






Abstract
Background. Neutrophils and monocytes are crucial for controlling bacterial infections. More-frequent bacterial infections are accordingly encountered in neutropenic patients undergoing chemotherapy. This is not the case for pegylated interferon α (IFN-α)–induced neutropenia. We hypothesized that IFN-α induces a compensatory innate antibacterial state that prevents bacterial infections despite the neutropenia.
Methods. To investigate whether patients with hepatitis C virus infection treated with IFN-α killed group A Streptococcus (GAS) better than before initiating therapy, whole blood was used to perform ex vivo GAS killing assays before, during, and after IFN-α therapy.
Results. We found that IFN-α therapy enhanced GAS killing in whole blood ex vivo despite the decreased neutrophil and monocyte numbers during IFN-α therapy. IFN-α also boosted neutrophil- and monocyte-mediated GAS killing in vitro. Underlying mechanisms included increased production of the antibacterial properdin, a regulator of the complement activation, as well as reactive oxygen species.
Conclusions. These findings help to explain the rather discrepant facts of neutropenia but preserved antibacterial immune defenses in patients treated with IFN-α.
Neutrophils and monocytes are crucial for controlling and constraining bacterial infections. Their importance is highlighted by patients with chemotherapy-induced neutropenia and those with hereditary neutrophil disorders, who often die from severe bacterial infections [1, 2]. Pegylated interferon α (IFN-α) is used for treating conditions such as hepatitis C virus (HCV) infection, multiple myeloma, and rheumatologic diseases. IFN-α treatment frequently causes neutropenia. However, in contrast to the neutropenic patients undergoing chemotherapy, an increased risk for bacterial infections has not been observed in patients with IFN-α–induced neutropenia [3–5].
IFN is known to interfere with viral replication [6]. Most eukaryotic cells secrete IFN-α in response to foreign and autologous nucleic acid stimuli. Double-stranded RNA activates the cytosolic receptors RNA helicases retinoic acid–inducible gene I and melanoma-differentiation associated gene 5, driving type I IFN secretion [7]. The extracellularly located transmembrane receptor Toll-like receptor 3 (TLR3) also induces type I IFN secretion in response to double-stranded RNA, as does TLR4 in response to lipopolysaccharide [8], endosomal receptor TLR7 in response to single-stranded RNA, and endosomal TLR9 in response to double-stranded DNA [9]. Secreted IFN upregulates transcription of hundreds of IFN-stimulated genes (ISGs). Although many of these ISGs are still not fully described, it is known that their products exert various antiviral functions, such as inhibiting viral replication and boosting viral clearance [10, 11].
IFN-α affects certain bacterial species. Lymphocytes are rendered more susceptible to listeriolysin O, for example, causing increased apoptosis during Listeria monocytogenes infection [12, 13]. In contrast, IFN-α inhibits the intracellular replication cycle of Chlamydia trachomatis and Legionella pneumophila [14, 15] and protects mice against Salmonella Typhimurium infections [16]. Similarly group B Streptococcus induces type I IFN via TLR7, resulting in resistance to infection [17, 18], and group A Streptococcus (GAS) induces type I IFN in macrophages [19].
Macrophages are key effectors for controlling GAS infections, in synergy with the complement system [20], and properdin was identified to be crucial for the activation of the alternative complement pathway. Thereby, properdin boosts opsonization and phagocytosis and increases formation of membrane attack complexes [21–23].
GAS has come up with ingenious ways to evade host recognition, allowing it to survive the constant challenge of the host's innate immune defenses. Recently, we showed that secretion of the bacterial DNases helps circumvent TLR9-dependent host recognition [24]. This resulted in diminished IFN-α levels at the site of infection and reduced GAS killing by murine macrophages [25]. These findings, together with the clinical observation that IFN-α–treated individuals have no increased risk for bacterial infections despite neutropenia, suggest that IFN-α may protect against bacterial infections [5].
We hypothesized that IFN-α induces a compensatory innate antibacterial state that decreases the neutropenia-mediated risk of bacterial infections. We thus investigated whether IFN-α enhances bacterial clearance in blood. We chose GAS as a model pathogen to analyze the impact of IFN-α on bacterial clearance ex vivo, by examining blood obtained from individuals undergoing IFN-α therapy, and in vitro, by evaluating IFN-α–treated neutrophils and monocytes.
MATERIAL AND METHODS
Bacterial Strains
All experiments were performed with the GAS strain M1T1 5448 [26] cultured in Todd Hewitt Broth (BD, Temse, Belgium) supplemented with 2% yeast extract (THY) if not otherwise stated. Green fluorescent protein (GFP) was expressed in GAS with a GFP-pDCerm vector. To maintain the plasmid GFP-pDCerm, GAS was cultured in THY supplemented with 5 µg/mL erythromycin.
Patient Treatment Regimens
All studies were conducted in compliance with the current version of the Declaration of Helsinki, as well as all national legal and regulatory requirements. Experiments were declared to and approved by the cantonal ethics committee Zurich (numbers 2010-01126/0, 2012-0033, and 2011-0340), and all patients and healthy volunteers were asked for and provided written informed consent.
The decision to initiate HCV therapy was made by the treating physicians, and consent to initiate therapy was given by the patient. Therapy was started with subcutaneous peg-IFN-α2a (Pegasys, Roche, Basel, Switzerland) at a dose of 180 μg once weekly plus oral ribavirin (Roche; 600 mg taken twice daily for HCV genotypes 1/4 or 400 mg taken twice daily for genotypes 2/3). Blood values were monitored weekly, and peg-IFN-α2a and ribavirin doses were adjusted when deemed necessary by the treating physician. HCV RNA levels were measured at regular intervals to monitor the response to HCV therapy. The HCV load became undetectable during IFN-α therapy. All patients were receiving highly active antiretroviral therapy for human immunodeficiency virus (HIV) infection resulting in nondetectable HIV loads.
Isolation of Neutrophils and Monocytes
Neutrophils were isolated as described previously [27]. CD14+ monocytes were isolated from buffy coats (obtained at a local blood donation center in Zurich), using MACS CD14 MicroBeads (Miltenyi Biotec, Bergish Gladbach, Germany) according to the manufacturer's instructions. Purity was verified by fluorescence-activated cell sorting (FACS) and monocyte depleted cells were used when indicated.
GAS Killing by Blood
Blood from healthy volunteers and from HCV-infected, HIV-positive patients after termination of IFN-α therapy were preincubated with IFN-α (Pegasys) for 30 minutes at 37°C and challenged with GAS (2 × 104–3 × 104 colony-forming units [CFU]/mL) for 15 minutes at 37°C.
GAS Killing by Plasma
Plasma from healthy volunteers or from patients’ whole blood was used as follows. GAS (104 CFU) was preincubated for 20 minutes with properdin neutralization antibody (mouse anti-human monoclonal properdin antibody; Quidel A233, San Diego, California) or control antibody (mouse immunoglobulin G control isotype; Biolegend, London, United Kingdom) before incubation with plasma and addition of purified properdin (Quidel 412, San Diego; final concentration, 25 µg/mL). The tubes were incubated for 30 minutes at 37°C, and serial dilutions were plated on THY plates to quantify GAS survival.
GAS Killing by Neutrophils and Monocytes
Where indicated, neutrophils and monocytes were preincubated with various IFN-α concentrations for 20 minutes (neutrophils) or 1 hour (monocytes) at 37°C. GAS was added at a multiplicity of infection (MOI) of 1 followed by incubation for 30 minutes (for neutrophils) or 1 hour (for monocytes) at 37°C. When stated, GAS was incubated with patient plasma and either control or properdin neutralization antibody. The plasma (final concentration, 10%) and antibody-opsonized GAS (MOI, 1) were added to monocytes, and bacterial survival was assessed as described above. Otherwise, antibody-opsonized GAS was added with purified properdin (25 µg/mL) to the monocytes to assess survival. The number of bacteria killed intracellularly by monocytes was established by coincubating monocytes with GAS (MOI, 10). After incubation for 1 hour at 37°C with 5% CO2, antibiotics (10 µg/mL penicillin and 100 µg/mL gentamicin) were added for 3 hours. For extracellular neutrophil or monocyte killing assays, neutrophils or monocytes were preincubated with the phagocytosis-inhibitor cytochalasin D (10 µg/mL; MP Biomedicals, Luzern, Switzerland) for 20 minutes. GAS was added at a MOI of 1 and incubated for 90 minutes at 37°C with 5% CO2.
GAS Monocyte Phagocytosis
Monocytes were preincubated with indicated IFN-α concentrations for 1 hour at 37°C or for 20 minutes with cytochalasin D (10 µg/mL). GFP-GAS was added at a MOI of 10, followed by incubation for 1 hour at 37°C with 5% CO2. Cells were harvested and prepared for FACS analysis with a BD FACScan. The percentage of GFP-positive cells was calculated by using FlowJo software for Windows (version 10.0.8).
Monocyte Viability
The viability of monocytes in the presence of various IFN-α concentrations and with or without GAS was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay as reported previously [28].
Reactive Oxygen Species (ROS) Production
After prestimulation with various IFN-α concentrations for 20 minutes (for neutrophils) or 1 hour (for monocytes) at 37°C, cells were challenged with GAS (MOI, 10) or left unstimulated. ROS production was measured as described elsewhere [24].
Properdin Levels
Properdin concentrations in patients' plasma samples or in supernatants from cultured cells were measured by enzyme-linked immunosorbent assay (HK344-02; Hycult Biotech, Uden, the Netherlands) in accordance with the manufacturer's instructions.
Statistical Analysis
All experiments were performed in a minimum of 3 independent replicates. Normally distributed groups were compared to the untreated group by using 1-way analysis of variance with the post-hoc Dunnett multiple comparisons test. If the number of individuals was unequal, an unpaired 2-tailed Student t test was used. P values were calculated by the statistics program supplied by the GraphPad Prism 5.01 software.
RESULTS
Enhanced GAS Clearance by Blood Specimens From Patients Undergoing Systemic IFN-α Therapy
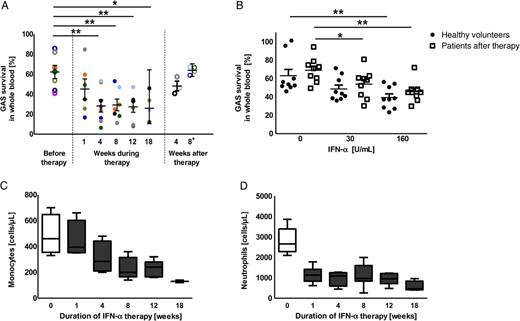
Enhanced group A Streptococcus (GAS) clearance by whole blood in the presence of interferon α (IFN-α). A, GAS clearance of blood from patients before, during, and after IFN-α therapy was assessed ex vivo. Patients were followed throughout the course of IFN-α therapy, with some time points missing owing to variable schedules of a few individuals. Open circles indicate untreated patients, and closed circles indicate samples obtained during IFN-α treatment. GAS survival in blood during therapy was compared to survival before therapy start. B, GAS clearance by blood taken from healthy volunteers (closed bars) or from patients 8 weeks after IFN-α therapy (open bars) after addition of IFN-α ex vivo. C and D, Absolut monocyte (C) and neutrophil (D) counts of patients before (open bars) and during (closed bars) IFN-α therapy are shown as box plots, with whiskers denoting 5th–95th percentiles. Experiments show data from a cohort of 8 patients, with 1 sample per patient per time point, or from 5 healthy volunteers. The experiment shown in panel B was repeated at least 3 times in triplicate with 3 individual healthy volunteers or 3 patients (data are means ± standard errors of the mean). Groups in panel A were compared by using the unpaired 2-tailed t test. Data in panel B were analyzed by using 1-way analysis of variance with the post hoc Dunnett multiple comparisons test. *P < .05 and **P < .01.
Ex vivo IFN-α administration resulted in the same killing efficiency in whole blood obtained from patients after termination of IFN-α therapy (Figure 1B) and from healthy volunteers. The enhanced clearance of GAS by whole blood from patients after termination of IFN-α therapy (Figure 1B) was similar to the GAS clearance observed during therapy (Figure 1A). To address a potential influence of HCV infection, we compared blood collected before and after HCV was cleared (and IFN-α therapy had been stopped). We found that survival of GAS in whole blood before therapy was similar to survival 8 weeks after therapy, suggesting that the HCV load has no effect on GAS clearance (Figure 1A). In view of the long half-life of IFN-α, we examined samples obtained 8 weeks after termination of IFN-α therapy, to eliminate potential interference by IFN-α.
To rule out the possibility that IFN-α affects GAS growth, we assessed GAS growth in the presence or absence of IFN-α. We found no evidence that IFN-α directly interfered with GAS growth (Supplementary Data) at IFN-α concentrations comparable to those in patients' serum [29].
Neutrophils and monocytes are the first immune cells to fight off bacterial infections. Despite the severe IFN-α–induced neutropenia and monocytopenia observed in all our patients (Figure 1C and 1D), IFN-α exerted an antibacterial effect in whole blood (Figure 1A). This further underscores that IFN-α boosts GAS clearance in whole blood, independent of the HCV load or monocyte and neutrophil cell count and without directly inhibiting bacterial growth.
Enhanced GAS Killing by Monocytes in the Presence of IFN-α
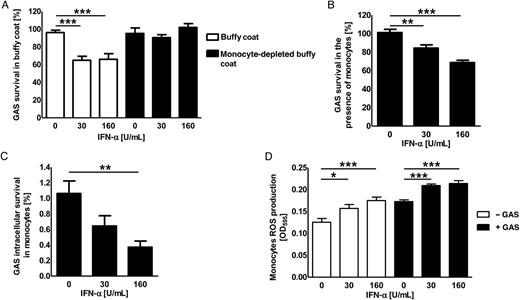
Enhanced intracellular killing of group A Streptococcus (GAS) by human monocytes in the presence of interferon α (IFN-α). A, Survival of GAS in buffy coats (open bars) or monocyte-depleted buffy coats (closed bars) derived from healthy donors after preincubation with IFN-α. B and C, Total (B) and intracellular (C) killing of GAS by monocytes induced by various concentration of IFN-α ex vivo. D, Reactive oxygen species (ROS) production by monocytes in the absence (open bars) or presence (closed bars) of GAS after induction with various concentrations of IFN-α ex vivo. Each experiment was performed in triplicate and was repeated 3 times (data are means ± standard errors of the mean). Groups were compared by using 1-way analysis of variance with the post hoc Dunnett multiple comparisons test. *P < .05, **P < .01, and ***P < .001.
Increased Levels of Properdin in Patients Undergoing IFN-α Therapy
Properdin, an activator of the complement pathway that supports opsonization, phagocytosis, and formation of the membrane attack complex [21–23], is also highly upregulated in peripheral blood cells treated with IFN-α ex vivo [30]. We hypothesized that increased properdin levels may lead to enhanced bacterial killing. To test this hypothesis, we quantified the properdin levels and assessed whether properdin results in enhanced GAS clearance.
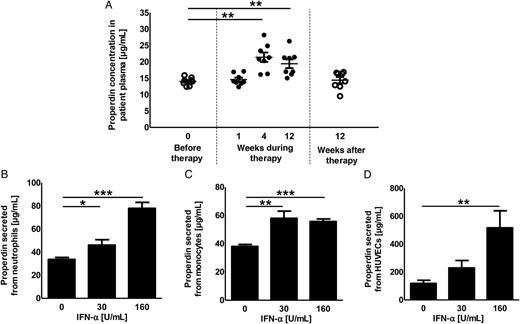
Influence of interferon α (IFN-α) treatment on plasma properdin concentrations. A, Properdin concentrations in plasma derived from patients before (open circles), during (closed circles), and after IFN-α therapy (open circles) was assessed by enzyme-linked immunosorbent assay. The properdin concentrations were compared to the concentration found in the plasma of patients before therapy start (open boxes). B–D, Properdin secretion by cultured human primary cells was measured by enzyme-linked immunosorbent assay. Data are for neutrophils (B), monocytes (C), and human umbilical vein endothelial cells (HUVECs; D). The experiment in panel A was performed twice in duplicate with plasma from 4 patients. The experiments in panels B–D were performed in triplicate and were repeated 3 times (data are means ± standard errors of the mean). Groups were compared by using 1-way analysis of variance with the post hoc Dunnett multiple comparisons test. *P < .05, **P < .01, and ***P < .001.
Properdin Supports IFN-α–Mediated GAS Killing by Plasma and Monocytes
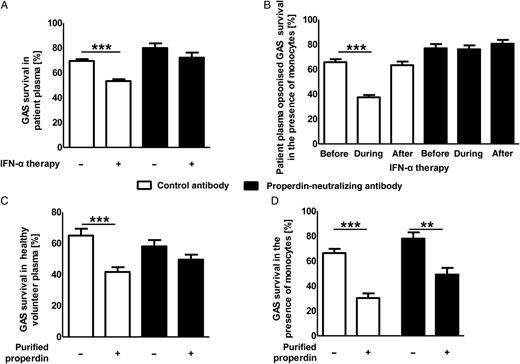
Group A Streptococcus (GAS) killing in the presence of endogenous or exogenous properdin. A, GAS killing by patient plasma obtained before or during interferon α (IFN-α) therapy in the presence (closed bars) or absence (open bars) of properdin-neutralizing antibodies. B, Plasma derived from patients before, during, and after IFN-α therapy was used to opsonize GAS. Killing of opsonized GAS by purified monocytes from healthy donors was assessed in the presence (closed bars) or absence (open bars) of properdin-neutralizing antibodies. C, GAS survival in plasma from healthy volunteers in the presence (closed bars) or absence (open bars) of purified properdin and/or properdin-neutralizing antibodies. D, Assessment of GAS killing by monocytes in the presence or absence of purified properdin and/or properdin-neutralizing antibodies. Each experiment was performed in triplicate and was repeated 3 times (data are means ± standard errors of the mean). Plasma from 3 patients (A and B) was used in triplicate. Groups were compared by using 1-way analysis of variance with the post hoc Dunnett multiple comparisons test. **P < .01 and ***P < .001.
To further confirm the role of properdin in controlling GAS infections, purified properdin was added to plasma from healthy volunteers. The addition of properdin led to increased killing of GAS (Figure 4C) in plasma, and again this effect was reduced by the addition of an antiproperdin neutralizing antibody. To investigate GAS killing mediated by monocytes in the presence of properdin, GAS together with purified properdin was added to the purified monocytes, and GAS killing was assessed. The addition of purified properdin led to increased killing of GAS by monocytes (Figure 4D). This effect was reduced by the addition of an antiproperdin neutralizing antibody. The minor but significant difference between the presence and the absence of purified properdin in combination with the antiproperdin neutralizing antibody might be linked to inefficiency of the antibody used or could reflect the properties of different physiological forms of properdin. In human plasma, properdin is found in dimers (P2), trimers (P3), or tetramers (P4), but it is not found as higher-order oligomers (Pn) [34]. Whether our observations are due to the differences in the ability and specificity of distinct properdin forms to bind and activate the complement system [35] or whether they emphasize the multifunctional property of monocytes for clearing bacterial infections remains to be clarified.
DISCUSSION
Our ex vivo studies, performed with blood collected from patients undergoing IFN-α therapy, showed enhanced clearance of GAS in whole blood already 1 week after initiating IFN-α therapy. GAS clearance increased over time and reached the maximum effect after 8–12 weeks of IFN-α therapy (2–3-fold increased killing). In parallel, we found increased properdin concentrations in the patients’ plasma. Increased properdin production by various cells under IFN-α promoted plasma-mediated GAS killing. Coincubation of phagocytic cells with IFN-α ex vivo activated ROS production, which can be a possible additional mechanism for increased GAS killing by IFN-α–treated blood. A third of the patients already had moderate neutropenia (500–1000 cells/µL) by week 1, half of all patients had moderate (500–1000 cells/µL) to severe (<500 cells/µL) neutropenia starting 4 weeks after therapy initiation, and the rest had mild neutropenia (>1000 cells/µL). Thus, we believe that enhanced neutrophil function induced by IFN-α and increased properdin levels help fighting infections in these patients. Taken together, these data help explain the clinical observation that patients undergoing IFN-α therapy are less subject to bacterial infections despite the observed IFN-α–induced neutropenia [36–38].
The complement system mediates bacterial clearance at both the early and late stages of the immune response. Upon treatment of peripheral blood cells with IFN-α, properdin was reported to be one of the most strongly induced genes [30]. IFN-α also upregulates the complement component C3 [39], which, along with increased properdin production, could also contribute to increased activation of the alternative pathway. Likewise, we found that patients undergoing IFN-α therapy had significantly increased properdin levels in plasma. Properdin is the key regulator of the alternative complement pathway, stabilizing C3bBb convertase. This leads to increased cleavage of C3 to C3b [40], which in turn binds to bacterial surfaces, acting as an opsonin. Properdin was previously reported to be important for increased opsonization with enhanced neutrophil/monocyte-mediated killing of bacteria, such as groups B and A Streptococcus [22, 41] and Neisseria species [42]. Taking a translational approach, our study went one step further, showing increased GAS killing by monocytes when opsonized with plasma obtained from patients during IFN-α therapy, compared to plasma obtained from patients before or after IFN-α therapy. We also showed that blood collected during IFN-α therapy, accompanied by increased properdin levels, led to enhanced bacterial clearance. Although various conformational forms of properdin have been found [34], it has so far not been described that their binding ability to live GAS has the potential to promote GAS killing. In this study, we showed that purified properdin, mainly consisting of Pn, efficiently contributed to GAS killing. Although we did not examine direct binding of physiological properdin to GAS, we showed that, by neutralizing properdin, GAS killing was reduced and thus indirectly showed that properdin might have a role in serum-mediated clearance of GAS. Despite the findings that both nonphysiological (Pn) and physiological (P2-P4) properdin play an important role in GAS killing, the detailed mechanisms require further exploration. In our ex vivo studies in which IFN-α was added to different cell types, we observed increased secretion of properdin not only from peripheral blood cells but also from vascular endothelial cells. This might explain the high concentration of properdin we found in blood collected from patients during IFN-α therapy. Further, the coexistence of properdin and IFN-α in patients' blood might explain the enhanced killing capacity observed. Blood obtained from patients during IFN-α therapy killed GAS better (27.4% bacterial survival at 12 weeks) than blood from healthy volunteers (67.8% bacterial survival). In addition, the duration of exposure to IFN-α may be another reason for the observed differences in GAS killing. Owing to the complexity of the immune system, it is probable that different mechanisms of bacterial killing are activated at different time points during IFN-α therapy, such as increased ROS production (during short exposure to IFN-α) and increased properdin expression (during longer exposure to IFN-α).
In summary, we found that both monocytes and the complement factor properdin are important components for controlling GAS infections in whole blood of patients undergoing IFN-α therapy, with enhanced properdin levels allowing compensation for IFN-α–induced neutropenia and monocytopenia.
Notes
Financial support. This work was supported by the Swiss National Foundation (grants 310030_130748/1 and 310030_146295 to A. S. Z. and grant PZ00P3_136639 to R. A. S.) and the Stiftung für Medizinische Forschung, University of Zurich (to A. S. Z.).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Presented in part: 8th International Symposium on Tonsils and Mucosal Barriers of the Upper Airways, Zurich, Switzerland, 18 July 2013; 5th Congress of European Microbiologists, Leipzig, Germany, 21–25 July 2013. Abstract 1427.
S. U. and N. K. contributed equally to this work.
Present affiliation: Department of Pediatrics, University of California–San Diego, La Jolla.


{kind=link}
{kind=link}
{kind=link}
{kind=link}