




Abstract
Our body is constantly exposed to pathogens and equipped with a highly elaborate immune system to fight against invading pathogens. The first line of defence is the innate immune system. It has evolved to detect conserved microbial molecular patterns, dubbed pathogen-associated molecular patterns (PAMPs), through pattern recognition receptors (PRRs). The binding of PRRs to PAMPs activates intracellular signalling cascades that lead to the expression of proinflammatory cytokines, Type I interferons and other antiviral proteins that all coordinate the elimination of pathogens and infected cells. PRRs can be classified as transmembrane receptors, including Toll-like receptors and some C-type lectin receptors, and as cytosolic receptors including retinoic acid-inducible gene-I-like receptors, nucleotide-binding domain and leucine-rich repeat-containing proteins, and cyclic GMP-AMP synthase. Studies have revealed that innate immune signals, including the ones activated by cytosolic PRRs, are triggered on organelle membranes. Here, we review the recent insights into how organelle membranes and their associated membrane lipids contribute to PRR-mediated innate immune signals.
Abbreviations
- C16
16-carbon
- 2-BP
2-bromopalmitate
- CARDs
caspase activation and recruitment domains
- CD
Crohn’s disease
- dsDNA
double-stranded DNA
- EOS
early-onset sarcoidosis
- ER
endoplasmic reticulum
- HIDS
hyper-IgD syndrome
- iE-DAP
γ-glutamyl-meso-diaminopimelic acid
- IRF3
interferon regulatory factor 3
- LGP2
laboratory of genetics and physiology 2
- MAM
mitochondria-associated ER membranes
- MAPK
mitogen-activated protein kinase
- MAVS
mitochondrial antiviral-signalling protein
- MDA5
melanoma differentiation-associated protein 5
- MDP
muramyl dipeptide
- MKD
Mevalonate kinase deficiency
- mtDNA
mitochondrial DNA
- MTOC
microtubule organizing centre
- MVK
mevalonate kinase
- NEK7
NIMA-related kinase 7
- NOD
nucleotide-binding oligomerization domain
- OSBPs
oxysterol-binding proteins
- PALM
photoactivated localization microscopy
- PM
plasma membrane
- PtdIns4P
phosphatidylinositol 4-phosphate
- SAVI
STING-associated vasculopathy with onset in infancy
- SLE
systemic lupus erythematosus
- SM
sphingomyelin
- SMS1
sphingomyelin synthase 1
- STING
stimulator of interferon genes
- TBK1
TANK1-binding kinase 1
- TFEB
transcription factor EB
- TGN
trans-Golgi network
- ZDHHC
zinc finger DHHC-type palmitoyl transferase
The innate immune system is an evolutionary ancient system to combat invading pathogens. It acts as the first line of defence with pattern recognition receptors (PRRs), detecting pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that are released upon cellular stress or tissue injury (1,). The ligand-bound PRRs activate intracellular signalling cascades that lead to the expression of proinflammatory cytokines, Type I interferons and other antiviral proteins that all coordinate the elimination of pathogens and infected cells. PRRs can be classified as transmembrane receptors including toll-like receptors (TLRs) (2,) and some C-type lectin receptors (CLRs) (3,), and as cytosolic receptors including retinoic acid-inducible gene-I (RIG-I)-like receptors (4,), NOD-like receptors (NLR) proteins (5,), pyrin (6,) and cGAS (7,). Several transmembrane-type PRRs are distributed at various organelles, such as early endosomes, late endosomes and recycling endosomes (REs). Some PRRs, upon their binding to ligands, migrate to other organelles by membrane traffic and trigger the innate immunity signals there (8, 9,). Not surprisingly, dysregulated membrane trafficking of PRRs underlies the pathogenesis of various autoinflammatory diseases and neurodegenerative diseases. The activation of innate immune responses, particularly in antigen-presenting cells such as dendritic cells, is essential for activating T cells through the PRR-dependent production of various cytokines and interferons, thus shaping the subsequent adaptive immune responses (10).
Protein S-palmitoylation (hereafter, simply referred to as protein palmitoylation) is a reversible lipid posttranslational modification to the sulfhydryl group of a cysteine (Cys) residue (11). Because of the hydrophobic nature of a 16-carbon (C16:0) palmitoyl group, protein palmitoylation enhances the hydrophobicity of the target proteins and contributes to their membrane association. Protein palmitoylation occurs not only to non-transmembrane proteins but to transmembrane proteins, suggesting that protein palmitoylation enhances their affinity to specific membrane domains, such as raft domains. Protein palmitoylation is also implicated in protein stability, conformation and homotypic and heterotypic interactions.
Recent studies have revealed that several PRRs, including cytosolic ones, exploit organelle membranes and their associated membrane lipids for triggering the innate immune signals (Fig. 1). In this review, the innate immune signals are grouped into three classes according to the organelles on which they are activated, namely, the Golgi apparatus, endosomes and mitochondria. We particularly focus on the engagement of palmitoylation of PRRs and membrane lipids with the innate immune signals.
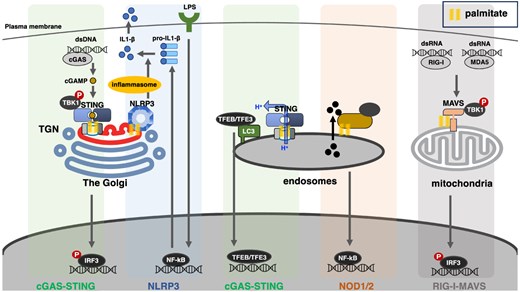
Various PRRs (cGAS, NLRP3, NOD1/2, RIG-I and MDA5) and their associated proteins (STING and MAVS) trigger the innate immune signals on the membrane of organelles that include the Golgi, endosomes and mitochondria. Protein palmitoylation emerges as an integral post-translational modification in the activation of the innate immune signals: it promotes the membrane association for cytosolic PRRs, or clustering of transmembrane-type PRRs and their associated proteins.
Innate Immune Signals from the Golgi Apparatus
The Golgi apparatus (hereafter, simply referred to as the Golgi) is a highly polarized organelle in animal cells and often locates near the nucleus and the microtubule organizing centre (MTOC) (12,). Newly synthesized proteins from the endoplasmic reticulum (ER) enter at the cis face of the Golgi, the cis-Golgi network, and then they move through the stack to the trans-Golgi network (TGN) where they are sorted to their different destinations both within and outside the cell (13,). Passage through the Golgi stack is often accompanied by post-translational modifications, such as glycosylation and sulfation of cargo proteins and lipids, synthesis of different lipid species and palmitoylation of proteins. In addition to the transport of proteins and lipids to and from the Golgi via vesicular and tubular trafficking intermediates, there is also extensive non-vesicular transport of lipids between the ER and TGN, making TGN a unique membrane compartment with specific lipid expression. Recent studies have identified the TGN as the site where cGAS and NLRP3 (a molecule belonging to the NLR family) activate the innate immune signals as described below. The pyrin pathway can be triggered on the Golgi by a disease-causative variant of CDC42 (CDC42R186C), which undergoes palmitoylation at its C-terminus (14). The detailed molecular mechanism of the CDC42-pyrin signal is described in another review article by Dr. Yasumi in this issue.
cGAS–STING pathway
Innate immune response can be triggered with double-stranded DNA (dsDNA) in the cytosol, which emerges, e.g. in the setting of infection of dsDNA viruses (15,). Two proteins, cGAS and stimulator of interferon genes (STING), have been identified as critical regulators of the response. cGAS is a cytosolic PRR and binds to cytosolic dsDNA (7,). The binding of dsDNA to cGAS activates the enzymatic activity of cGAS, leading to the generation of cGAMP from GTP and ATP (16). cGAMP diffuses throughout the cytosol and binds STING to trigger Type I interferon and inflammatory responses.
STING (17,), also known as endoplasmic reticulum interferon stimulator (MITA) (18,), endoplasmic reticulum interferon stimulator (ERIS) (19,), MPYS (20,) or TMEM173, is an ER-localized transmembrane protein. STING is proposed to act as a scaffold to activate TANK1-binding kinase 1 (TBK1), a cytosolic serine/threonine protein kinase (21,). The activated TBK1 then phosphorylates the transcription factor interferon regulatory factor 3 (IRF3). Phosphorylated IRF3 by TBK1 dimerizes and translocates into the nucleus to induce transcription of genes that encode Type I interferons such as interferon-beta (IFN-β) (22,). STING also induces proinflammatory response via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) by the activation of TBK1 and IκB kinase epsilon (IKKε) (23, 24,). While the cGAS–STING pathway is critical for successful protection against dsDNA viruses, the dysregulation of the pathway results in the excessive production of inflammatory mediators with detrimental effects on surrounding cells and tissues. In line with this, recent studies have revealed that the dysregulated STING activity is causative of multiple autoinflammatory diseases (Aicardi–Goutières syndrome, STING-associated vasculopathy with onset in infancy (SAVI) (25, 26,), coatomer protein complex subunit alpha (COPA) syndrome (27–31,)) and neurodegenerative diseases (Parkinson’s disease (32,), amyotrophic lateral sclerosis (33, 34,), multiple sclerosis (35)).
Mechanism of STING activation at TGN
After binding to cGAMP, STING immediately translocates from the ER to the Golgi (36–38,). The interference with the ER-to-Golgi traffic by a fungal metabolite brefeldin A or expression of Shigella effector IpaJ suppresses the STING-dependent signals, suggesting that STING activates TBK1 at the post-ER compartments (36, 38–41,). Indeed, high-resolution fluorescence microscopy reveals that TBK1 becomes associated with the Golgi, in particular, at the TGN after STING stimulation (42). In agreement with this, phosphorylated TBK1, a hallmark of the STING activation, is found specifically at the TGN.
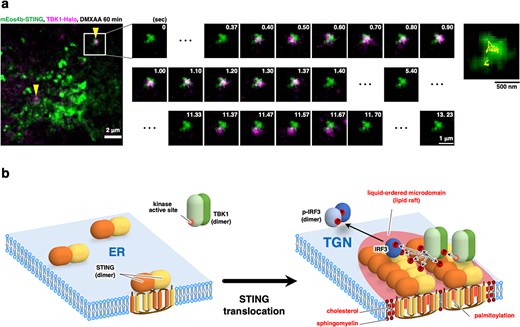
(A) mEos4b-STING and TBK1-HaloTag7 were stably expressed in STING/TBK1-double knockout MEFs. A typical overlapped image obtained by simultaneous observation of mEos4b-STING clusters (green, PALM image) and single molecules of TBK1-HaloTag7-SF650T (magenta) 60 min after stimulation with DMXAA. Arrowheads indicate the co-localization of STING clusters with single molecules of TBK1. (B) A graphical abstract illustrating cholesterol- and palmitoylation-dependent STING clustering at the TGN.
A study reveals that palmitoylation of STING is essential for activation of TBK1 and IRF3 (38). Treatment of cells with a pan-palmitoylation inhibitor 2-bromopalmitate (2-BP) inhibits Type I interferon response. Mutation of two membrane-proximal Cys residues (Cys88/91) of STING mostly abolishes its palmitoylation, and the palmitoylation-deficient STING variant loses the ability to induce the STING-dependent signal.
Protein palmitoylation is implicated in the clustering of proteins into lipid rafts, specific lipid microdomains consisting of cholesterol and sphingomyelin (SM) at the plasma membrane (PM) (43,). Notably, in addition to PM, TGN has been assumed to have lipid rafts, given that (i) there is non-vesicular direct cholesterol transport from the ER to TGN (44,) and (ii) sphingomyelin synthase 1 (SMS1) localizes primarily at TGN (45,). Interference with lipid rafts by addition of C6-ceramide, knockout of SMS1 or inhibition of oxysterol-binding proteins (OSBPs) essential for the cholesterol transport to TGN, inhibits the STING signal (38, 46,). The essential role of cholesterol and SM in the STING signal is further supported by a cell-free assay to assess whether endogenous STING in microsomes can be phosphorylated by recombinant TBK1 (47). Treating microsomes with sphingomyelinase or methyl-β-cyclodextrin, an agent to extract cholesterol from membranes, suppresses the phosphorylation of STING by TBK1.
A very recent study with photoactivated localization microscopy (PALM) proves the STING clustering at TGN (~20 STING molecules per cluster) (46,). The clustering requires STING palmitoylation and the TGN lipid raft. Single-molecule imaging of TBK1 reveals that STING clustering enhances the residential time of TBK1 on membranes (Fig. 2A: data are adapted from (46) under a Creative Commons Attribution 4.0 International License). The advantage of STING clustering in TBK1 recruitment may be explained as follows: once TBK1 is detached from STING, there are other TBK1-free STING molecules around when STING clusters, in addition to the STING molecule that TBK1 originally binds to. Therefore, STING clustering increases the encounter probability of TBK1 and STING. Clustering of STING may be beneficial for TBK1 activation/phosphorylation because two TBK1 dimers need to be placed close each other for their autophosphorylation/autoactivation.
STING phosphorylation by TBK1 is essential for IRF3 recruitment (21). Intriguingly, treatment of cells with MRT67307 (TBK1 inhibitor) reduced the residential time of TBK1 on membranes. The result suggested that STING phosphorylation by TBK1 facilitates the activation/phosphorylation of IRF3 by stabilizing the active TBK1 on STING.
After the exit of STING from the TGN, STING reaches REs where STING becomes K63-ubiquitinated, which is essential for STING degradation by lysosomes (48). These results emphasize the activation and inactivation of STING are highly compartmentalized processes with distinct post-translational modifications.
NLRP3 pathway
NLR family pyrin domain-containing 3 (NLRP3) is a cytosolic PRR that responds to multiple PAMPs/DAMPs and forms a multicomponent complex (the NLRP3 inflammasome) that promotes inflammation through the production of proinflammatory cytokines IL-1β and IL-18 (49). The activation of the NLRP3 inflammasome involves a priming step and an activation step. In the priming step, lipopolysaccharides (LPSs) or other PAMPs are recognized by PRRs and activate NF-κB to induce expression of NLRP3, pro-IL-1β and pro-IL-18. In the activation step, various PAMPs or DAMPs, including the bacterial pore-forming toxin nigericin and extracellular ATP, let NLRP3 bind to apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and NIMA-related kinase 7 (NEK7), leading to the formation of the inflammasome complex. The NLRP3 inflammasome then recruits and activates caspase-1, which cleaves pro-IL-1b and pro-IL-18 to generate mature IL-1β and IL-18. The activated caspase-1 also triggers pyroptosis via the cleavage of gasdermin D.
Modification of PRRs with palmitoylation
| Protein | Palmitoylated cysteine residues | Palmitoyl transferases |
|---|---|---|
| STING | Cys88, Cys91 | Not determined |
| NLRP3 | Cys130, Cys419, Cys837, Cys838, Cys844, Cys958 | ZDHHC1, ZDHHC5, ZDHHC7, ZDHHC12, ZDHHC17 |
| NOD1 | Cys558, Cys567, Cys952 | ZDHHC5 |
| NOD2 | Cys395, Cys1033 | ZDHHC5 |
| MAVS | Cys46, Cys79, Cys508 | ZDHHC4, ZDHHC7, ZDHHC24 |
| Protein | Palmitoylated cysteine residues | Palmitoyl transferases |
|---|---|---|
| STING | Cys88, Cys91 | Not determined |
| NLRP3 | Cys130, Cys419, Cys837, Cys838, Cys844, Cys958 | ZDHHC1, ZDHHC5, ZDHHC7, ZDHHC12, ZDHHC17 |
| NOD1 | Cys558, Cys567, Cys952 | ZDHHC5 |
| NOD2 | Cys395, Cys1033 | ZDHHC5 |
| MAVS | Cys46, Cys79, Cys508 | ZDHHC4, ZDHHC7, ZDHHC24 |
Modification of PRRs with palmitoylation
| Protein | Palmitoylated cysteine residues | Palmitoyl transferases |
|---|---|---|
| STING | Cys88, Cys91 | Not determined |
| NLRP3 | Cys130, Cys419, Cys837, Cys838, Cys844, Cys958 | ZDHHC1, ZDHHC5, ZDHHC7, ZDHHC12, ZDHHC17 |
| NOD1 | Cys558, Cys567, Cys952 | ZDHHC5 |
| NOD2 | Cys395, Cys1033 | ZDHHC5 |
| MAVS | Cys46, Cys79, Cys508 | ZDHHC4, ZDHHC7, ZDHHC24 |
| Protein | Palmitoylated cysteine residues | Palmitoyl transferases |
|---|---|---|
| STING | Cys88, Cys91 | Not determined |
| NLRP3 | Cys130, Cys419, Cys837, Cys838, Cys844, Cys958 | ZDHHC1, ZDHHC5, ZDHHC7, ZDHHC12, ZDHHC17 |
| NOD1 | Cys558, Cys567, Cys952 | ZDHHC5 |
| NOD2 | Cys395, Cys1033 | ZDHHC5 |
| MAVS | Cys46, Cys79, Cys508 | ZDHHC4, ZDHHC7, ZDHHC24 |
NLRP3 is involved in many inflammatory and autoimmune diseases. Gain-of-function NLRP3 mutations (50–52,) are relevant with cryopyrin-associated periodic syndromes, a group of rare heritable autoinflammatory diseases that includes familial cold autoinflammatory syndrome; chronic infantile neurological, cutaneous and articular syndrome; and Muckle–Wells syndrome. Hyperactivation of the NLRP3 inflammasome is associated with neurodegenerative diseases, gout and diabetes (53).
Mechanism of NLRP3 inflammasome activation at TGN
NLRP3 is recruited to multiple small puncta from the cytosol upon nigericin treatment (54,). The small puncta are positive with TGN38, a resident membrane protein at TGN, indicating the puncta are formed from TGN. The deletion of four consecutive lysine (Lys) residues (residues 127–130) abolishes the NLRP3 puncta formation and activation of the downstream signal (54,). In addition to cholesterol and SM, phosphatidylinositol 4-phosphate (PtdIns4P) is enriched in the TGN membrane (55,). Targeting a PtdIns4P phosphatase Sac1 or Sac2 to TGN abolishes NLRP3 recruitment (54), indicating that the negatively charged headgroup of PtdIns4P interacts directly with the polybasic region (Lys127–Lys130) of NLRP3.
A number of recent studies show that NLRP3 is subjected to palmitoylation on multiple Cys residues, which influences the NLRP3 signal (Table I). Cys130 (56,), Cys419 (57,), Cys837 (58,), Cys838 (58,), Cys844 (59,) or Cys958 (60) is palmitoylated by zinc finger DHHC-type palmitoyl transferase (ZDHHC) 7, ZDHHC17, ZDHHC5, ZDHHC12 or ZDHHC1, respectively. Palmitoylation of Cys130 and Cys958 increases the affinity of NLRP3 to the TGN membrane, whereas the one of Cys419, Cys837 and Cys838 enhances NLRP3–NEK7 interaction. Palmitoylation of Cys844 is critical for NLRP3 degradation by lysosomes and thus for termination of the signal from NLRP3 inflammasome.
Innate Immune Signals from Endosomes
Cells survey the extracellular environment for signs of infection, and in doing so, they run the risk of being infected and killed by microbes. For this reason, cells internalize microbes by endocytosis and deliver them to endosomes/lysosomes (endolysosomes). The harsh acidic environment of endolysosomes with various hydrolases active in acidic pH, ranging from 4 to 5, facilitates degradation of the pathogens, leading to the release of PAMPs to the lumen of endolysosomes and to the cytosol through specific transporters. Therefore, placing PRRs to endolysosomes is quite practical, ensuring a rapid immune response against invading pathogens (2,). Transmembrane-type PRRs including several TLRs and cytosolic ones including the nucleotide-binding oligomerization domain (NOD) proteins, NOD1 and NOD2, trigger the innate immune signals from endolysosomes. STING also signals from endosomes. In this section, we review the NOD1/2 and STING signals from endosomes. Regarding the TLR signals, please refer to the reviews by Zhang et al. (61,) and by Ruysschaert et al. (62): they discuss the contribution of palmitoylation of TLRs and lipid microdomains in the TLR signal.
NOD1/NOD2 pathway
NOD1 and NOD2 are crucial for innate immune responses to certain bacterial infections (63, 64,). They are cytosolic proteins that respond to fragments of bacterial peptidoglycan and that initiate NF-κB-dependent and mitogen-activated protein kinase (MAPK)-dependent gene transcription. NOD1 detects γ-glutamyl-meso-diaminopimelic acid (iE-DAP), which is a dipeptide that is present in a peptidoglycan that is primarily found in Gram-negative bacteria but also in select groups of Gram-positive bacteria. By contrast, NOD2 detects muramyl dipeptide (MDP) that is ubiquitously present in bacterial peptidoglycan. Several peptide transporters, including PepT1, PepT2, SLC15A3 and SLC15A4, mediate the egress of iE-DAP and MDP to the cytosol (65–67).
NOD1 and NOD2 polymorphisms are associated with a number of autoinflammatory diseases including Crohn’s disease (CD), ulcerative colitis, Blau syndrome, Behcet’s syndrome, early-onset sarcoidosis (EOS) and atopic diseases (68–70). CD-associated NOD2 loss-of-function mutations result in reduced NF-κB activity upon stimulation with MDP, whereas EOS and Blau syndrome are associated with apparent NOD2 gain-of-function mutations.
Mechanism of NOD1/2 activation at endosomes
Although soluble in the cytosol, NOD1/2 associate with the PM and endosomes (67, 71, 72,). The truncation mutants that lose membrane localization are defective in the induction of the downstream signal. Treatment of cells by 2-BP, a pan-palmitoytlransferase inhibitor, reduces the membrane localization of NOD1/2 and inhibits the activation of NF-κB and MAPK (73,). Cys558/Cys567/Cys952 in NOD1 and Cys395/Cys1033 in NOD2 are subjected to palmitoylation. Importantly, palmitoylation-deficient mutants fail to localize to bacteria-containing endosomes and to induce the downstream signal (73,). ZDHHC5 primarily mediates the palmitoylation of NOD1/2 (73). The NOD1/2 recruitment to endosomes may be beneficial for the rapid detection of iE-DAP and MDP that are released through transporters to the cytosolic face of endosomes. The concentration of NOD1/2 on endosomal membrane may also facilitate binding and autophosphorylation of receptor-interacting serine/threonine-protein kinase 2, the critical downstream molecule of NOD1/2.
A number of CD-associated loss-of-function NOD2 mutant proteins (3020insC, Arg702Trp, Leu248Arg, Ala612Thr, Ala755Val and Arg1019stop) display ~70–90% reduction in palmitoylation levels. In contrast, the EOS-associated NOD2 gain-of-function mutant Cys495Tyr displays a 2-fold increase in palmitoylation compared with that of wild-type NOD2 (73). These data suggest the relevance of NOD2 palmitoylation with NOD2-related inflammatory diseases.
cGAS–STING pathway
As aforementioned, STING is a mobile protein. After stimulation, STING exits the ER and translocates to the TGN where STING activates TBK1 and IRF3. STING further translocates to REs and to lysosomes where STING is degraded by microautophagic process (48, 74, 75,). During the post-Golgi traffic of STING to lysosomes, STING induces the lipidation of microtubule-associated protein 1 light chain 3 (LC3) (37,). In canonical autophagy, LC3 is conjugated to the polar head group of phosphatidylethanolamine in double-membrane autophagosomes, where it plays a pivotal role in autophagosome formation. In contrast, LC3-positive membranes emerged through STING stimulation are single-membrane structures (76). The lipidation of LC3 on single-membrane structures is referred to as ‘conjugation of ATG8 (LC3) to single membranes’ (CASM).
Two recent studies provide new insights into the molecular mechanism underlying CASM triggered by STING in the Golgi and endosomes (77, 78,). These studies show that STING is a proton channel and that STING de-acidifies the lumen of the Golgi and perinuclear endosomes. A chemical STING agonist, compound 53, which binds to the STING transmembrane domain (77), inhibits STING-mediated proton efflux in vitro, STING-dependent organelle de-acidification and CASM. Thus, CASM appears to depend on proton efflux through the STING pore.
Just recently, it is shown that STING activates two transcription factors, transcription factor EB (TFEB) and transcription factor binding to IGHM enhancer 3 (TFE3), which are central to lysosomal biogenesis and activity. Intriguingly, the activation of TFEB and TFE3 requires CASM, but not the activation of TBK1 (79). The upregulation of lysosomal function by STING may contribute to the clearance of invading pathogens and/or to cellular resistance to STING-triggered inflammation.
Innate Immune Signals from Mitochondria
Mitochondria play a critical role in cellular energy production by generating ATP through oxidative phosphorylation, and are a critical source of building blocks for biosynthetic pathways, including nucleotide synthesis, fatty acid and cholesterol synthesis, amino acid synthesis and glucose and haem synthesis (80,). Besides these fundamental functions, mitochondria also serve as key modulators of innate immunity and inflammation. For example, macrophages and neutrophils produce and employ mitochondrial reactive oxygen species as direct antimicrobial agents (81,). Release of mitochondrial DNA (mtDNA) to the cytosol activates the cGAS–STING pathway, triggering the inflammatory signal (32, 33,). The innate immune signal against dsRNA virus is triggered on mitochondrial outer membrane and requires cytosolic RIG-I-like receptors (4,) and mitochondrial antiviral-signalling protein (MAVS) (82,), a single transmembrane protein also known as IFN-beta promoter stimulator 1 (IPS-1) (83,), caspase recruitment domain adaptor inducing IFN-beta (CARDIF) (84,) or virus-induced signaling adaptor (VISA) (85).
Mechanism of RIG-I–MAVS activation at mitochondria
RIG-I-like receptors are dsRNA sensors that localize in the cytosol. This protein family includes three members: RIG-I, melanoma differentiation-associated protein 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). RIG-I binds to short dsRNA with a 5′-triphosphate end, while MDA5 binds to longer dsRNA. LGP2 is widely believed to regulate RIG-I and MDA5.
The binding of RIG-I and MDA5 to dsRNA induces conformational changes that expose and multimerize their caspase activation and recruitment domains (CARDs), which allows homotypic CARD–CARD interactions with MAVS. As a result, MAVS forms cluster on mitochondrial outer membrane, which acts as a scaffold to activate TBK1 and IKKε. Activated TBK1 and IKKε then activate IRF3, interferon regulatory factor 7 (IRF7) and NF-κB, leading to the induction of the expression of Type I interferons and inflammatory genes.
It has become clear that the activities of RIG-I/MDA5–MAVS pathway are dynamically controlled by ubiquitination (please refer to the comprehensive review by Rehwinkel (86,)). For example, RIG-I undergoes Lys63-linked polyubiquitination at Lys172 by an E3 ligase TRIM25, which contributes to effective RIG-I oligomerization and signal transduction (87,). Unanchored Lys63-linked polyubiquitin chains also facilitate RIG-I oligomerization and signal transduction (88, 89,). MAVS undergoes Lys63-linked polyubiquitination at Lys10, Lys311 and Lys461 by an E3 ligase TRIM31, which promotes MAVS oligomerization and signal transduction (90,). MAVS also undergoes Lys27-linked polyubiquitination at Lys325 by an E3 ligase TRIM21, promoting the recruitment of TBK1 to MAVS (91).
Quite recently, several studies independently show the critical contribution of palmitoylation of MAVS to its activation. ZDHHC4, ZDHHC7 or ZDHHC24 mediates palmitoylation of Cys79 (92,), Cys508 (93,) or Cys46/Cys79 (94) of MAVS, respectively, which facilitates MAVS clustering and its downstream signals. As discussed in the cGAS–STING pathway, the clustering of MAVS may be beneficial for the activation of TBK1. In contrast to TGN where STING becomes clustered, mitochondrial outer membrane has not been reported to be enriched in raft lipids. How palmitoylation of MAVS promotes its clustering in membrane needs further investigation.
Intriguingly, a loss-of-function MAVS variant (Cys79Phe), is associated with reduced Type I interferon production in a subset of systemic lupus erythematosus (SLE) patients (95, 96). Therefore, MAVS mutations that affect its palmitoylation may contribute to the development of specific SLE subtypes.
Regulation of innate immune signals by protein geranylgeranylation
Mevalonate kinase deficiency (MKD) or hyper-IgD syndrome (HIDS) is a periodic fever syndrome that is caused by loss-of-function mutations in mevalonate kinase (MVK), which converts mevalonic acid into 5-phosphomevalonic acid. This pathway is responsible for producing cholesterol as well as non-sterol isoprene compounds including farnesyl-pyrophosphate and geranylgeranyl-pyrophosphate. Geranylgeranyl-pyrophosphate is a precursor to geranylgeranylated proteins, which include small GTPases such as ras homolog family member A (RHOA) and ras-related C3 botulinum toxin substrate 1 (RAC1). The geranylgeranyl group, like the palmitoyl group, is sufficiently hydrophobic to confer stable membrane binding to the modified proteins.
Geranylgeranylated RHOA activates serine–threonine kinases N1 and N2 that phosphorylate Ser208 and Ser242 on pyrin. The phosphorylated pyrin can bind a 14-3-3 protein that, when bound to pyrin, keeps pyrin inactive (97, 98,). Defects in geranylgeranylation, seen in MKD or HIDS, lead to RHOA inactivation and consequent pyrin inflammasome activation and IL-1β release (97).
Geranylgeranylation of RAC1 regulates the RIG-I-MAVS pathway. Protein geranylgeranylation and subsequent palmitoylation promote RAC1 translocation into mitochondria-associated ER membranes (MAM) upon viral infection. MAM-localized RAC1 limits interaction of MAVS with E3 ligase TRIM31 and hence inhibits MAVS ubiquitination, oligomerization and activation. In line with these observations, mice with myeloid deficiency of protein geranylgeranylation show improved survival upon influenza A virus infection (99).
Concluding Remarks
Recent studies have revealed that various innate immune signals, even the ones from cytosolic PRRs, are triggered on organelle membranes. The clustering of PRRs or their associated proteins such as STING and MAVS at membrane is a common mechanism that activates the downstream signals. Protein palmitoylation also emerges as a common mechanism that may facilitate the clustering of PRRs or their associated proteins. Besides protein palmitoylation, protein geranylgeranylation contributes to the innate immune signals by assuring the proper subcellular localization of PRR regulators such as RHOA and RAC1.
Based mostly on the results of band shift observed in western blot and puncta formation observed in fluorescence microscopy, STING has been proposed to act as a protein scaffold to recruit and activate TBK1 and IRF3. However, these methods cannot rule out the possibility that STING binds some other proteins to generate protein aggregates. In this regard, PALM observation may become a complementary approach, by which the clustering of the molecule of interest itself can directly be quantitated. Indeed, homo-clustering of STING in live cells is proved by PALM and shown to be formed specifically at TGN membranes (46).
The finding that STING needs palmitoylation for its activity (38,) paves the way to develop or identify STING-specific antagonists, such as C-176 (100,), C-178 (100,), H-151 (100,), nitro-unsaturated fatty acids (101,) and 4-hydroxynonenal (102,), which all covalently bind to Cys88 and/or Cys91 of STING and thus prevent STING palmitoylation. Among them, C-176 and C-178 are shown not to interfere with palmitoylation of calnexin and transferrin receptors, suggesting their specificity to STING Cys91 (100,). The administration of C-176 or H-151 attenuates pathological features of autoinflammatory disease in mice (100,). Given the critical contribution of palmitoylation of PRRs and their associated proteins to innate immune signalling and inflammation, protein palmitoylation may be a promising target in drug development to cure autoinflammatory and neurodegenerative diseases (103).
Acknowledgements
We apologize to colleagues whose works we could not cite owing to length restrictions.
Funding
This work was supported by JSPS KAKENHI Grant Number JP24H00548 (to T.T.), JST CREST (JPMJCR21E4) (T.T.), The Takeda Science Foundation (to T.T.), The Uehara Memorial Foundation (to T.T.), The Graduate school of Life Sciences Research Encouragement Award 2024 (to Y.K.) and JSPS KAKENHI Grant Number (25 K18455) (to Y.K.).
Conflict of Interest
None declared.

{kind=link}
{kind=link}