α-hemolysin (HlyA) is particularly important and regulates multiple aspects of host cell biological processes, including the nuclear factor-κB (NFκB) signaling pathway [8–11], as well as the expression of genes encoding proinflammatory mediators [12–14]. However, the mechanisms underlying HlyA-mediated repression of inflammatory gene expression remain unclear.
Recently, a variety of studies have shed light on the mechanisms of metabolic processes regulating gene expression by histone acetylation [15, 16]. In this process, acetyl-coenzyme A (acetyl-CoA) is an essential substrate for histone acetylation. Hence, the availability of acetyl-CoA could regulate gene transcription by influencing histone acetylation levels [17]. In eukaryotic cells, the generation of nucleocytoplasmic acetyl-CoA is catalyzed mainly by 2 enzymes. The enzyme ATP-citrate lyase (ACLY), produces acetyl-CoA from mitochondrial-derived citrate, while the acyl-CoA synthase short-chain family member 2 (ACSS2) enzyme converts acetate into acetyl-CoA [18]. It is well established that the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling pathway plays a principal role in cellular metabolism [19]. AKT promotes ACLY phosphorylation, which in turn induces histone acetylation by increasing acetyl-CoA levels [20]. The level of phosphorylated AKT correlates positively with increased histone acetylation. Reciprocally, attenuation of AKT signaling significantly lowers histone acetylation [22]. The robust metabolic change in infected host cells is also largely driven by the AKT signaling pathway [22]. Bacteria including UPEC, Staphylococcus aureus, and Aeromonas species are known to attenuate signaling of the key cellular metabolic PI3K/AKT pathway [8, 10]. We recently showed that UPEC was also able to decrease histone acetylation to epigenetically modulate host gene expression [10], and in this study we wanted to establish whether these 2 lines of knowledge—regulation of proinflammatory gene expression and epigenetic modification—are linked.
In the current study, we aimed to establish how HlyA-mediated inhibition of inflammatory gene expression was achieved and whether epigenetic modifications were involved in the process. We found that UPEC infection induced HlyA-dependent inhibition of the metabolic enzyme ACLY, leading to low acetyl-CoA levels and aberrant histone deacetylation. This resulted in reduced NFκB p65 binding to the promoter regions of proinflammatory cytokine and chemokine genes. In vitro and in vivo these effects were ameliorated by exogenous acetate. Taken together, our study indicates that UPEC-induced dysregulation of host cell metabolism modulates histone acetylation leading to suboptimal innate immune responses.
METHODS
Bacterial Strains and Cell Line
A UPEC pyelonephritic strain 536 (National Center for Biotechnology Information, NC_008253, CP000247), prevalent in UTIs, and a mutant with deletion of both α-hemolysin (hlyA) genes (ECP_RS23150, ECP_RS19410) in the UPEC 536 strain (hlyA double mutant, UPEC 536 HDM) [23] was used for in vitro infections. Bacteria were grown static in Luria-Bertani (LB) medium 24 hours prior to use. The human bladder epithelial cell line 5637 (ATCC HTB-9) was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen), and incubated at 37°C in the presence of 5% CO2.
Histopathology and Hematoxylin and Eosin Staining
Bladders were fixed with Methacarn (60% methanol, 30% chloroform, 10% glacial acetic acid) and then embedded in paraffin blocks. Hematoxylin and eosin staining was performed on 5-µm bladder sections and histopathological scores of bladder sections were determined in a blinded fashion using a semiquantitative scoring system as previously described [24] (see Supplementary Materials).
Immunoblotting
Following treatment, uroepithelial 5637 cells were lysed in lysis buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100 [v/v], 0.25% Igepal CA-630 [v/v], 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride) containing protease inhibitor cocktail (Sigma) and phosphatase inhibitor (Thermo Scientific). The protein concentrations of each sample were determined by Bradford assay (Bio-Rad). Twenty micrograms of proteins were separated using 7.5%–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes using a semidry blotting technique (Hybond-ECL 0.2 µm; GE Healthcare). Membranes were probed with the indicated primary antibodies and corresponding peroxidase-conjugated secondary antibodies. The blots were developed by enhanced chemiluminescence (Thermo Scientific) and visualized by using the Fusion Imaging system (Peqlab). The relative levels of IκBα, acH4, H4K5ac, H4K20me3, acH3, H3K9ac, H3K9me3, and p-ACLY were determined by normalization to β-actin or ACLY by measuring the intensity of individual bands using ImageJ software (National Institutes of Health).
RESULTS
UPEC Infection Induces NFκB Activation But Does Not Result in Proinflammatory Cytokine/Chemokine Expression
To determine the mechanisms underlying the suppressed inflammatory response seen during UPEC infection [14], we infected the human bladder carcinoma cell line 5637 with UPEC strain 536 or the same strain in which both hlyA genes were deleted (HlyA double mutant, HDM) [23]. Cell cultures infected with UPEC did not significantly induce inflammatory gene expression, while in contrast, cultures infected with HDM exhibited high levels of expression of inflammatory genes including CCL2, CXCL2, CCL20, CXCL8, and IL1A (Figure 1A). Expression of these cytokines requires activation of the transcription factor NFκB. Therefore, we next asked whether the difference in induction of inflammatory gene expression between UPEC and HDM was due to differences in NFκB activation. We observed that both UPEC and HDM induced degradation of the NFκB-inhibitor IκBα (Figure 1B), and translocation of the NFκB p65 subunit to the nucleus (Figure 1C). Taken together, these results indicate that the UPEC virulence factor HlyA is required for suppression of the expression of chemokine and cytokine genes during infection, but that this suppression is not mediated by inhibition of NFκB activation or translocation to the nucleus.
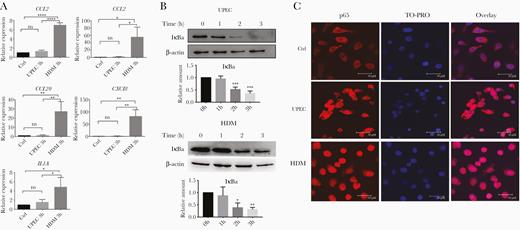
UPEC infection induces NFκB activation but does not result in proinflammatory cytokine/chemokine expression. A, Human bladder carcinoma 5637 cells were incubated with UPEC, HDM, or medium alone (Ctrl) for 3 hours. mRNA expression of indicated genes was quantified by RT-qPCR. Shown are the means ± SD of 3 independent experiments. Data were analyzed by 1-way ANOVA with multiple comparisons test. B, 5637 cells were infected with UPEC or HDM for indicated time points, and then analyzed for the presence of IκBα and β-actin proteins by immunoblotting. Representative images of 3 independent experiments are shown. Relative amounts of IκBα were quantified as described in “Methods.” Data were analyzed by 1-way ANOVA with multiple comparisons test (means ± SD of 3 independent experiments). C, Immunofluorescence labelling of NFκB p65 in 5637 cells incubated for 3 hours with UPEC, HDM, or medium alone (Ctrl). Nuclei were stained with TO-PRO nuclear dye. Representative images of each sample are shown. Experiments were repeated 3 times. Scale bar = 30 µm. *P < .05, **P < .01, ***P < .001, ****P < .0001. Abbreviations: ANOVA, analysis of variance; Ctrl, control; HDM, α-hemolysin double mutant; TO-PRO, TO-PRO™-3 Iodide; IκBα, inhibitor of κBα; NFκB, nuclear factor-κB; ns, nonsignificant; RT-qPCR, quantitative reverse transcription polymerase chain reaction; UPEC, uropathogenic Escherichia coli.
UPEC Infection Induces HlyA-Dependent Histone Deacetylation by Inhibiting ACLY Signaling
As the HlyA-mediated suppression of inflammatory gene expression was not the result of an impaired activation of NFκB signaling, we next asked whether chromatin remodeling processes employed by UPEC are responsible for the observed suppression.
We infected 5637 cells with UPEC or HDM and assessed the levels of histone acetylation and methylation at H2, H3, and H4 by western blot analysis of cell lysates. We found that overall acetylation of H3 and H4 decreased from 1 hour after UPEC infection (Figure 2A) but remained unchanged during infection with HDM (Figure 2B), indicating the requirement for HlyA. We also saw deacetylation of H2A and H2B following UPEC infection, while acetylation levels were reduced with HDM (Supplementary Figure 1A and 1B). Histone methylation was unaffected by either bacterial strain (Figure 2A and 2B and Supplementary Figure 1A and 1B). We then used site-specific acetyl lysine antibodies to more clearly identify the locations of reduced histone acetylation after UPEC infection and found a marked HlyA-dependent decrease in acetylation at H4K5 and H3K9 (Figure 2A).
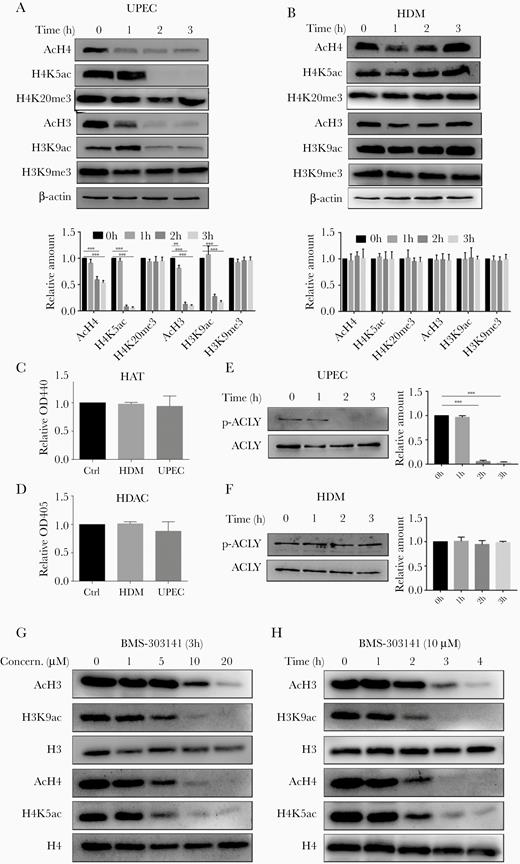
UPEC specifically reduces histone acetylation via AKT/ACLY signaling. A and B, Histone acetylation or methylation in (A) UPEC- and (B) HDM-infected 5637 cells were assessed by immunoblot analyses. C and D, 5637 cells were incubated with UPEC, HDM, or medium alone (Ctrl) for 3 hours before measurement of HAT and HDAC enzymatic activity. Data were analyzed by 1-way ANOVA with multiple comparisons test (mean ± SD, n = 3). E and F, 5637 cells were infected with (E) UPEC or (F) HDM for the indicated time periods. Immunoblots were probed with antibodies against ACLY and p-ACLY. G and H, 5637 cells were treated with the ACLY inhibitor BMS-303141 across a range of (G) doses and (H) times. Immunoblots of homogenates were probed with the indicated antibodies. A, B, E–H, Representative western blots are shown. Experiments were performed 3 times. A, B, E, F, Bar graphs represent the ratio of respective antibodies to β-actin (n = 3). Abbreviations: ACLY, ATP-citrate lyase; AKT, protein kinase B; ANOVA, analysis of variance; Ctrl, control; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDM, α-hemolysin double mutant; OD, optical density; UPEC, uropathogenic Escherichia coli.
Histone acetylation/deacetylation is executed by histone acetyltransferase (HAT) and histone deacetylase (HDAC). We therefore compared the activity of these enzymes in control, UPEC-infected, and HDM-infected cells. Neither UPEC nor HDM significantly changed the activities of either HAT or HDAC (Figure 2C and 2D).
We therefore asked whether metabolic changes induced by UPEC infection were linked to the observed patterns of histone acetylation. HlyA-containing UPEC strains are known to attenuate AKT signaling [10, 25], which is required for phosphorylation and increased enzymatic activity of ACLY [21]. Thus, we performed immunoblot analyses of UPEC- and HDM-infected cells and saw that the level of AKT/ACLY phosphorylation was decreased by UPEC but remained unchanged by HDM (Figure 2E and 2F and Supplementary Figure 1C). To establish whether an inhibition of ACLY would indeed lead to reduced histone acetylation, we treated 5637 cells with the ACLY inhibitor BMS-303141 and subsequently assessed histone acetylation levels by immunoblot. Similar to UPEC infection, BMS-303141 treatment resulted in lower H3 and H4 acetylation in a time- and dose-dependent manner (Figure 2G and 2H). Importantly, similar to UPEC infection, BMS-303141 treatment also suppressed HDM-mediated cytokine expression (Supplementary Figure 1D).
Collectively, these results indicate that HylA-dependent decreases in acetylation at H4K5 and H3K9 during UPEC infection could be mediated by decreased phosphorylation of ACLY.
Exogenous Acetate Increases Cellular Acetyl-CoA and Ameliorates UPEC-Mediated Histone Deacetylation
Because ACLY is involved in histone acetylation through regulation of acetyl-CoA abundance [18], we next measured acetyl-CoA levels during UPEC and HDM infection. We uncovered significantly lower levels of acetyl-CoA in UPEC-infected, but not HDM-infected, 5637 cells (Figure 3A). When acetyl-CoA levels were restored by supplementation with extracellular acetate (Figure 3A), we saw that levels of H3 and H4 acetylation returned to levels comparable to untreated controls (Figure 3B).
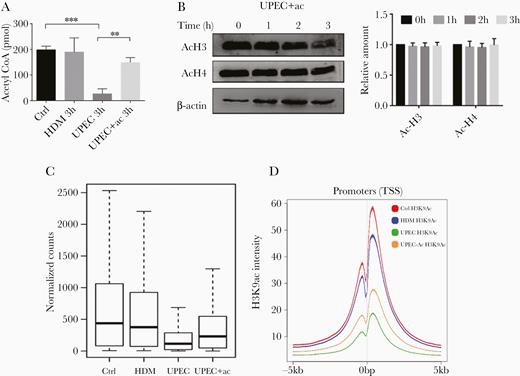
Genome-wide reduction of H3K9ac upon infection with UPEC is partially reversed by supplementation of acetate. A, 5637 cells were incubated with medium (Ctrl), HDM, UPEC, or UPEC plus 5 mM sodium acetate for 3 hours before levels of acetyl-CoA were measured. Data were analyzed by 1-way ANOVA with multiple comparisons test (mean ± SD; *P < .05, **P < .01; n = 3). B, 5637 cells were incubated with UPEC plus 5 mM sodium acetate for the indicated times before cells were lysed for immunoblot analysis using antibodies specific for acetylated H3 and H4. β-actin was used as a loading control. Representative western blots of 3 independent experiments and ratio of acH3/acH4 to β-actin are shown. C, Boxplot depicts normalized read counts across all H3K9ac peaks as detected in the indicated treatment group. D, Genome-wide H3K9ac signals in promoter regions (TSS = 0 bp) of indicated treatments. Abbreviations: ac, acetate; ANOVA, analysis of variance; Ctrl, control; HDM, α-hemolysin double mutant; TSS, transcription start site; UPEC, uropathogenic Escherichia coli.
Genome-Wide Reduction of H3K9ac Upon Infection With UPEC Is Partially Reversed by Supplementation of Acetate
Our results reported above showed that UPEC infection caused reduced histone acetylation with concomitant suppression of inflammatory genes, and that these effects were rescued by supplementation of acetate. Therefore, we next measured the effects of decreased histone acetylation on genome-wide gene expression. We performed chromatin immunoprecipitation (ChIP) with a H3K9ac antibody followed by deep sequencing in 5637 cells treated with UPEC, HDM, and UPEC supplemented with acetate. We focused on H3K9ac modification, because H3K9ac is generally associated with transcriptionally active genes [26, 27]. In addition, akin to acetylated H3 and H4, levels of acetylated H3K9ac were decreased by UPEC infection (Figure 2A). Consistent with immunoblot analyses (Figure 2A), we saw a dramatic depletion of H3K9ac peaks in UPEC-infected cells, which was partially rescued by supplementation of acetate. Similarly, we identified 21 834 merged peak regions in untreated controls; however, UPEC treatment only resulted in a significantly decreased signal across the merged peak regions, an effect that was partially rescued by addition of acetate (Figure 3C and Supplementary Figure 2). Reduced acetylation and its rescue by addition of acetate occurred globally and affected the vast majority of H3K9ac sites throughout the genome (Figure 3C). This observation holds true for different classes of cis-regulatory elements. Analysis of average binding across all human transcriptional start sites further revealed that UPEC treatment also decreased H3K9ac signals at promoter regions, which was similarly rescued by supplementation of acetate (Figure 3D).
Exogenous Acetate Increases Histone Acetylation and p65 Binding to the CXCL8 Promotor During UPEC Infection
If UPEC-induced acetyl-CoA reductions were causing decreased inflammatory gene expression due to decreased histone acetylation, it should be possible to reinstate normal histone acetylation at these gene regions by treatment with exogenous acetate. Indeed, treating UPEC-infected cells with acetate markedly increased the signal of H3K9ac at promoter transcription start sites (TSS) of inflammatory genes such as CXCL8 (Figure 4A), CXCL2 (Supplementary Figure 3A), and IL1A (Supplementary Figure 3B) compared to UPEC infection without acetate supplementation. It needs to be noted though that restoration levels were lower than those of HDM infection or noninfection control.
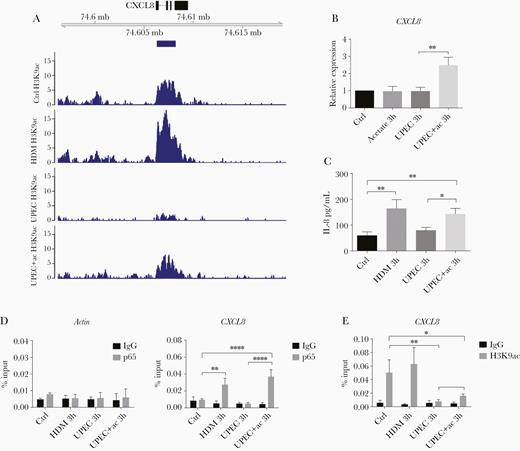
Acetate recovers UPEC-induced CXCL8 suppression by promotion of histone acetylation. A, Above: Genome browser snap-shot depicting the genomic region of the CXCL8 gene. Below: H3K9ac profiles indicating the coverage peaks obtained after spike-in normalization. Blue bar below the gene model indicates the H3K9ac peaks detected under control conditions. B, Relative CXCL8 mRNA expression and (C) IL-8 protein concentration in 5637 cells 3 hours after the indicated treatments, measured by RT-qPCR and ELISA, respectively. Data were analyzed by 1-way ANOVA with multiple comparisons test (mean ± SD of 3 independent experiments). D, The enrichment of p65 at the CXCL8 promoter was assessed by ChIP-qPCR in 5637 cells incubated with medium (Ctrl), HDM, UPEC, or UPEC plus sodium acetate for 3 hours. The actin promoter was used as negative control (mean ± SD, n = 3). E, H3K9ac ChIP-qPCR analyzing the CXCL8 promoter region (mean ± SD, n = 3). D and E, Data were analyzed by 2-way ANOVA with multiple comparisons test. *P < .05, **P < .01, ****P < .0001. Abbreviations: ac, acetate; ANOVA, analysis of variance; ChIP, chromatin immunoprecipitation; ELISA, enzyme-linked immunosorbent assay; HDM, α-hemolysin double mutant; IgG, immunoglobulin G; IL-8, interleukin-8; RT-qPCR, quantitative reverse transcription polymerase chain reaction; UPEC, uropathogenic Escherichia coli.
Because neutrophils play a particularly important role in the innate immune response against UPEC infection and CXCL8 is a key mediator of neutrophil recruitment [28], we next focused on investigating the mechanism underlying how UPEC can epigenetically suppress CXCL8 expression. In this regard, we first measured the expression levels of CXCL8 in UPEC-infected cells with and without acetate supplementation. As expected, while HDM infection increased the mRNA expression (Figure 1A and Figure 4B) and protein production (Figure 4C) of CXCL8/IL-8, UPEC infection did not (Figure 1A and Figure 4B and 4C). However, supplementation of UPEC-infected cultures with acetate significantly increased the mRNA expression of CXCL8 with concomitant production of IL-8 protein, compared to UPEC infection alone (Figure 4B and 4C). ChIP-qPCR further revealed that acetate supplementation of UPEC-infected cultures significantly increased p65 binding to the CXCL8 promoter region relative to UPEC alone (Figure 4D), and significantly elevated the level of H3K9 acetylation at the CXCL8 promoter region (Figure 4E).
Taken together, these data show that UPEC-mediated reductions in acetyl-CoA suppress H3K9 acetylation within the promoter region of inflammatory cytokine genes, leading to reduced NFκB p65 binding and decreased gene expression. These effects could be overcome in vitro by addition of exogenous acetate.
Acetate Treatment Ameliorates UPEC-Elicited Cystitis In Vivo
We then asked whether acetate supplementation could similarly ameliorate UPEC-induced effects on inflammatory gene expression in vivo. In a UPEC-elicited murine cystitis model with and without acetate supplementation (Figure 5A), we found that UPEC infection reduced the levels of p-ACLY, overall acetylation of H4, and acetylation at H3K9 in the bladder of these mice, while H3K9 methylation was unaffected (Figure 5B), recapitulating the patterns seen in 5637 cells in vitro (Figure 2A and 2E). When we pretreated animals with acetate 1 day prior to infection, by day 1 postinfection we saw clear increases in p-ACLY, overall acetylation of H4, and acetylation at H3K9 in the bladder, while H3K9 methylation was again unchanged (Figure 5B). Administration of acetate also increased the expression of Cxcl1 (the murine ortholog of human CXCL8) at day 1 postinfection (Figure 5C). As Cxcl1 plays a crucial role in recruiting neutrophils, we measured the neutrophil population in the infected urinary bladder of these mice. At day 1 postinfection, the number of neutrophils were significantly higher in the bladders of HDM-infected mice compared to the UPEC-infected group, while administration of acetate increased the number of neutrophils present in UPEC-infected bladders (Figure 5D and 5E and Supplementary Figure 4). Neutrophil numbers gradually decreased in both HDM and UPEC plus acetate treated groups, whereas they failed to be cleared even at day 7 postinfection in UPEC-infected urinary bladder, indicative of a late-resolving immune response and chronic inflammation (Figure 5E).
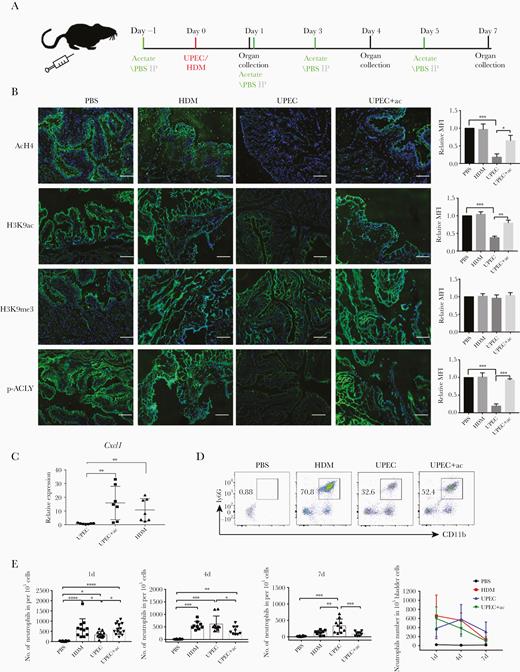
Acetate treatment alters bladder inflammation kinetics in UPEC-elicited mouse cystitis by inducing Cxcl1 expression. A, C57BL/6J mice received intraperitoneal injection of 500 mg/kg acetate or PBS 1 day before transurethral injection of PBS, or 1 × 108 UPEC or HDM, followed by intraperitoneal injections of acetate or PBS every 2 days. Urinary bladder was collected at indicated days. B, Representative images of acH4, H3K9ac, H3K9me3, and p-ACLY immunolabeling of urinary bladder sections from C57BL/6J mice 1 day postinfection. Scale bar = 100 µm. The bar graphs represent the MFI of each group (n = 3). C, RT-qPCR analysis of Cxcl1 mRNA expression from urinary bladder cell homogenates after 1 day of indicated treatments. Experiments were conducted 3 times. Data were analyzed by 1-way ANOVA with multiple comparisons test (mean ± SD, n = 7). D, Flow cytometry analysis of the abundance of neutrophils (CD11b+Ly6G+) isolated from bladder homogenates after 1 day of the indicated treatments. Plots are representative of 3 independent experiments. E, Number of neutrophils per 1 × 105 single cells from bladders after 1, 4, or 7 days of the indicated treatments (n = 6–10/group pooled from 3 independent experiments). Data were analyzed by 1-way ANOVA with multiple comparisons test. *P < .05, **P < .01, ***P < .001. Abbreviations: ac, acetate; ACLY, ATP-citrate lyase; ANOVA, analysis of variance; HDM, α-hemolysin double mutant; IP, intraperitoneal; MFI, mean fluorescence intensity; PBS, phosphate-buffered saline; RT-qPCR, quantitative reverse transcription polymerase chain reaction; UPEC, uropathogenic Escherichia coli.
Given the evidence of poor clearance of UPEC (Supplementary Figure 4D) analyzed the histology of infected bladders at different time points postinfection (Figure 6A). Consistent with the trends in neutrophil numbers, the histopathological score of the HDM-infected group was highest at day 1 with a subsequent steady decrease, whilst for the UPEC group the score gradually increased and remained persistently high at day 7 (Figure 6B). Moreover, prior and intermittent administration of acetate ameliorated the pathological changes at all time points investigated as was evidenced by lower numbers of infiltrating intraepithelial or subepithelial inflammatory cells and loss of surface epithelium (Figure 6A), all of which led to significantly reduced histopathological scores (Figure 6B).
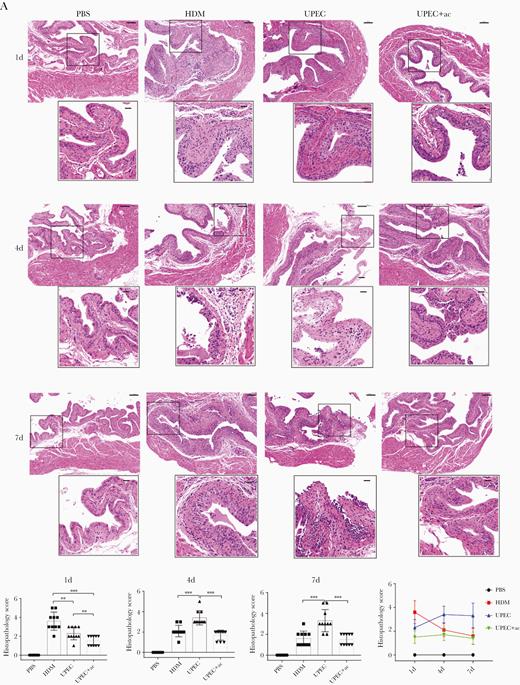
Acetate treatment ameliorates UPCE-elicited cystitis. A, Representative images of hematoxylin and eosin-stained bladder sections from mice treated with PBS, HDM, UPEC, or UPEC plus acetate at the indicated timepoints are shown. Scale bars = 100 µm (upper) and 25 µm (lower). B, Histopathology scores were assessed; n = 6–10/group of 3 independent experiments were conducted. Data points represent values for each individual mouse, bars indicate median values. Data were analyzed by 1-way ANOVA with multiple comparisons test. **P < .01, ***P < .001. Abbreviations: ac, acetate; ANOVA, analysis of variance; HDM, α-hemolysin double mutant; PBS, phosphate-buffered saline; UPCE, uropathogenic Escherichia coli.
DISCUSSION
Viruses and intracellular bacteria trigger an energy-dependent inflammatory response to eliminate pathogens, and on the other hand utilize host cell nutrients and other cellular metabolites to propagate and to avoid host innate immune responses [29, 30]. The complex molecular and biological interplay between host and pathogen can be challenging to dissect: infection with intracellular bacteria such as UPEC results in altered host metabolic processes, chromatin remodeling, and manipulation of the immune responses directed against them [12, 31–33]. While previous studies have demonstrated these alterations, they have typically focused on them in isolation [28, 32, 34] and a coherent picture linking these features of UPEC infection has been lacking. In this study, we showed that UPEC infection initially stimulates activation of the NFκB pathway but does not result in increased transcription of proinflammatory cytokines and chemokines. Instead, the virulence factor HlyA is responsible for inhibition of ACLY phosphorylation, leading to decreased acetyl-CoA levels and aberrant histone deacetylation. Moreover, a decreased level of H3K9ac in infected cells correlated with downregulated inflammatory gene expression, including IL-8, by hindrance of p65 binding to the promoter region. As these effects were driven by low levels of acetyl-CoA, supplementation with acetate ameliorated UPEC-mediated suppression of IL-8 expression by increasing H3K9ac at the promoter region to facilitate p65 binding (Figure 7). The same phenomena were present in vivo in a murine model of UPEC cystitis and were similarly rescued by treatment with acetate. These results demonstrate at a mechanistic level how UPEC manipulates cellular metabolism to suppress innate immune responses.
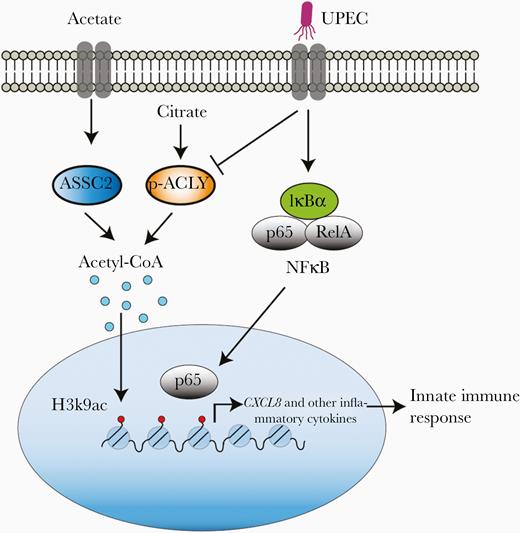
Acetate treatment rescues UPEC-mediated suppression of inflammatory gene expression, such as CXCL8, by increasing H3K9ac level. UPEC infection activates NFκB signaling to initiate innate immune response; however, simultaneously, UPEC-induced decrease in ACLY phosphorylation limits cellular acetyl-CoA production, reducing H3K9ac level and hindering p65 binding to the promoter of inflammatory genes such as CXCL8. This strategy enables UPEC to avoid the host innate immune response. Acetate treatment can rescue UPEC-mediated decreased inflammatory gene expression by increasing the acetyl-CoA pool and reinstating H3K9ac levels. Abbreviations: ASSC2, acyl-CoA synthase short-chain family member 2; ACLY, ATP-citrate lyase; IκBα, inhibitor of κBα; IL-8, interleukin-8; NFκB, nuclear factor-κB; UPEC, uropathogenic Escherichia coli.
The UPEC virulence factor HlyA is known to regulate multiple aspects of host cell biology, including the NFκB signaling pathway [8–11], and to attenuate production and secretion of proinflammatory cytokines and chemokines [12, 35–37]. Unlike other proposed mechanisms such as stabilizing Iκ-Bα [38], inhibition of the TIR domain [39], or induction of degradation of p65 [40], we found UPEC can impair NF-κB signaling by hinderance of p65 binding via the action of HlyA. Our genome-wide transcriptome analyses confirmed this (unpublished data). However, unlike other bacterial infections, where suppressed inflammatory gene expression results from inactivation of the NFκB signaling pathway [31], we found here clear evidence that UPEC initiates NFκB signaling by the degradation of its inhibitor IκBα and subsequent translocation of the NFκB p65 subunit to the nucleus.
Besides NFκB signaling, epigenetic mechanisms play an essential role in regulating the expression of inflammatory cytokines [41]. Pathogens similarly employ epigenetic mechanisms such as DNA methylation, histone modifications, and chromatin remodeling to modulate gene expression and escape the innate immune response of host cells [30, 34, 42]. As an example, Listeria monocytogenes and Streptococcus pneumonia reduce H3 and H4 acetylation with decreased expression of proinflammatory genes as a consequence [43]. In the case of UPEC, a putative link between pathogen-mediated reduction in histone acetylation and expression of host genes has only recently been investigated [10]. In general, histone acetylation is catalyzed by HAT utilizing acetyl-CoA as an essential cofactor [17], whereas histones are deacetylated by HDAC. In the present study, despite evident changes to histone acetylation, we did not find any alteration in the activities of HAT or HDAC during UPEC infection of the bladder epithelial cell line 5637. Hence, reduced levels of histone acetylation were not dependent on the enzymatic activity of HAT/HDAC.
It is known that metabolic reprogramming, such as that seen during UPEC infection, can also induce epigenetic deregulation through modulating the availability of key substrates [44]. In the case of histone acetylation, availability of the tricarboxylic acid cycle intermediate acetyl-CoA is required [45]. Recent data have shown that Toll-like receptor 4 (TLR4) activation of macrophages by lipopolysaccharide (LPS) causes redirection of metabolic fluxes to generate increased acetyl-CoA, leading to augmented histone acetylation via activation of ACLY [46]. Moreover, inhibiting this activation of ACLY significantly reduced LPS-induced inflammatory cytokine and chemokine gene expression [46]. Similarly, our results show that the acetyl-CoA level was significantly decreased after infection with an HlyA-producing UPEC strain by inhibiting the level of ACLY, which coincided with reduced histone acetylation and suppressed expression of inflammatory genes. In addition, our ChIP-seq data demonstrate that acetylation of H3K9 is strongly reduced at the TSS-promoter region of specific genes, including those encoding chemokines and cytokines. Importantly, adding acetate restored both the level of histone acetylation and expression of inflammatory genes in infected cells in vitro. Altogether, our study indicates that the UPEC pore-forming toxin hlyA inactivates AKT/ACYL signaling pathways, which resulted in low levels of acetyl-CoA and in turn reduced histone acetylation. Of note, the inactivation of AKT/signaling pathways by UPEC HylA is not specific to UPEC. Other intracellular bacteria with pore-forming toxins, including Aeromonas species and S. aureus, attenuate PI3K/AKT signaling pathways [8]. Strikingly, these bacteria also induce a dramatic dephosphorylation of H3 and deacetylation of H4, which correlates with reduced transcriptional activity of a subset of host genes, including key immunity genes [43]. Based on these previously published studies we speculate that the observed effects are not specific to UPEC but it may hold true for all pore-forming toxins, a speculation that needs to be further investigated.
Acetate is an important biological mediator in the host defense against infection and in cancer. Acetate accumulates in serum following systemic bacterial infection, which leads to increased glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzyme acetylation in CD8-expressing memory T cells via an expanded cellular acetyl-CoA pool, a process necessary for optimal memory function [47]. Acetate also promotes T-cell effector function in a glucose-restricted environment, such as in cancer cells and tissues [48]. Moreover, acetate binds the G-protein–coupled receptor 43 (GPR43) to increase neutrophil migration and aid in the resolution of inflammatory responses [49]. Similarly, our results demonstrate that acetate improves recovery from UPEC-elicited cystitis by enhancing the innate immune response and by promoting immune resolution, but whether by stimulation of GPR43 or by increasing histone acetylation still needs further investigation. Acetate treatment has predominantly an effect on H3K9 acetylation both in vitro and in vivo, which is a marker for active gene transcription. Therefore, it potentially may be of use in rescuing for rescuing histone deacetylation and regulating gene expression, especially in cancer and autoimmune diseases. Collectively, our data suggest that acetyl-CoA is a connecting link between metabolic and innate immune responses in UPEC infection and that targeting pathogen-regulated host metabolism could be utilized as an innovative therapeutic strategy for the treatment of UTI.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank Mr Matthew Jones for language editing of the manuscript, and Dr Lucy Robinson of Insight Editing London for critical review and editing of the manuscript.
Financial support. This work was supported by the Deutsche Forschungsgemeinschaft (grant number BH93/1–4); National Natural Science Foundation of China (grant numbers 81700609 to Z. Z. and 81901466 to M. W.); China Scholarship Council (fellowship to Z. Z. and M. W.); and Justus-Liebig-University of Giessen (to Z. Z., M. W., S. B., A. M., and S. B. H.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Z. Z. and M. W. contributed equally.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}