






Abstract
There is growing evidence that Zika virus (ZIKV) can cause devastating infant brain defects and other neurological disorders in humans. However, no specific antiviral therapy is available at present. We tested a series of 2′-C- or 2′-O-methyl–substituted nucleosides, 2′-C-fluoro-2′-C-methyl–substituted nucleosides, 3′-O-methyl–substituted nucleosides, 3′-deoxynucleosides, derivatives with 4′-C-azido substitution, heterobase-modified nucleosides, and neplanocins for their ability to inhibit ZIKV replication in cell culture. Antiviral activity was identified when 2′-C–methylated nucleosides were tested, suggesting that these compounds might represent promising lead candidates for further development of specific antivirals against ZIKV.
On 1 February 2016, the World Health Organization declared a public health emergency of international concern regarding neurological disorders associated with the rapid emergence of Zika virus (ZIKV) in Oceania and the Americas [1]. Previously, ZIKV was a relatively neglected mosquito-borne arbovirus in the genus Flavivirus, family Flaviviridae. ZIKV infections have been known in Africa and Asia since the 1940s. During the last years, the virus caused several outbreaks of infection across Oceania [2]. In May 2015, a ZIKV outbreak was first reported in Brazil, and within months most countries in Latin America and the Caribbean had reported local transmission of the virus [1, 3]. Until recently, ZIKV was associated with benign infection in humans, with common symptoms that include fever, rash, joint pain, and conjunctivitis. The illness was usually mild, with symptoms lasting for several days. However, there is growing evidence in Oceania and the Americas that ZIKV can cause devastating brain birth defects, most prominently microcephaly [4], and neurological disorders in adults, including Guillain-Barré syndrome, meningoencephalitis [5], and myelitis [6]. At present, neither vaccination nor specific antiviral therapies are available to prevent or treat ZIKV infections, making a search for effective viral inhibitors an international research priority.
Nucleoside analogues are an important class of antiviral agents now commonly used as therapeutics for human viral infections, including AIDS and hepatitis B virus, cytomegalovirus, and herpes simplex virus infections [7]. These agents are generally safe and well tolerated since they target viral but not cellular polymerases and cause premature termination of viral nucleic acid synthesis [7]. In the present study, we evaluated 2′-C- and 2′-O-methyl–substituted nucleosides, 2′-C-fluoro-2′-C-methyl–substituted nucleosides, 3′-O-methyl–substituted nucleosides, 3′-deoxynucleosides, derivatives with a 4′-C-azido substitution, heterobase-modified nucleosides, and neplanocins for their ability to inhibit ZIKV replication in cell culture, with the objective of identifying promising lead candidates for further development of specific antivirals against ZIKV.
METHODS
Vero cells (ATCC CCL-81, African Green Monkey, adult kidney, epithelial) were used for determining ZIKV multiplication, for antiviral assays, and for conducting plaque assays. The cells were cultured at 37°C in 5% CO2 in Dulbecco′s modified Eagle′s medium supplemented with 10% fetal bovine serum and a 1 % mixture of antibiotics (Sigma-Aldrich, Prague, Czech Republic).
ZIKV strain MR766 (prototype strain, isolated from blood from experimental forest sentinel rhesus monkey, Uganda, 1947; GenBank accession no. AY632535) from the collection of the National Institute of Health Dr Ricardo Jorge– CEVDI/INSA (Águas de Moura, Portugal) and from the European Virus Archive was used for evaluation of the antiviral activity of the test compounds. The virus was passaged >100 times in suckling mice and/or in Vero cells prior to this study.
The following nucleoside analogues were purchased: 2′-C-methyl–, 2′-O-methyl–, and 3′-O-methyl–substituted nucleosides, 3′-deoxynucleosides, sofosbuvir, and 6-azauridine from Carbosynth (Compton, United Kingdom); 4′-azidocytidine, balapiravir, and RO-9187 from Medchemexpress (Stockholm, Sweden); neplanocin A from Cayman Chemical (Ann Arbor, Michigan); 3-deazaneplanocin A from Selleckchem (Munich, Germany); mericitabine from ChemScene (Monmouth Junction, New Jersey); PSI-6206 from ApexBio (Boston, Massachusetts); and tubercidin, toyocamycin, sangivamycin, ribavirin, and 2′-deoxynucleosides from Sigma-Aldrich (Prague, Czech Republic); rigid amphipathic. The test compounds were solubilized in 100% dimethyl sulfoxide (DMSO) to yield 10 mM stock solutions.
A viral infectivity inhibition assay was performed to measure the antiviral efficacy of nucleoside analogues in cell culture. Vero cells were seeded in 96-well plates (approximately 2 × 104 cells/well) and incubated without the presence of the drug for 24 hours, to form a confluent monolayer. Following incubation, the medium was aspirated from the wells and replaced with 200 µL of fresh medium containing 50 µM of the test compound (3 wells/compound), which was inoculated with ZIKV at a multiplicity of infection (MOI) of 0.1 plaque-forming units (PFU) at the same time as the test compound were added. As a negative control, DMSO was added to virus- and mock-infected cells at a final concentration of 0.5% (v/v). Culture medium was monitored for 5 days after infection to yield a 70%–90% cytopathic effect (CPE) in virus control wells, using the Olympus BX-5 microscope equipped with an Olympus DP-70 CCD camera. To quantify the CPE, culture media were collected at the end of the experiment (ie, day 5 after infection), and cell death was determined using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega; Madison, Wisconsin) according to the manufacturer′s instructions as described previously [8]. Viral titers were determined by plaque assay and expressed as PFU/milliliter [9]. For dose-response studies, Vero cell monolayers were cultured with 200 µL of medium containing the test compounds over the concentration range of 0–100 µM and ZIKV at a MOI of 0.1. Drug addition and virus infection was done at the same time. The medium was collected from the wells at 2 and 3 days after infection, and the viral titers were determined by plaque assay and used to construct ZIKV dose-response curves. The dose-response curves on day 2 after infection were used to estimate the 50% effective concentration (EC50). To measure the compound-induced inhibition of viral surface antigen expression, a cell-based flavivirus immunostaining assay was performed as previously described [8]. For determination of nucleoside analogue cytotoxicity, a colorimetric assay using Dojindo′s highly water-soluble tetrazolium salt (Cell Counting Kit-8, Dojindo Molecular Technologies; Rockville, Maryland) and the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega; Madison, Wisconsin) were used. The concentration of compound that reduced cell viability by 50% was considered as the 50% cytotoxic concentration (CC50).
RESULTS
A series of 29 nucleoside analogues (Supplementary Figure 1) was tested at a concentration of 50 µM for their ability to inhibit CPE mediated by ZIKV infection on Vero cells. Inhibition of ZIKV-induced CPE was monitored by light microscopy from days 1 to 5 after infection and quantified at the end of the experiment, using the colorimetric cell death in an in vitro assay. Five of the nucleoside analogues, 7-deaza-2′-C-methyladenosine (7-deaza-2′-CMA), 2′-C-methyladenosine (2′-CMA), 2′-C-methylcytidine (2′-CMC), 2′-C-methylguanosine (2′-CMG), and 2′-C-methyluridine (2′-CMU), were found to inhibit ZIKV-mediated CPE in cell culture at a concentration of 50 µM and to reduce significantly the cell death ratio in the test wells when compared with mock-treated ZIKV-infected cells (P < .05, by a 2-tailed Student t test). All other compounds had no or little effect on ZIKV-induced CPE and cell death. Tubercidin, toyocamycin, and sangivamycin were found to be cytotoxic, causing cell death in all cells at micromolar concentrations. On the basis of these preliminary results, nucleosides with a methyl moiety at the 2′-C position of the ribose ring (7-deaza-2′-CMA, 2′-CMA, 2′-CMC, 2′-CMG, and 2′-CMU) were selected for further testing.
2′-C–methylated derivatives reduced the viral titer in a dose-dependent manner (mean EC50 [±SD] of 5.26 ± 0.12 µM for 2′-CMA, 8.92 ± 3.32 µM for 7-deaza-2′-CMA, 22.25 ± 0.03 µM for 2′-CMG, 10.51 ± 0.02 µM for 2′-CMC, and 45.45 ± 0.64 µM for 2′-CMU; Figure 1A and Table 1). Dose-response curves for the 2′-C–methylated nucleosides were characterized by a typical sigmoidal shape with relatively steep slopes both on days 2 and 3 after infection (Supplementary Figure 2). Interestingly, 2′-CMU exhibited a relatively weak anti-ZIKV effect, treatment with 100 µM of the compound reduced virus titers 102-fold as compared to a mock-treated culture. The anti-ZIKV activity of 2′-C–methylated nucleosides was further confirmed by a cell-based flavivirus immunostaining assay. 2′-C–methylated nucleosides exhibited a dose-dependent inhibition of the expression of ZIKV surface E antigen in Vero cells at day 2 after infection (Figure 1C). A strong inhibition of ZIKV antigen expression was observed in 2′-CMA, 7-deaza-2′-CMA, and 2′-CMC as compared to 2′-CMG and 2′-CMU, for which relatively high EC50 values were calculated.
ZIKV Inhibition and Cytotoxicity Characteristics of Selected Nucleoside Analogues
| Compound | Structure | EC50, µM, Mean ± SDa,b | Apparent EC50, µM, Mean ± SDa,c | CC50, µMa | SId |
|---|---|---|---|---|---|
| 2′-C-methyladenosine |  | 5.26 ± 0.12 | 26.41 ± 1.14 | >100f | >19.01 |
| 7-deaza-2′-C-methyladenosine |  | 8.92 ± 3.32 | 42.87 ± 1.88 | >100 | >11.21 |
| 2′-C-methylguanosine |  | 22.25 ± 0.03 | 71.23 ± 4.11 | >100 | >4.49 |
| 2′-C-methylcytidine |  | 10.51 ± 0.02 | 61.93 ± 3.76 | >100g | >9.51 |
| 2′-C-methyluridine |  | 45.45 ± 0.64 | >100e | >100 | >2.20 |
| Compound | Structure | EC50, µM, Mean ± SDa,b | Apparent EC50, µM, Mean ± SDa,c | CC50, µMa | SId |
|---|---|---|---|---|---|
| 2′-C-methyladenosine | | 5.26 ± 0.12 | 26.41 ± 1.14 | >100f | >19.01 |
| 7-deaza-2′-C-methyladenosine | | 8.92 ± 3.32 | 42.87 ± 1.88 | >100 | >11.21 |
| 2′-C-methylguanosine | | 22.25 ± 0.03 | 71.23 ± 4.11 | >100 | >4.49 |
| 2′-C-methylcytidine | | 10.51 ± 0.02 | 61.93 ± 3.76 | >100g | >9.51 |
| 2′-C-methyluridine | | 45.45 ± 0.64 | >100e | >100 | >2.20 |
Abbreviations: CC50, 50% cytotoxic concentration; EC50, 50% effective concentration; SD, standard deviation; ZIKV, Zika virus.
a Determined from 3 independent experiments.
b Calculated as a 50% reduction of virus titers, using the Reed-Muench method.
c Calculated as a point of inflection from dose-response curves, using log-transformed viral titers.
d The selectivity index (SI) is calculated as CC50/EC50.
e Treatment with 100 µM of 2′-CMU reduced virus titers 102-fold compared to a mock-treated culture.
f Treatment of cell culture with 2′-CMA at a concentration of 100 µM led to a reduction in cell viability to 79.1%.
g Treatment of cell culture with 2′-CMC at concentration of 100 µM led to a reduction in cell viability to 69.3%.
ZIKV Inhibition and Cytotoxicity Characteristics of Selected Nucleoside Analogues
| Compound | Structure | EC50, µM, Mean ± SDa,b | Apparent EC50, µM, Mean ± SDa,c | CC50, µMa | SId |
|---|---|---|---|---|---|
| 2′-C-methyladenosine | | 5.26 ± 0.12 | 26.41 ± 1.14 | >100f | >19.01 |
| 7-deaza-2′-C-methyladenosine | | 8.92 ± 3.32 | 42.87 ± 1.88 | >100 | >11.21 |
| 2′-C-methylguanosine | | 22.25 ± 0.03 | 71.23 ± 4.11 | >100 | >4.49 |
| 2′-C-methylcytidine | | 10.51 ± 0.02 | 61.93 ± 3.76 | >100g | >9.51 |
| 2′-C-methyluridine | | 45.45 ± 0.64 | >100e | >100 | >2.20 |
| Compound | Structure | EC50, µM, Mean ± SDa,b | Apparent EC50, µM, Mean ± SDa,c | CC50, µMa | SId |
|---|---|---|---|---|---|
| 2′-C-methyladenosine | | 5.26 ± 0.12 | 26.41 ± 1.14 | >100f | >19.01 |
| 7-deaza-2′-C-methyladenosine | | 8.92 ± 3.32 | 42.87 ± 1.88 | >100 | >11.21 |
| 2′-C-methylguanosine | | 22.25 ± 0.03 | 71.23 ± 4.11 | >100 | >4.49 |
| 2′-C-methylcytidine | | 10.51 ± 0.02 | 61.93 ± 3.76 | >100g | >9.51 |
| 2′-C-methyluridine | | 45.45 ± 0.64 | >100e | >100 | >2.20 |
Abbreviations: CC50, 50% cytotoxic concentration; EC50, 50% effective concentration; SD, standard deviation; ZIKV, Zika virus.
a Determined from 3 independent experiments.
b Calculated as a 50% reduction of virus titers, using the Reed-Muench method.
c Calculated as a point of inflection from dose-response curves, using log-transformed viral titers.
d The selectivity index (SI) is calculated as CC50/EC50.
e Treatment with 100 µM of 2′-CMU reduced virus titers 102-fold compared to a mock-treated culture.
f Treatment of cell culture with 2′-CMA at a concentration of 100 µM led to a reduction in cell viability to 79.1%.
g Treatment of cell culture with 2′-CMC at concentration of 100 µM led to a reduction in cell viability to 69.3%.
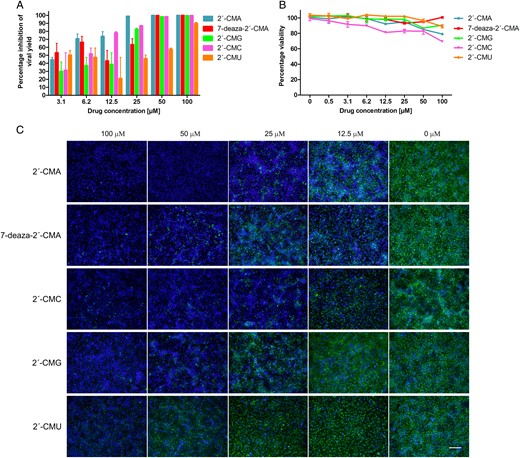
A, Dose-dependent inhibition of Zika virus (ZIKV) replication by 2′-C–methylated nucleosides in Vero cells. Vero cells were treated with different concentrations of 2′-C–methylated nucleosides and at the same time infected with ZIKV. Culture supernatants were collected at 48 hours after infection and analyzed for ZIKV infectivity by plaque assay. B, Viability of Vero cells treated by 2′-C–methylated nucleosides at a range of concentrations of 0–100 µM. Vero cells were exposed to the tested compounds for 72 hours. Viability was measured by a colorimetric assay, using Dojindo′s highly water-soluble tetrazolium salt test (triplicate setting) and confirmed by optical microscopy. C, Inhibition of ZIKV antigen expression by nucleoside inhibitors. ZIKV-infected Vero cells were fixed on slides at 48 hours after infection and stained with flavivirus-specific antibody labeled with FITC (green) and counterstained with DAPI (blue). Scale bar, 50 µm.
The cytotoxicity of 2′-C–methylated nucleosides was assessed in Vero cells. Except for 2′-CMC, which exerted a weak cytotoxic effect on Vero cells (treatment of the cell culture at a compound concentration of 100 µM led to a reduction in cell viability to 69.3%), all 2′-C–methylated nucleosides tested showed no cytotoxic effects at the highest tested concentration of 100 µM with no detectable effect on cell proliferation (Table 1 and Figure 1B).
DISCUSSION
ZIKV, a previously neglected mosquito-borne virus, is prompting worldwide concern because of its alarming connection to a neurological birth disorder. In most cases, ZIKV causes a benign disease, but in some patients the infection can manifest as myelitis or meningoencephalitis, or it can trigger Guillain-Barré syndrome, a severe neurological disorder characterized by progressive muscle weakness that can result in respiratory failure [1, 3–6]. No effective therapy for ZIKV infection is available at present; therefore, research on possible antiviral compounds active against ZIKV has become an international priority. Because human cells lack RNA-dependent RNA polymerase (RdRp), this class of enzymes appears to be one of the most promising targets for antivirals against flaviviruses that use RdRp for replication. Nucleoside analogs can target viral RdRp to terminate viral RNA replication after incorporation into the viral nascent RNA chain [10–12]. Here, we analyzed a library of nucleoside analogues for their in vitro activity against ZIKV.
Evaluation of various modifications at the nucleoside 2′-position revealed that the introduction of the 2′-C-methyl substituent to the nucleoside β-face resulted in inhibition of ZIKV replication in vitro. Different EC50 values for individual 2′-C–methylated nucleosides (ranging from 5.26 to 45.45 µM) indicate that their antiviral activities were substantially affected also by the identity of the heterocyclic base moiety. Introduction of guanine or especially uridine into the nucleoside molecule considerably reduced the anti-ZIKV activity in vitro. The cytotoxicity of the 2′-C–methylated nucleosides was observed to be either zero or negligible, except for 2′-CMC. The cytotoxicity of the studied compounds was assessed also in human neuroblastoma cells UKF-NB-4 (data not shown) and in porcine kidney cells (PS) [8] with results comparable to those for Vero cells. This is in a sharp contrast with high (submicromolar) cytotoxicity of tubercidin, toyocamycin, and sangivamycin, chemically related structures devoid of the 2′-C-methyl group. Taken together, the 2′-C-methyl substituent appeared to be an important structural element for highly selective ZIKV inhibition and reduced cytotoxicity. We can speculate that the 2′-C-methyl substituent does not prevent binding to the ZIKV RdRp active site and allows the viral RNA chain termination. During the peer review of this manuscript, another group published further evidence that 7-deaza-2’-CMA exhibits anti-ZIKV activity [13].
Interestingly, no or negligible anti-ZIKV activity was observed for 2′-α-fluoro-2′-β-methyl– or 2′-O-methyl–substituted nucleosides, as well as for 3′-O–modified nucleosides. The introduction of the fluoro- moiety into the C2′ position or the methyl moiety into the O2′ or O3′ position probably eliminates the 2′-α-hydroxy or 3′-α-hydroxy hydrogen bond donor/acceptor, which could result in complete abrogation of the nucleoside inhibitory activity. The observed inactivity could be also explained by inefficient cellular uptake and metabolism to convert the nucleoside molecule to the corresponding triphosphate form [14]. A detailed study of the compound uptake and metabolic conversion goes beyond the scope of this brief report and will be a subject of a future report. Neither nucleosides with 4′-azido modification nor nucleosides with chemically modified heterobase moiety exerted anti-ZIKV activity.
In conclusion, we have demonstrated that 2′-C–methylated nucleosides exerted activity against ZIKV under in vitro conditions. These compounds provide a basis for structure-based optimization and rational design of effective prodrugs, which will be further tested in rodent models for therapy of ZIKV infection.
Notes
Financialsupport. This work was supported by the Czech Science Foundation (grantGA14–29256S); the Ministry of Education, Youth, and Sports of the Czech Republic, under the NPU I program (grant LO1218); the National Subvention for the Development of Research Organizations (grant RVO:61388963); the European Virus Archive Goes Global project, which has received funding from the European Union's Horizon 2020 research and innovation program (grant 653316).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References


{kind=link}