






Abstract
Background. Middle East respiratory syndrome coronavirus (MERS-CoV) causes severe respiratory illness in humans. Fundamental questions about circulating viruses and transmission routes remain.
Methods. We assessed routinely collected epidemiologic data for MERS-CoV cases reported in Saudi Arabia during 1 January–30 June 2015 and conducted a more detailed investigation of cases reported during February 2015. Available respiratory specimens were obtained for sequencing.
Results. During the study period, 216 MERS-CoV cases were reported. Full genome (n = 17) or spike gene sequences (n = 82) were obtained from 99 individuals. Most sequences (72 of 99 [73%]) formed a discrete, novel recombinant subclade (NRC-2015), which was detected in 6 regions and became predominant by June 2015. No clinical differences were noted between clades. Among 87 cases reported during February 2015, 13 had no recognized risks for secondary acquisition; 12 of these 13 also denied camel contact. Most viruses (8 of 9) from these 13 individuals belonged to NRC-2015.
Discussions. Our findings document the spread and eventual predominance of NRC-2015 in humans in Saudi Arabia during the first half of 2015. Our identification of cases without recognized risk factors but with similar virus sequences indicates the need for better understanding of risk factors for MERS-CoV transmission.
Middle East respiratory syndrome coronavirus (MERS-CoV) is known to cause severe respiratory illness in humans, with deaths recorded in 35%–40% of cases reported globally [1]. Since the first recognition of MERS in 2012, all cases reported to the World Health Organization have been linked to the Arabian Peninsula, with >85% of cases reported from Saudi Arabia [1]. Camels (Camelus dromedarius) have been suspected as a reservoir for MERS-CoV, based on case investigations [2], serologic studies [3], and the isolation of virus from camels [4–8]. Direct camel contact has also been identified as a risk factor for human illness [9]. Secondary human transmission has been demonstrated among close contacts of symptomatic cases, primarily following healthcare-associated exposures [10–12] and, to a lesser degree, household exposures [13]. There is no definitive evidence of sustained human-to-human transmission in the community [14].
MERS-CoV infection can exhibit a wide range of clinical manifestations, including mild or limited symptoms among those identified through contact tracing [11]. Prolonged viral shedding from the respiratory tract of those without obvious symptoms has been demonstrated [15], and transmission related to unrecognized cases has been suggested [12, 16] but not documented.
MERS-CoV sequences obtained to date suggest periodic introductions of the virus into human populations, presumably from an animal reservoir, with subsequent limited chains of transmission in households and healthcare settings. The temporal persistence of identified viral clades appears limited, consistent with an R0 of <1 [17, 18]. Intervals between the beginning and end of the circulation of a clade vary, with longer intervals suggesting the existence of undetected human cases [19]. Cases and clusters continue to be reported from countries in or near the Arabian Peninsula, presenting an ongoing threat for broader transmission [20].
To assess the epidemiologic and clinical features of the disease, we investigated all cases reported by the Saudi Arabia Ministry of Health (MoH) during January–June 2015, and we attempted genetic sequencing on all available specimens.
METHODS
This investigation was part of an emergency public health response and was determined to be nonresearch by the MoH and Centers for Disease Control and Prevention (CDC) and therefore not subject to institutional review board review.
Epidemiologic Investigation
MERS-CoV Case Definition in Saudi Arabia
At the time of this investigation, reporting in Saudi Arabia was required for all patients with clinical or radiologic evidence of MERS-CoV infection and a positive real-time reverse transcription–polymerase chain reaction (rRT-PCR) test result [21]. All rRT-PCR–positive cases identified at non-MoH facilities required confirmation at MoH laboratories.
January–June 2015
We assessed the routinely collected epidemiologic information for all MERS-CoV cases reported by the MoH during 1 January–30 June 2015, to provide a basic epidemiologic description. For this analysis, we included only individuals who met the case definition described above (ie, symptomatic cases).
February 2015
February 2015 was a period of increased reporting. To perform a more in-depth analysis, we collected additional information for all individuals with laboratory-confirmed MERS-CoV infections during February 2015. This included all cases meeting the case definition as described above, as well as those identified as having a laboratory-confirmed case but no recognized symptoms; individuals not meeting the case definition [21] were typically identified through contact tracing. We reviewed available MoH case investigation records and data reported through the MoH Health Electronic Surveillance Network. We collected demographic information, medical history, outcome information, and treatment location. We assessed the likelihood of acquisition from another person (secondary acquisition) by determining whether a patient (1) was a healthcare professional (HCP), (2) had been admitted to a healthcare facility 2–14 days before illness onset, (3) had visited any healthcare facility in the 14 days before illness onset, or (4) had direct contact with either another documented case of MERS-CoV infection or with someone with an acute respiratory illness of unknown cause in the 14 days prior to illness. When it was not possible to determine the criteria described above by using available information, we conducted telephone interviews (in Arabic) to collect additional exposure information. Proxies (a close friend or immediate family member who was familiar with the patient's activities during this period) were interviewed if the case was deceased, still hospitalized, or too ill to participate. Among cases without any of the aforementioned risk factors for secondary acquisition (hereafter referred to as sporadic cases), we asked during telephone interviews about the history of exposure to camels [9]. Interviewees were prompted to describe examples of camel exposures, including direct contact or visiting a live market, slaughterhouse, or race where camels were present. We also assessed travel history.
Statistical Analysis
Demographic and clinical characteristics were reported, and differences were assessed for significance by using χ2, Wilcoxon rank sum, and Kruskal–Wallis tests, where appropriate. Data were analyzed using SAS, version 9.3 (SAS Institute, Cary, North Carolina).
Molecular Detection and Sequencing
Molecular testing was performed on all respiratory specimens available during January–June 2015.
Specimens and Molecular Testing at the MoH
Respiratory specimens, including nasopharyngeal and oral pharyngeal swabs, both separate and combined, nasopharyngeal and tracheal aspirates, and sputa collected from suspected MERS cases were tested at MOH laboratories by upE and ORF1a rRT-PCR assays [22]. Available specimen aliquots (or RNA extracts) that tested positive for MERS-CoV by both assays were shipped on dry ice to the CDC (Atlanta, Georgia) for sequencing.
Molecular Testing at the CDC
Sample aliquots (200–300 µL, if available) were extracted on a NucliSens EasyMAG (BioMerieux), and 100 µL of total nucleic acid elutes were recovered. The specimen extract were retested by MERS-CoV N2 and/or N3 rRT-PCR assays [23], and sequencing was attempted on confirmed positive samples. Overlapping nested primer sets were used for amplification and Sanger sequencing of the MERS-CoV spike genes and selected genomes (Supplementary Table 1). Amplicon sequencing was performed in both directions, using sequencing and internal amplification primers, with the BigDye Terminator v3.1 Cycle Sequencing Kit on a 3730xl Genetic Analyzer (Thermo Fisher Scientific). Sequencher 5.3 software (Gene Codes) was used for sequence assembly and editing.
Phylogenetic Analyses
Nucleotide sequences were aligned using Clustal X, version 1.83, implemented in BioEdit, version 7.2.5. Phylogenies were estimated using neighbor-joining and maximum likelihood methods implemented in Molecular Evolutionary Genetics Analysis, version 6.0621 [24], and Bayesian inference, using MrBayes v3.2.6 [25]. The neighbor-joining method used maximum composite likelihood distance estimation and maximum likelihood used general time reversible (GTR) model of nucleotide substitution with γ-distributed rate variation and a proportion of invariant sites (GTR + G + I). MrBayes was performed under a GTR model of nucleotide substitution with 4 categories of γ-distributed rate heterogeneity and a proportion of invariant sites (GTR + 4 + I).
Genetic Recombination and Ancestral Analysis
Putative recombination events were identified using Recombination Detection Program software, version 4.70 (RDP4; available at: http://web.cbio.uct.ac.za/~darren/rdp.html), with the default settings [26]. The complete genome sequence of each of the viruses in the NRC-2015 clade was aligned with the genomes outside the clade. The multiple sequence alignment was then imported into the RDP software for detection of recombination. The software uses several algorithms, including GENECONV, BootScan, MaxChi, Chimaera, SiScan, and 3Seq, to detect putative recombination events. The potential minor and major parental sequences and the beginning and end breakpoints of the potential recombinant sequences were also defined by RDP4 software. Putative recombinant events were considered significant when a P value of ≤ .05 was observed for the same event, using ≥4 algorithms.
Time estimates to the most recent ancestor were calculated using the Bayesian Markov Chain Monte Carlo (MCMC) method implemented in BEAST v1.8.2 [27]. The coding regions (ORF1ab, S, ORF3, ORF4a, ORF4b, ORF5, E, M, and N) in the genomes grouping within NRC-2015 were concatenated, and the HKY+ Γ4 substitution model was used with independent rates for each of the positions in the codon. A lognormal relaxed molecular clock (uncorrelated) was used with Gaussian Markov random field Bayesian skyride coalescent. Bayesian MCMC analysis was run for 25 million steps. Parameters for tMRCA, rate, and trees were sampled every 5000 steps, with the first 10% removed as burn-in. Time estimate values thus obtained were also compared with strict and exponential relaxed clock models.
RESULTS
During 1 January–30 June 2015, 216 MERS-CoV cases from 10 of the 13 regions of Saudi Arabia were reported by the MoH; MERS-CoV–positive individuals with no recognized symptoms, and who therefore did not meet the case definition, were not included. The longest period between case reports was 11 days. Among these 216 cases, 214 were hospitalized, and 102 (47%) died. Most patients were male (161 [75%]) and of Saudi nationality (147 [68%]). Median age was 56 years (range, 20–93 years).
Molecular Analysis of MERS-CoV Strains
Of the 216 symptomatic cases reported during the study period, 124 had respiratory specimens available for further testing at the CDC; 1 specimen was also available from an individual with no recognized symptoms who did not meet the case definition. Of the 125 available respiratory specimens collected during 6 January–3 June 2015, spike gene sequences were obtained from 99 (Supplementary Table 2). Phylogenetic analysis of the 99 spike sequences with 251 previously published sequences (Figure 1) revealed a novel subclade (henceforth referred to as NRC-2015) within clade B [18]; this subclade contained most (72 of 99 [72.7%]) of the 2015 sequences and included sequences recently reported from travel-associated MERS cases in the Republic of Korea (accession number KT029139), China (KT006149), and Thailand (KT225476). The neighbor joining tree bootstrap value supporting NRC-2015 was low (<70%), which may reflect the recombinant character of the clade sequences (see below) or the lower number of informative sites in the spike gene. However, similar tree topologies reproducing NRC-2015 were obtained with the spike gene sequences by both maximum likelihood and Bayesian analyses, supporting this finding (data not shown). To more fully characterize NRC-2015, 17 MERS-CoV–positive samples with sufficient available volume were selected for genome sequencing, including 12 NRC-2015 sequences from different time points and 5 from outside of the subclade.
![Phylogeny of Middle East respiratory syndrome coronavirus (MERS-CoV) spike sequences. Phylogenetic analysis of the MERS-CoV spike gene coding region (4062 nucleotides) obtained from 99 cases in this study and 251 previously published sequences available in GenBank. Major clades A and B are indicated by vertical bars. Figure 1 continued. The novel subclade (NRC-2015) is repositioned for clarity. Spike gene sequences obtained in this study are marked with solid circles; red circles indicate samples with available genome sequences (Supplementary Figure 1). The tree was constructed by the neighbor-joining method, and bootstrap resampling values (1000 replicates) ≥70% are indicated above the respective nodes. Numbers in brackets following some strain identifiers are the number of identical sequences with the same location and sample collection time. The scale bar shows the genetic distance as nucleotide substitutions per site. To compare persistence and geographic distribution of NRC-2015 with other viruses identified during 2012–2015, we assigned virus sequences to different subclades (depicted in color), based on previously described clades [19, 28–30].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/214/5/10.1093_infdis_jiw236/2/m_jiw23601.jpeg?Expires=1749907238&Signature=aiTMviBkzo-2DaMyw4La~vPeh9TEheKf5uSxe~ugv0kQHStkSy1cDk0j7Wc-RRZ14PHVxdfKIF5kcr88VlxxIyV7rQ2Lyg18Ie5obnRYRIMk2gifvQcOUoMne91BUAWnm-P~2cKB5ju16kM6m0NkKMKip4V4SVqKVmN2hfnGt6qO7hmS1qsyX6LI1fMaPVf~GpBuKwwVEiePCiQrZFCO620nUqnugBcJ8hLZ9cDxZZgDlDLQbCpzsEfMJ4bY9gRGcDWbW1petXmCizClh6bAn2VUd2x-hYwGsXkuFQ-xcG1EYH06Y87w9qspT5iLWwVcKn2OY4ZZbyW1A~CejMfgnQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Phylogeny of Middle East respiratory syndrome coronavirus (MERS-CoV) spike sequences. Phylogenetic analysis of the MERS-CoV spike gene coding region (4062 nucleotides) obtained from 99 cases in this study and 251 previously published sequences available in GenBank. Major clades A and B are indicated by vertical bars. Figure 1 continued. The novel subclade (NRC-2015) is repositioned for clarity. Spike gene sequences obtained in this study are marked with solid circles; red circles indicate samples with available genome sequences (Supplementary Figure 1). The tree was constructed by the neighbor-joining method, and bootstrap resampling values (1000 replicates) ≥70% are indicated above the respective nodes. Numbers in brackets following some strain identifiers are the number of identical sequences with the same location and sample collection time. The scale bar shows the genetic distance as nucleotide substitutions per site. To compare persistence and geographic distribution of NRC-2015 with other viruses identified during 2012–2015, we assigned virus sequences to different subclades (depicted in color), based on previously described clades [19, 28–30].
Phylogenetic analysis of these full genomes with genome sequences available on GenBank reproduced NRC-2015 with high support values (Supplementary Figure 1). Mutational analysis of the 12 new NRC-2015 genomes with previously published NRC-2015 genomes obtained from human samples collected in Saudi Arabia (n = 2), Korea/China (n = 2), and Thailand (n = 1) revealed 9 unique nucleotide substitutions common to all NRC-2015 members, of which 3 were nonsynonymous, with predicted amino acid changes in the ORF1a (n = 2) and matrix (n = 1) proteins (Supplementary Table 3). Recombination analysis on the newly available genome sequences from NRC-2015 identified 2 possible recombination events involving sequences from outside the clade as potential minor and major parental strains. The first event had a predicted breakpoint at nucleotide position 17 475 (99% confidence interval [CI], 13 502–19 074), located in ORF1ab, and the second event had a predicted breakpoint at 23 976 (99% CI, 23 571–24 862), located in the spike gene. Recombination analysis was performed using RDP software, and events detected with a P value of ≤ .05 were considered evidence of true recombination (Supplementary Table 4).
To date the emergence of NRC-2015, MCMC analysis was performed on the concatenated coding regions of the genomes grouping within NRC-2015, using BEAST. The most recent common ancestor of the virus was approximately 0.85 years (95% CI, .45–1.34) before sample KT225476.2 (17 June 2015), or August 2014 (Supplementary Table 5).
Epidemiologic Investigation
Among the 216 cases reported during 1 January–30 June 2015, NRC-2015 was first detected in a case with onset in mid-January 2015 (Figure 2A). During the study period, NRC-2015 viruses were detected in 6 regions of Saudi Arabia (Figure 3), and the proportion of patients identified with NRC-2015 increased steadily over time (Figure 2B).
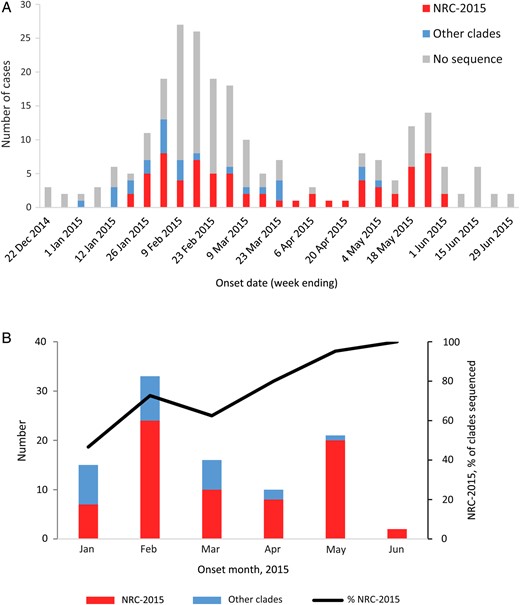
Middle East respiratory syndrome coronavirus (MERS-CoV) cases reported by the Saudi Arabia Ministry of Health (MoH) during 1 January–30 June 2015. A, MERS-CoV cases, by clade and week of illness onset. B, Sequenced viruses, by clade and month of illness onset. The 216 cases reported by the MoH during 1 January–30 June 2015 are included. These graphs do not include the single sequence from the individual with no recognized symptoms who did not meet the case definition. NRC-2015 was defined using the spike gene phylogenies (Figure 1).

Map of Middle East respiratory syndrome coronavirus (MERS-CoV) cases, by subclade and region of Saudi Arabia. The 216 cases reported by the Saudi Arabia Ministry of Health during 1 January–30 June 2015, are included. This map does not include the single sequence from the individual with no recognized symptoms who did not meet the case definition. NRC-2015 was defined using the spike gene phylogenies (Figure 1).
NRC-2015 was next compared to past and present subclades within clade B, using sequences available in GenBank (Figure 4). NRC-2015 was more widely distributed geographically than any other identified members of clade B. The duration of circulation of recognized subclades ranged from 16 to 665 days. At the conclusion of our investigation period, NRC-2015 had been circulating for 135 days, which was longer than 7 of 9 other identified subclades. In our analysis, the longest circulating subclade reported was Riyadh_KKUH-1_2014, which was first detected in July 2013 and was still circulating as of May 2015. During our investigation period, 10 of 99 sequenced viruses belonged to Riyadh_KKUH-1_2014. No viruses belonging to clade A were detected.
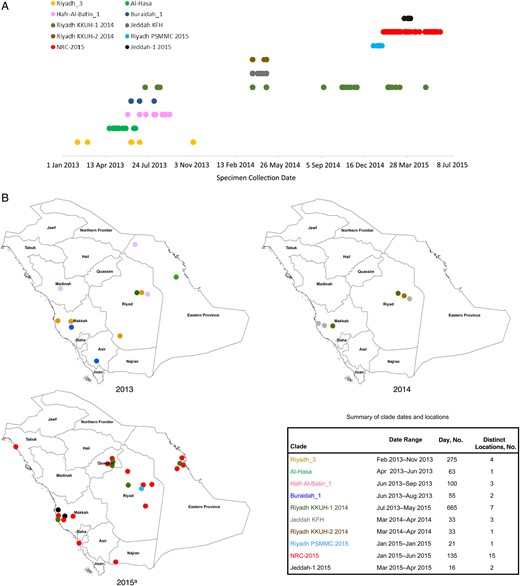
Geotemporal distribution of Middle East respiratory syndrome coronavirus (MERS-CoV) clades in Saudi Arabia during 1 January 2013–30 June 2015. For human-derived sequences in each subclade within the broader clade B (as defined using the spike gene phylogenetic analysis in Figure 1), we used GenBank to determine length of persistence, using the earliest and most recent date of detection (A), and we determined geographic distribution, based on city of detection within Saudi Arabia (B). a2015 depicts clades detected through 30 June 2015.
A comparison of patients infected with NRC-2015 versus other circulating viruses revealed no significant differences in age, sex, rate of mortality, time between onset of symptoms and death, or length of hospital stay among fatal cases (Supplementary Table 6). There was also no difference in mean cycle threshold values, a proxy for virus load, with respiratory specimens containing NRC-2015 versus other clades, although these were not adjusted for timing of specimen collection (Supplementary Table 6).
For our more detailed analysis of cases reported during 1–28 February 2015, we identified 87 MERS-CoV–positive patients (Table 1). Of these, 77 patients (89%) satisfied the case definition for routine reporting and required hospitalization; the remaining 10 individuals (11%) had no recognized symptoms (and did not satisfy the case definition) but are included in this analysis. The 87 patients were reported from 35 different healthcare facilities across 7 regions in Saudi Arabia; 17 of these facilities reported ≥2 cases within the same 14-day period. Of these 87 patients, sequences could be obtained from 34, of which 24 (71%) were associated with NRC-2015. No clinical differences were apparent when comparing NRC-2015 to other circulating viruses (Table 1).
Epidemiologic and Clinical Characteristics of Patients Identified With Laboratory-Confirmed Middle East Respiratory Syndrome Coronavirus Infection in February 2015 in Saudi Arabia
| Characteristic | Sequenced | No Sequence, No. (%) (n = 53) | Total, No. (%) (n = 87) | |
|---|---|---|---|---|
| NRC-2015 Clade, No. (%) (n = 24) | Other Clades, No. (%) (n = 10) | |||
| Sex | ||||
| Male | 17 (71) | 6 (60) | 39 (74) | 62 (71) |
| Female | 7 (29) | 4 (40) | 14 (26) | 25 (29) |
| Age group, y | ||||
| 18–34 | 2 (8) | 1 (10) | 13 (25) | 16 (18) |
| 35–64 | 14 (58) | 9 (90) | 24 (45) | 47 (54) |
| ≥65 | 8 (33) | 0 (0) | 15 (28) | 23 (26) |
| Unknown | 0 (0) | 0 (0) | 1 (2) | 1 (1) |
| Nationality | ||||
| Saudi | 14 (58) | 6 (60) | 36 (68) | 56 (64) |
| Non-Saudi | 10 (42) | 4 (40) | 17 (32) | 31 (36) |
| Underlying illness | ||||
| Any | 14 (58) | 7 (70) | 30 (57) | 51 (59) |
| Diabetes | 10 (42) | 4 (40) | 16 (30) | 30 (34) |
| Heart disease | 1 (4) | 1 (10) | 3 (6) | 5 (6) |
| Hypertension | 10 (42) | 1 (10) | 16 (30) | 27 (31) |
| Exposure classification | ||||
| Household | 1 (4) | 2 (20) | 10 (19) | 13 (15) |
| Healthcare provider | 4 (17) | 1 (10) | 9 (17) | 14 (16) |
| Inpatient | 3 (13) | 3 (30) | 15 (28) | 21 (24) |
| Hospital visitor | 6 (25) | 2 (20) | 8 (15) | 16 (18) |
| Sporadica | 8 (33) | 1 (10) | 4 (8) | 13 (15) |
| Undeterminedb | 2 (8) | 1 (10) | 7 (13) | 10 (11) |
| Hospitalized | ||||
| No | 0 (0) | 0 (0) | 10 (19) | 10 (11) |
| Yes, survived | 10 (42) | 4 (40) | 22 (42) | 36 (41) |
| Yes, died | 14 (58) | 6 (60) | 21 (40) | 41 (47) |
| Characteristic | Sequenced | No Sequence, No. (%) (n = 53) | Total, No. (%) (n = 87) | |
|---|---|---|---|---|
| NRC-2015 Clade, No. (%) (n = 24) | Other Clades, No. (%) (n = 10) | |||
| Sex | ||||
| Male | 17 (71) | 6 (60) | 39 (74) | 62 (71) |
| Female | 7 (29) | 4 (40) | 14 (26) | 25 (29) |
| Age group, y | ||||
| 18–34 | 2 (8) | 1 (10) | 13 (25) | 16 (18) |
| 35–64 | 14 (58) | 9 (90) | 24 (45) | 47 (54) |
| ≥65 | 8 (33) | 0 (0) | 15 (28) | 23 (26) |
| Unknown | 0 (0) | 0 (0) | 1 (2) | 1 (1) |
| Nationality | ||||
| Saudi | 14 (58) | 6 (60) | 36 (68) | 56 (64) |
| Non-Saudi | 10 (42) | 4 (40) | 17 (32) | 31 (36) |
| Underlying illness | ||||
| Any | 14 (58) | 7 (70) | 30 (57) | 51 (59) |
| Diabetes | 10 (42) | 4 (40) | 16 (30) | 30 (34) |
| Heart disease | 1 (4) | 1 (10) | 3 (6) | 5 (6) |
| Hypertension | 10 (42) | 1 (10) | 16 (30) | 27 (31) |
| Exposure classification | ||||
| Household | 1 (4) | 2 (20) | 10 (19) | 13 (15) |
| Healthcare provider | 4 (17) | 1 (10) | 9 (17) | 14 (16) |
| Inpatient | 3 (13) | 3 (30) | 15 (28) | 21 (24) |
| Hospital visitor | 6 (25) | 2 (20) | 8 (15) | 16 (18) |
| Sporadica | 8 (33) | 1 (10) | 4 (8) | 13 (15) |
| Undeterminedb | 2 (8) | 1 (10) | 7 (13) | 10 (11) |
| Hospitalized | ||||
| No | 0 (0) | 0 (0) | 10 (19) | 10 (11) |
| Yes, survived | 10 (42) | 4 (40) | 22 (42) | 36 (41) |
| Yes, died | 14 (58) | 6 (60) | 21 (40) | 41 (47) |
a Patients who were not a contact of a case or a person with acute respiratory illness and did not report exposure to a healthcare facility in the 2 weeks prior to illness onset.
b Patients for whom sufficient data were not available.
Epidemiologic and Clinical Characteristics of Patients Identified With Laboratory-Confirmed Middle East Respiratory Syndrome Coronavirus Infection in February 2015 in Saudi Arabia
| Characteristic | Sequenced | No Sequence, No. (%) (n = 53) | Total, No. (%) (n = 87) | |
|---|---|---|---|---|
| NRC-2015 Clade, No. (%) (n = 24) | Other Clades, No. (%) (n = 10) | |||
| Sex | ||||
| Male | 17 (71) | 6 (60) | 39 (74) | 62 (71) |
| Female | 7 (29) | 4 (40) | 14 (26) | 25 (29) |
| Age group, y | ||||
| 18–34 | 2 (8) | 1 (10) | 13 (25) | 16 (18) |
| 35–64 | 14 (58) | 9 (90) | 24 (45) | 47 (54) |
| ≥65 | 8 (33) | 0 (0) | 15 (28) | 23 (26) |
| Unknown | 0 (0) | 0 (0) | 1 (2) | 1 (1) |
| Nationality | ||||
| Saudi | 14 (58) | 6 (60) | 36 (68) | 56 (64) |
| Non-Saudi | 10 (42) | 4 (40) | 17 (32) | 31 (36) |
| Underlying illness | ||||
| Any | 14 (58) | 7 (70) | 30 (57) | 51 (59) |
| Diabetes | 10 (42) | 4 (40) | 16 (30) | 30 (34) |
| Heart disease | 1 (4) | 1 (10) | 3 (6) | 5 (6) |
| Hypertension | 10 (42) | 1 (10) | 16 (30) | 27 (31) |
| Exposure classification | ||||
| Household | 1 (4) | 2 (20) | 10 (19) | 13 (15) |
| Healthcare provider | 4 (17) | 1 (10) | 9 (17) | 14 (16) |
| Inpatient | 3 (13) | 3 (30) | 15 (28) | 21 (24) |
| Hospital visitor | 6 (25) | 2 (20) | 8 (15) | 16 (18) |
| Sporadica | 8 (33) | 1 (10) | 4 (8) | 13 (15) |
| Undeterminedb | 2 (8) | 1 (10) | 7 (13) | 10 (11) |
| Hospitalized | ||||
| No | 0 (0) | 0 (0) | 10 (19) | 10 (11) |
| Yes, survived | 10 (42) | 4 (40) | 22 (42) | 36 (41) |
| Yes, died | 14 (58) | 6 (60) | 21 (40) | 41 (47) |
| Characteristic | Sequenced | No Sequence, No. (%) (n = 53) | Total, No. (%) (n = 87) | |
|---|---|---|---|---|
| NRC-2015 Clade, No. (%) (n = 24) | Other Clades, No. (%) (n = 10) | |||
| Sex | ||||
| Male | 17 (71) | 6 (60) | 39 (74) | 62 (71) |
| Female | 7 (29) | 4 (40) | 14 (26) | 25 (29) |
| Age group, y | ||||
| 18–34 | 2 (8) | 1 (10) | 13 (25) | 16 (18) |
| 35–64 | 14 (58) | 9 (90) | 24 (45) | 47 (54) |
| ≥65 | 8 (33) | 0 (0) | 15 (28) | 23 (26) |
| Unknown | 0 (0) | 0 (0) | 1 (2) | 1 (1) |
| Nationality | ||||
| Saudi | 14 (58) | 6 (60) | 36 (68) | 56 (64) |
| Non-Saudi | 10 (42) | 4 (40) | 17 (32) | 31 (36) |
| Underlying illness | ||||
| Any | 14 (58) | 7 (70) | 30 (57) | 51 (59) |
| Diabetes | 10 (42) | 4 (40) | 16 (30) | 30 (34) |
| Heart disease | 1 (4) | 1 (10) | 3 (6) | 5 (6) |
| Hypertension | 10 (42) | 1 (10) | 16 (30) | 27 (31) |
| Exposure classification | ||||
| Household | 1 (4) | 2 (20) | 10 (19) | 13 (15) |
| Healthcare provider | 4 (17) | 1 (10) | 9 (17) | 14 (16) |
| Inpatient | 3 (13) | 3 (30) | 15 (28) | 21 (24) |
| Hospital visitor | 6 (25) | 2 (20) | 8 (15) | 16 (18) |
| Sporadica | 8 (33) | 1 (10) | 4 (8) | 13 (15) |
| Undeterminedb | 2 (8) | 1 (10) | 7 (13) | 10 (11) |
| Hospitalized | ||||
| No | 0 (0) | 0 (0) | 10 (19) | 10 (11) |
| Yes, survived | 10 (42) | 4 (40) | 22 (42) | 36 (41) |
| Yes, died | 14 (58) | 6 (60) | 21 (40) | 41 (47) |
a Patients who were not a contact of a case or a person with acute respiratory illness and did not report exposure to a healthcare facility in the 2 weeks prior to illness onset.
b Patients for whom sufficient data were not available.
The 87 patients with laboratory-confirmed disease reported during February were also classified according to their reported exposures during the 2 weeks before illness onset. Record review and interviews were conducted during 11–25 March 2015. Among the 87 cases, 51 were classifiable using information obtained by the initial case investigation. Interviews were attempted for the remaining 36 patients. Of these, 28 (78%) were interviewed; 1 individual refused to participate, and 7 patients were not available. Proxy interviews were conducted for 22 of 28 interviews, including for 18 patients who were deceased and for 4 of 10 patients who survived.
Among the 87 patients, 13 (15%) were determined to have had household contact with a confirmed MERS-CoV case, 14 (16%) were HCPs, 21 (24%) were inpatients in a healthcare facility, 16 (18%) were hospital visitors, and 10 (11%) were unable to be classified owing to a lack of available information (Table 1). Notably, 13 patients (15%) denied exposure to a healthcare facility or to a person with acute respiratory illness in the 2 weeks before illness onset and were classified as sporadic cases (Tables 1 and 2); among these, 1 individual reported visiting a camel farm in the 2 weeks before illness onset. Among the 13 sporadic cases, 2 were available for interview, and 11 were interviewed by proxy. Among the 11 interviewed by proxy, 9 were deceased and 2 were too ill to participate in the interview. Sequences were obtained for 9 sporadic cases, and 8 (89%) were NRC-2015, including the individual who had visited the camel farm.
Sporadic Middle East Respiratory Syndrome (MERS) Coronavirus Cases Reported in Saudi Arabia During February, 2015
| Patient | Interview | Clade (GenBanka) | Region | Age, y | Sex | Saudi Nationality | Survived | Underlying Illness | Ever Smoker | Occupation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proxy | NRC-2015G (KT806053) | Riyadh | 56 | F | Yes | No | Yes | Unknown | Unemployed |
| 2 | Self | NRC-2015G (KT806052) | Riyadh | 26 | M | Yes | Yes | Yes | Yes | Security |
| 3 | Proxy | NRC-2015G (KT806048) | Eastern | 49 | M | Yes | No | Yes | Yes | Sales |
| 4 | Proxy | NRC-2015G (KT806044) | Jeddah | 73 | F | No | No | Yes | No | Unemployed |
| 5 | Proxy | NS | Madinah | 70 | M | Yes | No | Yes | No | Retired |
| 6 | Proxy | NS | Riyadh | 79 | M | Yes | No | Yes | Yes | Unemployed |
| 7 | Proxy | NRC-2015S (KT806050) | Riyadh | 49 | F | Yes | Yes | Yes | No | Unemployed |
| 8 | Proxy | NRC-2015G (KT806054) | Najran | 47 | M | No | No | Yes | No | Blacksmith |
| 9 | Proxy | NS | Eastern | 41 | M | Yes | Yes | No | No | Unemployed |
| 10 | Proxy | OtherS (KT805966) | Riyadh | 51 | M | Yes | No | Yes | Yes | Retired |
| 11 | Proxy | NRC-2015G (KT806045) | Jeddah | 65 | F | No | No | Yes | No | Unemployed |
| 12 | Proxy | NS | Qasim | 45 | M | No | No | Yes | No | Joiner/blacksmith |
| 13b | Self | NRC-2015S (KT806002) | Qasim | 42 | M | No | Yes | No | Yes | Driver |
| Patient | Interview | Clade (GenBanka) | Region | Age, y | Sex | Saudi Nationality | Survived | Underlying Illness | Ever Smoker | Occupation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proxy | NRC-2015G (KT806053) | Riyadh | 56 | F | Yes | No | Yes | Unknown | Unemployed |
| 2 | Self | NRC-2015G (KT806052) | Riyadh | 26 | M | Yes | Yes | Yes | Yes | Security |
| 3 | Proxy | NRC-2015G (KT806048) | Eastern | 49 | M | Yes | No | Yes | Yes | Sales |
| 4 | Proxy | NRC-2015G (KT806044) | Jeddah | 73 | F | No | No | Yes | No | Unemployed |
| 5 | Proxy | NS | Madinah | 70 | M | Yes | No | Yes | No | Retired |
| 6 | Proxy | NS | Riyadh | 79 | M | Yes | No | Yes | Yes | Unemployed |
| 7 | Proxy | NRC-2015S (KT806050) | Riyadh | 49 | F | Yes | Yes | Yes | No | Unemployed |
| 8 | Proxy | NRC-2015G (KT806054) | Najran | 47 | M | No | No | Yes | No | Blacksmith |
| 9 | Proxy | NS | Eastern | 41 | M | Yes | Yes | No | No | Unemployed |
| 10 | Proxy | OtherS (KT805966) | Riyadh | 51 | M | Yes | No | Yes | Yes | Retired |
| 11 | Proxy | NRC-2015G (KT806045) | Jeddah | 65 | F | No | No | Yes | No | Unemployed |
| 12 | Proxy | NS | Qasim | 45 | M | No | No | Yes | No | Joiner/blacksmith |
| 13b | Self | NRC-2015S (KT806002) | Qasim | 42 | M | No | Yes | No | Yes | Driver |
In the 2 weeks before becoming ill, none had known contact with a patient with MERS, a person with severe respiratory illness, and anyone mildly ill, and none reported working in or visiting a healthcare facility.
Abbreviations: G, full genome sequence obtained; S, spike region sequence obtained; NS, not sequenced.
a GenBank accession number.
b Visited a camel farm.
Sporadic Middle East Respiratory Syndrome (MERS) Coronavirus Cases Reported in Saudi Arabia During February, 2015
| Patient | Interview | Clade (GenBanka) | Region | Age, y | Sex | Saudi Nationality | Survived | Underlying Illness | Ever Smoker | Occupation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proxy | NRC-2015G (KT806053) | Riyadh | 56 | F | Yes | No | Yes | Unknown | Unemployed |
| 2 | Self | NRC-2015G (KT806052) | Riyadh | 26 | M | Yes | Yes | Yes | Yes | Security |
| 3 | Proxy | NRC-2015G (KT806048) | Eastern | 49 | M | Yes | No | Yes | Yes | Sales |
| 4 | Proxy | NRC-2015G (KT806044) | Jeddah | 73 | F | No | No | Yes | No | Unemployed |
| 5 | Proxy | NS | Madinah | 70 | M | Yes | No | Yes | No | Retired |
| 6 | Proxy | NS | Riyadh | 79 | M | Yes | No | Yes | Yes | Unemployed |
| 7 | Proxy | NRC-2015S (KT806050) | Riyadh | 49 | F | Yes | Yes | Yes | No | Unemployed |
| 8 | Proxy | NRC-2015G (KT806054) | Najran | 47 | M | No | No | Yes | No | Blacksmith |
| 9 | Proxy | NS | Eastern | 41 | M | Yes | Yes | No | No | Unemployed |
| 10 | Proxy | OtherS (KT805966) | Riyadh | 51 | M | Yes | No | Yes | Yes | Retired |
| 11 | Proxy | NRC-2015G (KT806045) | Jeddah | 65 | F | No | No | Yes | No | Unemployed |
| 12 | Proxy | NS | Qasim | 45 | M | No | No | Yes | No | Joiner/blacksmith |
| 13b | Self | NRC-2015S (KT806002) | Qasim | 42 | M | No | Yes | No | Yes | Driver |
| Patient | Interview | Clade (GenBanka) | Region | Age, y | Sex | Saudi Nationality | Survived | Underlying Illness | Ever Smoker | Occupation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proxy | NRC-2015G (KT806053) | Riyadh | 56 | F | Yes | No | Yes | Unknown | Unemployed |
| 2 | Self | NRC-2015G (KT806052) | Riyadh | 26 | M | Yes | Yes | Yes | Yes | Security |
| 3 | Proxy | NRC-2015G (KT806048) | Eastern | 49 | M | Yes | No | Yes | Yes | Sales |
| 4 | Proxy | NRC-2015G (KT806044) | Jeddah | 73 | F | No | No | Yes | No | Unemployed |
| 5 | Proxy | NS | Madinah | 70 | M | Yes | No | Yes | No | Retired |
| 6 | Proxy | NS | Riyadh | 79 | M | Yes | No | Yes | Yes | Unemployed |
| 7 | Proxy | NRC-2015S (KT806050) | Riyadh | 49 | F | Yes | Yes | Yes | No | Unemployed |
| 8 | Proxy | NRC-2015G (KT806054) | Najran | 47 | M | No | No | Yes | No | Blacksmith |
| 9 | Proxy | NS | Eastern | 41 | M | Yes | Yes | No | No | Unemployed |
| 10 | Proxy | OtherS (KT805966) | Riyadh | 51 | M | Yes | No | Yes | Yes | Retired |
| 11 | Proxy | NRC-2015G (KT806045) | Jeddah | 65 | F | No | No | Yes | No | Unemployed |
| 12 | Proxy | NS | Qasim | 45 | M | No | No | Yes | No | Joiner/blacksmith |
| 13b | Self | NRC-2015S (KT806002) | Qasim | 42 | M | No | Yes | No | Yes | Driver |
In the 2 weeks before becoming ill, none had known contact with a patient with MERS, a person with severe respiratory illness, and anyone mildly ill, and none reported working in or visiting a healthcare facility.
Abbreviations: G, full genome sequence obtained; S, spike region sequence obtained; NS, not sequenced.
a GenBank accession number.
b Visited a camel farm.
DISCUSSION
Our study provides the first nationwide epidemiologic and phylogenetic description of MERS-CoV in humans in Saudi Arabia, allowing for a better understanding of transmission during a period of continuous, frequent reporting of new cases. Our investigation demonstrates the emergence, persistent circulation, and eventual predominance of a recently identified recombinant MERS-CoV clade in humans that includes viruses detected in the Republic of Korea [31, 32], Thailand (accession number KT225476), and China in 2015 [31, 33]. Previous documentation of the duration of circulation in humans of 4 different MERS-CoV clades in Saudi Arabia during 2012–2013 noted an average detection time of 98 days [19]. In contrast, we demonstrate that NRC-2015 has persisted longer than most previously documented clades. NRC-2015 was found to eventually predominate over the 6-month study period and attain a wide geographic distribution in a comparatively short period. While this apparent emergence and clade displacement is suggestive of greater epidemiologic fitness [34], we observed no clinical differences between NRC-2015 and other clades; the implications for virus replication and transmission need further study. During preparation of this manuscript, sequences obtained from camels in Oman in May 2015 [35] and Saudi Arabia during July 2014–April 2015 [36] were reported that showed similar recombination features and phylogenetic association with NRC-2015. In camels, NRC-2015 (referred to as lineage 5 [36]) was first detected in July 2014 and became predominant in Saudi Arabia during a period that overlaps with our study, corroborating our findings of an increased prevalence in humans relative to other clades.
Recombination has been documented among CoVs [37] and has been linked to the emergence of more-pathogenic strains of some animal CoVs [38–40]. Evidence of intraspecies recombination has also been found with the human CoVs HKU1 [41], NL63 [42], OC43 [43], and, more recently, MERS-CoV [44]. Genome analysis of human MERS-CoV strains from Saudi Arabia in 2015 and the recent outbreak in South Korea/China [31–33] and camels as noted above [35, 36] revealed a probable signature recombination event between 2 different parental clade B viruses involving a region of the ORF1ab and spike genes. We confirmed this finding and documented an increasing prevalence of this virus in humans among samples collected since January 2015 from geographically distant communities in Saudi Arabia. Similar to recent reports [33], we estimate that this recombinant virus emerged sometime in mid-to-late 2014. Based on recently available sequence data from camels in Saudi Arabia, NRC-2015 (lineage 5) was predicted to have diverged between December 2013 and June 2014 [36].
In our study, further analysis of 87 patients with laboratory-confirmed MERS-CoV reported in February revealed 13 individuals with no recognized risks for secondary acquisition; none of these 13 reported direct camel contact, although 1 individual reported visiting a camel farm. Of those sequenced, most were infected with genetically very similar viruses, suggesting a potential for limited transmission from those with unrecognized MERS-CoV infection. These findings highlight the importance of strengthened epidemiologic and laboratory surveillance.
Most cases identified in Saudi Arabia in February had documented exposure to healthcare facilities, a well-demonstrated risk factor for MERS-CoV infection [10–12]. Seventeen of 35 affected facilities in Saudi Arabia in February experienced MERS-CoV infection clusters. Moreover, 16 of 87 patients in February (18%) were visitors to healthcare facilities. This is similar to the 2014 Jeddah outbreak, where 17% of investigated cases were visitors [11]. Recommendations to limit visitation in facilities with ongoing MERS-CoV transmission should be reinforced to limit these exposures.
Our investigation, which was performed as part of an emergency public health response, is subject to several limitations. First, specimens were not available for all cases during the study period, meaning that many viruses remained untyped; however, we observed no demographic differences between cases who had specimens sequenced and those who did not. Second, since its emergence in 2012, surveillance and sequencing of MERS-CoV strains has been incomplete; variations in sequence availability and documentation might have influenced the extent of persistence and geographic spread that we have determined for past circulating virus strains. Case definitions, testing practices, and testing locations have also changed during this period. Third, although we were able to obtain full genome sequences from 12 NRC-2015 samples, all of which possessed the expected recombinant signal, our sequencing was mostly limited to the spike gene alone, which poses the risk of misclassifying recombinant viruses as belonging to NRC-2015. This is illustrated in the recent study by Sabir et al [36], which reported multiple novel recombinant viruses in camels, including recombinants between NRC-2015 (lineage 5) and other virus clades. Fourth, because of the high morbidity and mortality of MERS-CoV infection, interviews with cases were not always possible, necessitating the use of proxies. It is possible that, combined with issues of recall, the quality of the information collected varied. Of particular consideration, 11 of 13 sporadic cases were classified on the basis of interviews with proxies, and pre-illness exposures might not have been accurately recognized and reported. Fifth, some camel exposures may have gone unrecognized because of disincentives for reporting camel exposures, given their cultural and economic significance in Saudi Arabia. Sixth, given the existing evidence of association between MERS-CoV illness and pre-illness healthcare exposure or exposure to sick individuals [10, 11, 13], our risk classification was hierarchical; that is, reported exposure to a setting where secondary acquisition was likely took precedence over reported exposure to camels. As such, we did not assess camel exposures in individuals with recognized risks for secondary acquisition.
Finally, although we have attempted to link the results of our epidemiologic investigation with MERS-CoV sequences obtained from investigated cases, we cannot fully assess the possible role of virus introductions from nonhuman sources. Recent phylogeny of MERS-CoV sequences from camels in Saudi Arabia indicated that the novel recombinant subclade (referred to as NRC-2015 in our manuscript) was also predominant in camels during a period overlapping with our study [36]. As such, our detection of closely related viruses in humans might in part reflect multiple introductions from camels with similar strains. Virus introductions from other currently unidentified sources might also be factor. Virus transmission dynamics within and between human and nonhuman sources of MERS-CoV will likely influence transmission routes in ways not yet fully understood.
This investigation describes the emergence, persistence, and widespread circulation of a novel recombinant MERS-CoV in Saudi Arabia. A lack of clearly defined epidemiologic links in some cases highlights the need for ongoing intensive epidemiologic and laboratory surveillance to better understand MERS-CoV transmission and to focus infection prevention and control efforts.
Notes
Acknowledgments. We thank Shifaq Kamili, for logistics of specimen shipment; and Laura Wright of the Centers for Disease Control and Prevention (CDC) Geospatial Research Analysis and Services Program.
Disclaimer. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC.
Financial support. This work was funded by the Saudi Arabia Ministry of Health and the CDC as part of an emergency response.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}