






Abstract
Protein kinase R (PKR) is a key antiviral protein involved in sensing and restricting viral infections. Here we analyzed the ability of Marburg virus (MARV) viral protein 35 (VP35) to inhibit PKR activation in human and bat cells. Similar to the related Ebola and Lloviu viruses, MARV VP35 was able to inhibit PKR activation in 293T cells. In contrast, we found that MARV VP35 did not inhibit human or bat PKR activation in human glioblastoma U-251-MG cells or a Rousettus aegyptiacus cell line. Additional experiments revealed that PACT, a known PKR regulator, was insufficient to rescue the ability of VP35 to inhibit PKR activation in these cells. Taken together, this study indicates that the ability of VP35 to inhibit PKR is cell type specific, potentially explaining discrepancies between the ability of filoviruses to potently block innate immune responses, and the high levels of interferon and interferon-stimulated genes observed in filovirus patients.
The filovirus family is subdivided into 3 genera: Ebolavirus, Marburgvirus, and Cuevavirus [1]. Most filoviruses are pathogenic to humans and are associated with high rates of morbidity and mortality [2]. Whereas the identification of animal reservoirs for ebolaviruses has remained elusive, the Egyptian rousette (Rousettus aegyptiacus) is a natural reservoir for Marburg virus (MARV) [3]. Interestingly, experimental infection of Egyptian rousettes with MARV leads to a controlled, asymptomatic infection and low levels of viral replication [4–6]. This raises the question of whether the disparity in pathogenicity observed in humans and bats is due, at least in part, to differences in the ability of MARV to inhibit innate immunity pathways in human and bat cells.
One key weapon in the cell’s antiviral arsenal is the protein kinase R (PKR) pathway. PKR is a serine threonine kinase that becomes activated upon binding to double-stranded RNA (dsRNA), leading to autophosphorylation, including the key residue threonine 451 (Thr451) [7]. Antibody detection of phosphorylated Thr451 is frequently used as a marker for PKR activation. In addition to dsRNA binding [8], overexpression of PKR induces its activation [9]. Activated PKR phosphorylates a number of cellular targets, with the alpha subunit of the eukaryotic initiation factor 2 (eIF2α) being the most well studied [10]. Phosphorylation of eIF2α leads to an inhibition of AUG codon-initiated cellular translational machinery, an extreme measure used by the cell to inhibit viral replication. The importance of this antiviral pathway in host defense is highlighted by the fact that many viruses have evolved countermeasures to block PKR activation (reviewed in [10]).
In addition to the role of viral protein 35 (VP35) in viral transcription and genome replication, all filoviruses use VP35 to inhibit interferon (IFN) induction in infected cells via multiple mechanisms (reviewed in [11, 12]). Both MARV and Ebola virus (EBOV) VP35 proteins contribute to IFN inhibition by competitively binding and sequestering dsRNA, thereby blocking the activation of dsRNA sensors [13–17]. EBOV VP35 was also shown to block PKR-activating protein (PACT)–mediated retinoic acid-inducible gene I (RIG-I) activation [18]. Despite similar functions among filovirus VP35 proteins, differences do exist. For example, while both EBOV and MARV VP35 are able to inhibit RIG-I activation, there are differences in how they bind dsRNA and the potency at which they inhibit RIG-I activation. This has been proposed as a mechanism to explain the observed disparity in the efficiency of EBOV and MARV VP35 to inhibit IFN induction [17]. While EBOV and Lloviu virus VP35 proteins have been shown to inhibit PKR activation [19–22], it has not been reported if MARV VP35 has the same ability. The scope of this study is to dissect possible MARV VP35-PKR interactions and functional consequences in human and bat cells.
Based on the functional conservation of the filoviral VP35 proteins, we anticipated that MARV VP35 would also be able to inhibit PKR activation. In this study, human PKR activation was achieved by overexpression and determined by phosphorylation. To test whether MARV VP35 is able to inhibit PKR activation, 293T cells were transfected with a plasmid encoding human PKR both with and without wild-type MARV VP35. A MARV VP35 mutant, R294A/K298A/R301A (MARV VP35-3A), that is analogous to one from EBOV VP35 that greatly reduces its ability to block PKR activation [20] (Figure 1A) was also tested. As predicted, MARV VP35 was able to inhibit the activation of human PKR in human cells as shown by reduced PKR phosphorylation levels in the presence of wild-type MARV VP35 (Figure 1B). Similar to the EBOV mutant, the MARV VP35-3A mutant was unable to inhibit human PKR (Figure 1B), implying that the mechanism of PKR inactivation by EBOV and MARV VP35 likely occurs through a similar mechanism [20]. We next sought to determine whether there is a difference in the ability of MARV VP35 to inhibit Egyptian rousette PKR. To investigate this possibility, the PKR gene sequence of Egyptian rousettes was determined (GenBank accession number MH665271), amplified using messenger RNA from the R. aegyptiacus kidney cell line RoNi/7.1 [23] and cloned into the pCAGGS expression vector. PKR cloned from RoNi/7.1 cells contained 6 amino acid differences from the published R. aegyptiacus PKR sequence, likely due to the highly diverse nature of Egyptian rousettes [24]. A hemagglutinin (HA) tag was added to the N-terminus of the bat PKR gene because there are no antibodies available targeting this protein. The PKR genes from humans and Egyptian rousettes are only moderately conserved, with 60% identity and 79% similarity. Antibodies detecting the phosphorylation of human PKR at residue threonine 451 were not able to detect the phosphorylated form of bat PKR overexpressed in 293T cells or endogenous bat PKR in RoNi/7.1 cells (data not shown). Fortuitously, however, overexpressed bat PKR was observed to migrate on a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel as 2 bands. SDS-PAGE analysis of cell lysates immunoprecipitated using an HA antibody exhibited a similar double band. Calf intestine phosphatase treatment of these samples revealed that the slower-migrating form is phosphorylated (Figure 1C). Staining of these samples with a phosphothreonine antibody showed that overexpressed bat PKR is phosphorylated on threonine residues, evidence that it is likely activated (Figure 1C). Based on these data, bat PKR migration on SDS-PAGE was used as an indication of phosphorylation and activation. To determine whether MARV VP35 inhibits bat PKR, we performed an analogous analysis as was performed with human PKR in 293T cells. Similar to what was seen with human PKR, the phosphorylation of bat PKR was inhibited by wild-type MARV VP35, as indicated by the disappearance of the upper HA-tagged bat PKR band, but not by MARV VP35-3A (Figure 1D).
![Marburg virus (MARV) viral protein 35 (VP35) inhibits the activation of human and bat protein kinase R (PKR) in 293T cells. A, Alignment of the region containing the mutations within the Ebola virus (EBOV) VP35-3A mutant [20] to MARV VP35. Residues in bold and underlined indicate amino acids that were mutated to alanine in the 3A mutants. B, 293T cells were transfected with the indicated amounts of hemagglutinin (HA)–tagged human protein kinase R (HA-hPKR) along with MARV VP35 (HA-35M) or MARV VP35-3A mutant (HA-35M-3A), and empty vector to normalize the amount of DNA transfected into all samples (done for all transfections unless no transfection is indicated). At 2 days posttransfection, cell lysates were harvested and analyzed by Western blot (WB). Western blots were visualized and quantified with the LI-COR Odyssey imaging system. All transfections using human cells were performed with TransIT-LT1 (Mirus) per the manufacturer’s instructions. Quantification of hPKR activation was calculated by dividing the signal of phosphorylated protein kinase R (pPKR-T451) by the total PKR signal (HA [hPKR]). Below, graph of the quantification of hPKR activation from 3 independent experiments, displayed as mean values with standard deviation (SD) error bars. White bar, hPKR alone; black bars, hPKR + 35M; gray bars, hPKR + 35M-3A. All statistics were determined using GraphPad Prism software. C, A plasmid encoding HA-tagged bat PKR (HA-bPKR) was transfected into 293T cells at the indicated amounts. Cell lysates were harvested and immunoprecipitated (IP) using an anti-HA antibody. A portion of each IP was then treated with calf intestinal phosphatase (+ CIP). Untreated and CIP-treated IP and cell lysates were analyzed by WB. pThr, antibody that recognizes phosphorylated threonine residues. Data are representative of 3 independent experiments. D, 293T cells were transfected as in (B) but with bPKR instead of hPKR, as indicated. Quantification of bPKR activation was calculated by dividing the signal of phosphorylated bPKR (slower migrating band of HA [bPKR]) by the total bPKR signal (both bands of HA [bPKR]) from 3 independent experiments, displayed as mean values with SD error bars. White bar, bPKR alone; black bars, bPKR + 35M; gray bars, bPKR + 35M-3A. E, 293T cells and a 293T cell line constitutively expressing HA-35M (293T-35M) were transfected with the indicated amounts of plasmids and analyzed and graphed as in (D). Data are representative of 3 independent experiments. Color code for bars is as shown in (D), with black striped bars representing constitutively expressed 35M. *P < .05, **P < .01, ***P < .001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/218/suppl_5/10.1093_infdis_jiy473/1/m_jiy47301.jpeg?Expires=1750084518&Signature=MyAY2iWI55oyp65LvcBj-5s0EcSL3FD2rqfuk0ayBx4W~CPfYHvbrmgc04qVgLdgDxFpW56BWdDWTOSmzG7q-7~zbyyAOwPzVhpUgEpl~luTO9vcMfcdrwFjQyJZ7ntFy3stBbrVKP8e83zKBrxGEfc8qvLk7zQcudBdvKn0AJnk6uZmryfclTEfYd~62eBH1hDUa3xIPMXg3RoL4FNkCevRoUBQePIstM4Z66DSR4Qu7kQqSUh0-owuvBkKxrfGu0lRCJrkNPkC5O50hSUA3M1DqJSoP9Ej4a9O90lrCmdQr2zeX2vNEDs4VqXqBqAfHMtC8ep~SCiEu7eJQSvmJQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Marburg virus (MARV) viral protein 35 (VP35) inhibits the activation of human and bat protein kinase R (PKR) in 293T cells. A, Alignment of the region containing the mutations within the Ebola virus (EBOV) VP35-3A mutant [20] to MARV VP35. Residues in bold and underlined indicate amino acids that were mutated to alanine in the 3A mutants. B, 293T cells were transfected with the indicated amounts of hemagglutinin (HA)–tagged human protein kinase R (HA-hPKR) along with MARV VP35 (HA-35M) or MARV VP35-3A mutant (HA-35M-3A), and empty vector to normalize the amount of DNA transfected into all samples (done for all transfections unless no transfection is indicated). At 2 days posttransfection, cell lysates were harvested and analyzed by Western blot (WB). Western blots were visualized and quantified with the LI-COR Odyssey imaging system. All transfections using human cells were performed with TransIT-LT1 (Mirus) per the manufacturer’s instructions. Quantification of hPKR activation was calculated by dividing the signal of phosphorylated protein kinase R (pPKR-T451) by the total PKR signal (HA [hPKR]). Below, graph of the quantification of hPKR activation from 3 independent experiments, displayed as mean values with standard deviation (SD) error bars. White bar, hPKR alone; black bars, hPKR + 35M; gray bars, hPKR + 35M-3A. All statistics were determined using GraphPad Prism software. C, A plasmid encoding HA-tagged bat PKR (HA-bPKR) was transfected into 293T cells at the indicated amounts. Cell lysates were harvested and immunoprecipitated (IP) using an anti-HA antibody. A portion of each IP was then treated with calf intestinal phosphatase (+ CIP). Untreated and CIP-treated IP and cell lysates were analyzed by WB. pThr, antibody that recognizes phosphorylated threonine residues. Data are representative of 3 independent experiments. D, 293T cells were transfected as in (B) but with bPKR instead of hPKR, as indicated. Quantification of bPKR activation was calculated by dividing the signal of phosphorylated bPKR (slower migrating band of HA [bPKR]) by the total bPKR signal (both bands of HA [bPKR]) from 3 independent experiments, displayed as mean values with SD error bars. White bar, bPKR alone; black bars, bPKR + 35M; gray bars, bPKR + 35M-3A. E, 293T cells and a 293T cell line constitutively expressing HA-35M (293T-35M) were transfected with the indicated amounts of plasmids and analyzed and graphed as in (D). Data are representative of 3 independent experiments. Color code for bars is as shown in (D), with black striped bars representing constitutively expressed 35M. *P < .05, **P < .01, ***P < .001.
We next sought to determine whether the ability of MARV VP35 to inhibit PKR extends to RoNi/7.1 cells. In contrast to what was seen in human cells, MARV VP35 was unable to inhibit either human or bat PKR activation when expressed in RoNi/7.1 cells (Figure 2A and 2B). Expression levels of both PKR and MARV VP35 were low in transfected RoNi/7.1 cells due to difficulties in transfecting these cells [25]. To confirm that the inability of MARV VP35 to inhibit PKR was not due to low VP35 expression levels in RoNi/7.1 cells, we generated 293T and RoNi/7.1 cell lines constitutively expressing HA-tagged MARV VP35 (293T-35M and RoNi-35M, respectively) using the pHAGE lentivirus system. Using these cell lines, we confirmed the results from the initial transfection studies; MARV VP35 was able to inhibit both human and bat PKR activation in 293T-35M but not RoNi-35M cells, despite high expression rates of MARV VP35 in the RoNi-35M cells (Figures 1E and 2C). We next tested influenza A virus (IAV) nonstructural protein 1 (NS1), a well-known PKR inhibitor (reviewed in [26]), as a control to confirm that PKR inactivation can be achieved in RoNi/7.1 cells. Unlike MARV VP35, IAV NS1 was able to inhibit PKR in a dose-dependent manner in transfected RoNi/7.1 cells, as indicated by the reduction of the upper bat PKR band (Figure 2D).
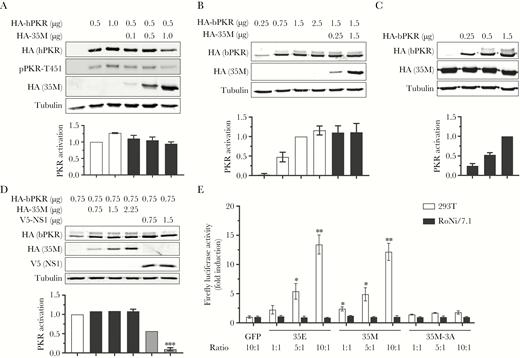
Marburg virus (MARV) viral protein 35 (VP35) does not inhibit the activation of human or bat protein kinase R (PKR) in RoNi/7.1 cells. A, RoNi/7.1 cells were transfected with the indicated amounts of hemagglutinin (HA)–tagged human PKR (HA-hPKR) and MARV VP35 (HA-35M) plasmids and analyzed and graphed as in Figure 1B. pPKR-T451, human PKR phosphorylated on residue threonine 451. Color code is as shown in Figure 1B. All transfections of bat cells were performed with lipofectamine LTX (Thermo Fisher Scientific). Data are representative of 3 independent experiments. B, RoNi/7.1 cells were transfected, analyzed, and graphed as in (A) but with HA-tagged bat PKR (HA-bPKR) instead of hPKR, as indicated. Data are representative of 3 independent experiments. Color code is as shown in Figure 1D. C, A RoNi/7.1 cell line constitutively expressing HA-35M (RoNi-35M) was transfected with the indicated amounts of plasmids and analyzed and graphed as in (B). Data are representative of 3 independent experiments. Color code is as shown in Figure 1D. D, RoNi/7.1 cells were transfected with the indicated amounts of plasmid encoding HA-bPKR along with HA-35M or V5-tagged influenza A virus nonstructural protein 1. Samples were analyzed and graphed as in (B). Data are representative of 3 independent experiments. Color code is as shown in Figure 1D except gray bars, V5-NS1. E, 293T and RoNi/7.1 cells were transfected with a vector that constitutively expresses firefly luciferase (pGL3-Control, Promega) plus either a plasmid encoding green fluorescent protein (GFP) or increasing amounts of plasmids encoding Ebola virus VP35 (35E), 35M, or 35M-3A (ratio refers to the ratio of these plasmids to pGL3-Control). At 3 days posttransfection, cell lysates were harvested and analyzed with the Luciferase Assay System (Promega) and LUMIstar Omega luminometer (BMG LabTech). Data are representative of 3 independent experiments. *P < .05, **P < .01, ***P<0.001.
The ability of EBOV VP35 to block PKR activation has been shown to lead to increased production of ectopically expressed proteins [20, 21]. Following a similar pattern, coexpression of a luciferase reporter gene with EBOV or MARV VP35 but not the MARV VP35-3A mutant in 293T cells resulted in increased reporter activity (Figure 2E, white bars). Similar transfections in RoNi/7.1 cells, however, had no effect on luciferase activity, mirroring the inability of filoviral VP35 to inhibit PKR activation in these cells (Figure 2E, black bars).
While binding to and sequestering dsRNA may be one of the mechanisms by which VP35 inhibits the activation of PKR and other dsRNA sensors, the K309A mutant of EBOV VP35 fails to bind dsRNA [27] but retains the ability to block PKR activation [20]. This indicates that VP35 is able to inhibit PKR phosphorylation via a mechanism other than by dsRNA sequestration. Direct interaction with PKR could serve as a mechanism for VP35 to inhibit the function of PKR. However, no interaction was detected despite repeated immunoprecipitation attempts (data not shown). One other mechanism by which VP35 could potentially inhibit PKR activation is through its ability to interact with PACT, a known PKR regulator that has previously been shown to bind EBOV VP35 [18, 28]. PACT and the related HIV-1 TAR RNA binding protein (TRBP) protein, which form both homodimers and heterodimers, are well-described regulators of PKR function. Whereas TRBP homodimers and PACT-TRBP heterodimers have been shown to inhibit PKR phosphorylation, PACT homodimers induce the phosphorylation of PKR [29, 30]. One possibility is that MARV VP35 could inhibit PKR activation by sequestering PACT into heterodimers. To test this hypothesis, we first performed immunoprecipitation analysis to determine if MARV VP35 is able to bind human PACT and TRBP, using EBOV VP35 as a positive control. In addition to confirming previous results that EBOV VP35 is able to bind to both human PACT and TRBP [18, 28], we observed that MARV VP35 is able to bind both of these proteins as well (Figure 3A).
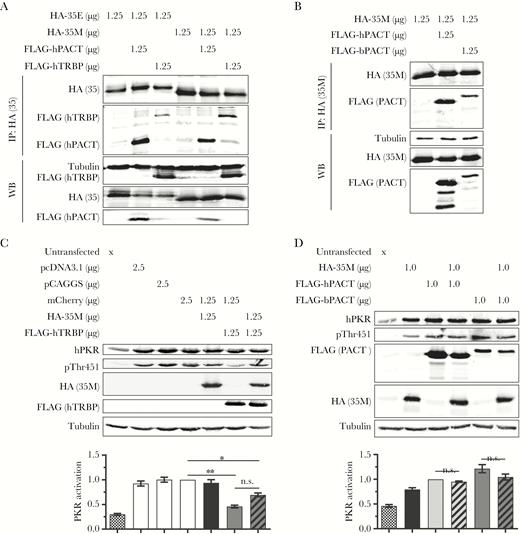
Human and bat PKR-activating protein (PACT) (hPACT and bPACT, respectively) bind Marburg virus (MARV) viral protein 35 (VP35), but are not sufficient to restore VP35-mediated protein kinase R (PKR) inhibition in U-251 MG cells. A, Coimmunoprecipitation analysis was performed in 293T cells to determine whether hemagglutinin (HA)–tagged Ebola virus VP35 (HA-35E) or MARV VP35 (HA-35M) are able to bind to FLAG–tagged hPACT (FLAG-hPACT) or human HIV-1 TAR RNA binding protein (TRBP) (FLAG-hTRBP). Cell lysates were harvested and immunoprecipitated (IP) using an anti-HA antibody and were analyzed as in Figure 1C. WB, Western blots. Data are representative of 3 independent experiments. B, Coimmunoprecipitation analysis was performed in 293T cells to determine whether HA-35M is able to bind to FLAG-hPACT or FLAG-tagged bPACT (FLAG-bPACT). Cell lysates were harvested and immunoprecipitated using an anti-HA antibody and were analyzed as in (A). Data are representative of 3 independent experiments. C and D, U-251 MG cells were not transfected (x) or transfected with the indicated plasmid amounts. At 2 days posttransfection, cell lysates were harvested and analyzed as in Figure 1B. hPKR, human PKR; pPKR-T451, human PKR phosphorylated on residue threonine 451. Data were analyzed and graphed as shown in Figure 1B and are representative of 3 independent experiments. Checkered bars, untransfected; white bars, empty vector or mCherry; black bars, 35M + mCherry (C) or 35M (D); light gray bar, hPACT; striped light gray bar, hPACT + 35M; dark gray bars, hTRBP + mCherry (C) or bPACT (D); striped dark gray bars, hTRBP + 35M (C) or bPACT + 35M (D). 35, 35E or 35M: PACT, hPACT or bPACT. Not significant (n.s.): P > .05. *P < .05, **P < .01.
If the mechanism of VP35-induced PKR inhibition was mediated by binding to PACT, a further prediction would be that the differences observed in VP35 function in the human and bat cells could be explained by the inability of MARV VP35 to bind to bat PACT. To test this hypothesis, we performed additional immunoprecipitation analyses in transfected 293T cells. A plasmid encoding a FLAG-tagged version of R. aegyptiacus PACT (GenBank accession number MH665272) was cloned in a similar manner as R. aegyptiacus PKR. The PACT genes from humans and Egyptian rousettes are fairly well conserved, with 89% identity and 90% similarity. MARV VP35 was observed to also bind to bat PACT (Figure 3B). While these data rule out PACT binding as the determinant for the differential inactivation of PKR in 293T and RoNi/7.1 cells, it remained possible that these interactions had different functional consequences. To further explore this possibility, we utilized the human glioblastoma cell line U-251 MG, which naturally expresses low levels of TRBP [31, 32] and which has been used previously to study PACT-mediated PKR activation [9, 29]. This is in contrast to 293T cells in which both PACT and TRBP are endogenously expressed [9]. Transfection of U-251 MG cells with any plasmid DNA was sufficient to activate endogenous PKR in U-251 MG cells (Figure 3C, compare lane 1 to lanes 2–4). As a positive control, human TRBP was shown to be sufficient to inhibit PKR activation in U-251 MG cells (Figure 3C, lane 6). Intriguingly, MARV VP35 was unable to inhibit PKR activation in these cells (Figure 3C, lane 5), suggesting that inhibition of PKR activation by MARV VP35 is cell type specific and not merely a species-specific difference. Coexpression of MARV VP35 and human TRBP in U-251 MG cells did not result in additional inhibition of PKR activation beyond what was seen with TRBP alone (Figure 3C, lanes 6 and 7). Although determining whether TRBP plays a role in the VP35-mediated inhibition of PKR is greatly complicated by the fact that TRBP alone is sufficient to inhibit PKR activation, these data indicate that VP35M does not enhance the ability of hTRBP to inhibit PKR. To test whether PACT plays a role in the ability of MARV VP35 to inhibit PKR activation, we co-transfected U-251 MG cells with MARV VP35 plasmid DNA along with human or bat PACT. Expression of neither human nor bat PACT facilitated MARV VP35-mediated PKR inhibition, indicating that PACT is not sufficient to facilitate this function (Figure 3D). An interesting possibility is that PACT and MARV VP35 fail to interact in U-251 MG and RoNi/7.1 cells due to cell type–specific differences in posttranslational modifications of these proteins, or due to the presence or absence of other cellular factors.
The inability of MARV VP35 to inhibit PKR in Egyptian rousette cells fits well with previous data that MARV infection leads to the induction of host innate immunity genes, mostly IFN-stimulated genes, in R. aegyptiacus cell lines [33]. It is also interesting considering a previous study reporting that PKR activation in different cells correlated with lower EBOV infection rates [34]. This raises the question of whether PKR activation plays a role in the reduced ability of filoviruses to replicate in bats [4, 35]. It is noteworthy that the observed difference in the ability of MARV VP35 to inhibit PKR activation appears not to be merely a species-specific effect, but also a cell type–specific effect. This observation might help to explain the discrepancy between potent inhibition of IFN induction by EBOV and MARV proteins, including VP35, and the strong activation of innate immune responses in the circulating immune cells of EBOV disease patients and in monkeys experimentally infected with MARV [36–38]. A species- and cell type–specific ability of filoviruses to inhibit antiviral responses could therefore be an important piece to understanding both species-specific morbidity of filovirus infection as well as the susceptibility of individual human cell types and organs to infection, which could have major implications in our attempts to control viral pathogenicity.
Notes
Acknowledgments. The authors thank M. A. Müller and C. Drosten, Charité-Universitätsmedizin Berlin, Germany, for providing RoNi/7.1 cells; W. P. Duprex, Boston University, Massachusetts, for providing U251 MG cells; and A. Garcia-Sastre, Icahn School of Medicine at Mount Sinai, New York, for providing plasmid pCAGGS-PR8 V5-NS1. The authors are grateful to S. S. Pavlovich and T. B. Kepler (both at Boston University) for providing R. aegyptiacus sequences, and J. R. Pacheco (Boston University) for excellent technical assistance.
Financial support. This work was supported by the Defense Threat Reduction Agency (grant number HDTRA1-14-1-0016, G. Palacios, principal investigator) and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (award number R21-AI126457 to E. M.).
Potential conflicts of interest. Both authors: No reported conflicts of interest. Both authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}