





Abstract
Staphylococcus aureus is a major human pathogen causing a vast array of infections with significant mortality. Its versatile physiology enables it to adapt to various environments. Specific physiological changes are thought to underlie the frequent failure of antimicrobial therapy despite susceptibility in standard microbiological assays. Bacteria capable of surviving high antibiotic concentrations despite having a genetically susceptible background are described as ‘antibiotic tolerant’. In this review, we put current knowledge on environmental triggers and molecular mechanisms of increased antibiotic survival of S. aureus into its clinical context. We discuss animal and clinical evidence of its significance and outline strategies to overcome infections with antibiotic-tolerant S. aureus.
Introduction
Staphylococcus aureus is one of the most versatile pathogens infecting humans. It harmlessly colonizes one-third of the human population in the nares or throat or on the skin,1 but can also lead to a vast array of potentially lethal invasive infections. S. aureus can infect virtually any tissue of the body such as skin, muscles, bones and joints, lungs, kidneys, heart or even the eyes or brain.2,3 Moreover, it readily grows on artificial surfaces of implanted foreign bodies such as orthopaedic devices, prosthetic heart valves or intravascular and urinary catheters. More than three decades ago, S. aureus was named the ‘persistent pathogen’4 due to its unusual recalcitrance towards antibiotic treatment. This has remained, to date, a major clinical challenge. S. aureus can persist for several days in the blood of up to one-third of patients despite correct antibiotic treatment;5–9 this is by far more common than with other bacteria.8,10 The associated mortality rate of 15%–50% leads to a calculated annual 2–10 deaths/100 000 population.11 Finally, current antibiotics have limited capacity to eradicate S. aureus in abscesses or in implant infections without the support of surgery.11–14 Physicians choose antibiotics according to a strain’s susceptibility measured as the MIC in the microbiology laboratory. When treatment nevertheless fails, this can have several clinical or pharmacological reasons (Table 1), many of which are preventable through optimized application of the antibiotics. However, evidence is emerging that the ability of S. aureus to become antibiotic tolerant represents a major microbiological reason for the high rate of treatment failure.
Reasons for treatment failure despite correct susceptibility-guided antibiotic therapy of the infecting S. aureus measured by the MIC
| Delayed initiation of antibiotic treatment |
| Low serum concentration of antibiotic |
| low oral bioavailability |
| long interval between dosing |
| augmented hepatic or renal clearance |
| dosage not adjusted to body weight |
| non-compliance of the patient |
| Insufficient antibiotic penetration to the site of infection |
| infections of niches such as bone or cerebrospinal fluid |
| intracellular infections |
| infection of necrotic tissue |
| Physicochemical inactivation of antibiotics at the site of infection |
| low pH |
| interaction with debris |
| interaction with biofilm matrix |
| Heteroresistant bacterial subpopulation |
| Bacterial tolerance to the applied antibiotic(s) |
| Delayed initiation of antibiotic treatment |
| Low serum concentration of antibiotic |
| low oral bioavailability |
| long interval between dosing |
| augmented hepatic or renal clearance |
| dosage not adjusted to body weight |
| non-compliance of the patient |
| Insufficient antibiotic penetration to the site of infection |
| infections of niches such as bone or cerebrospinal fluid |
| intracellular infections |
| infection of necrotic tissue |
| Physicochemical inactivation of antibiotics at the site of infection |
| low pH |
| interaction with debris |
| interaction with biofilm matrix |
| Heteroresistant bacterial subpopulation |
| Bacterial tolerance to the applied antibiotic(s) |
Reasons for treatment failure despite correct susceptibility-guided antibiotic therapy of the infecting S. aureus measured by the MIC
| Delayed initiation of antibiotic treatment |
| Low serum concentration of antibiotic |
| low oral bioavailability |
| long interval between dosing |
| augmented hepatic or renal clearance |
| dosage not adjusted to body weight |
| non-compliance of the patient |
| Insufficient antibiotic penetration to the site of infection |
| infections of niches such as bone or cerebrospinal fluid |
| intracellular infections |
| infection of necrotic tissue |
| Physicochemical inactivation of antibiotics at the site of infection |
| low pH |
| interaction with debris |
| interaction with biofilm matrix |
| Heteroresistant bacterial subpopulation |
| Bacterial tolerance to the applied antibiotic(s) |
| Delayed initiation of antibiotic treatment |
| Low serum concentration of antibiotic |
| low oral bioavailability |
| long interval between dosing |
| augmented hepatic or renal clearance |
| dosage not adjusted to body weight |
| non-compliance of the patient |
| Insufficient antibiotic penetration to the site of infection |
| infections of niches such as bone or cerebrospinal fluid |
| intracellular infections |
| infection of necrotic tissue |
| Physicochemical inactivation of antibiotics at the site of infection |
| low pH |
| interaction with debris |
| interaction with biofilm matrix |
| Heteroresistant bacterial subpopulation |
| Bacterial tolerance to the applied antibiotic(s) |
Earlier studies arbitrarily defined antibiotic tolerance as in vitro measurement of a ratio of MBC to MIC of ≥32. This definition, however, does not encompass the various populations and mechanisms of tolerance and its measurement is fraught with considerable variability.15,16 Therefore, more recently, leading experts have provided clarification about the terminology defining the effect of antibiotics on bacterial growth and survival (for details, see Balaban et al.17). Accordingly, tolerance describes the bacterial ability to survive usually lethal concentrations of a bactericidal antibiotic without change of the MIC.17,18 This can classically be visualized and measured in a slower killing rate in in vitro time–kill curves. However, so far, no new classification or measurable cut-offs have been introduced that may be applicable to clinical practice. From a population-level perspective, the term tolerance applies if an entire bacterial population shows increased survival whereas the term persistence is used if only a small fraction of a population (named persisters) exhibit a tolerant phenotype. Although the clinical relevance of this differentiation is not clear, we will use the term ‘increased antibiotic survival’19,20 to encompass both tolerance and persistence.
In this review, we put mechanisms and triggers of increased antibiotic survival into the context of various types of infection and life forms of S. aureus. By taking a broad clinical perspective, we extend the scope of previous reviews on tolerance and persistence in general17,18,21 as well as on S. aureus in particular.22,23 With the goal of translating basic research to clinical practice, we provide an overview of highly discussed as well as rather neglected studies of in vitro, in vivo and clinical aspects of antibiotic-tolerant S. aureus. We discuss approaches of how to overcome infections with antibiotic-tolerant S. aureus and identify future research questions and open gaps in knowledge.
Environmental triggers of increased antibiotic survival of S. aureus
Development of tolerant phenotypes can be interpreted as a survival strategy of a bacterial population in quickly changing and potentially toxic environments.24 Tolerant phenotypes can develop due to genetic traits,25–29 can evolve stochastically30,31 or can be triggered by specific environmental factors18 in both Gram-positive and Gram-negative bacteria. The majority of studies on increased antibiotic survival have focused on Gram-negative bacteria.32 Basic mechanisms of becoming tolerant are probably common for both Gram-positive and Gram-negative bacteria.20,21,33,34 However, not only do the identified molecular targets leading to tolerant phenotypes differ but S. aureus also stands out through the variety of evolved life forms causing infections associated with characteristics of increased antibiotic survival. The scientific pioneer Bigger had already noticed by 1944 that growth-inhibiting conditions induce increased antibiotic survival in S. aureus.35 Renewed research on the topic has now confirmed these observations and found several environmental factors such as low oxygen level,36 low pH37 or low amount of nutrients38 as responsible for triggering tolerant phenotypes (Figure 1).
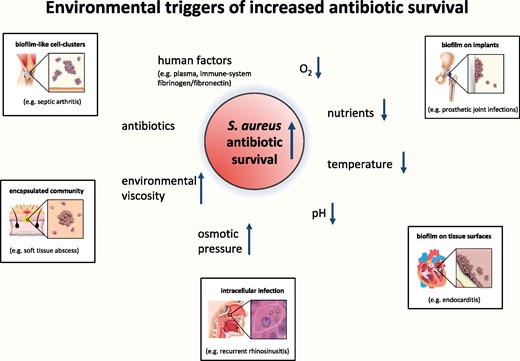
Examples of environmental triggers shown to increase S. aureus antibiotic survival. The insets show examples of situations associated with these triggers and with increased antibiotic survival. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Most mechanistic in vitro studies have focused on the free-floating planktonic single-cell life form of S. aureus in liquid culture. High planktonic population density is typically strongly associated with development of increased antibiotic survival. This is due to the exhaustion of growth-sustaining factors and accumulation of metabolic waste products or bacterial signalling molecules.39 However, it is not known which role such tolerant planktonic forms of S. aureus play in the clinical context. Single cells or small aggregates of S. aureus dividing in the interstitial space are most likely responsible for several acute infections such as pneumonia or skin and soft tissue infections. Short antibiotic regimens generally cure such infections12 and recurrence is uncommon. Hence, even if tolerant planktonic subpopulations survive the exposure to antibiotic treatment in these acute infections, they presumably do not withstand killing by the immune system in the unprotected environment of the interstitial space.
S. aureus increases its survival in vivo through the formation of multicellular conglomerates. Indeed, aggregating into cell clusters seems to be an intrinsic growth pattern of S. aureus,41 a phenotype constantly observed since its first discovery in the 1880s and serving as inspiration for its microbiological name (Greek: σταφνλοσ meaning grape cluster).42 The tendency to cluster dramatically increases in the presence of fibrinogen/fibronectin, which is abundant in synovial fluid of joints (e.g. in septic arthritis) or in intravascular fibrin and platelet clots (e.g. in endocarditis).43 These cell clusters can survive antibiotic exposures that would usually be bactericidal even without attachment to a surface,43,44 requiring prolonged antibiotic treatment to cure the respective infections. Inside a tissue, staphylococcal cell clusters can evolve into abscesses by digesting and transforming the surrounding tissue into a protective capsule of fibrin and necrotic debris.45 Antibiotics in the absence of surgery generally fail to eradicate S. aureus in abscesses.46 This has been attributed to reduced antibiotic penetration and physicochemical or enzymatic inactivation.47 However, since several antibiotics can achieve adequate concentrations inside an abscess,47 tolerant S. aureus in the acidic and low-oxygen environment of an abscess may play an important role in their recalcitrance against antibiotics. Concordantly, tolerant subpopulations have been isolated from abscesses manifesting as non-stable small colony variants (SCVs) with extended lag periods before regrowth.48
S. aureus can further increase its survival by forming multicellular biofilms encased by a self-produced matrix on organic or inorganic surfaces.49,50 Prototypical examples of such biofilm infections include endocarditis or implant-related infections.51,52 These are clinically relevant due to their ability to survive more than 100–1000-fold higher antibiotic concentrations compared with planktonic S. aureus.53–56 In such cases, increased survival cannot be explained by lack of biofilm penetration since even large antibiotics such as vancomycin,57,58 daptomycin59 or telavancin60 easily penetrate S. aureus biofilms. Gradients of oxygen and nutrients inside staphylococcal biofilms lead to spatial patterns of diverse physiological states with different levels of cellular respiration, DNA replication and protein synthesis61 and therefore potentially diverse levels of increased survival. Non-growing, tolerant fractions of biofilm bacteria are thought to be responsible for the prolonged survival of biofilms during exposure to bactericidal antibiotics requiring extended treatment and responsible for relapses once treatment ends.62 The number of tolerant cells in a biofilm increases with age and cell density,63,64 supporting the clinical concept that after a certain time of biofilm maturation, prosthetic implants need to be surgically removed to achieve cure.65,66 Moreover, biofilm-derived detached cell clusters exhibit a higher level of antibiotic survival than purely single-cell planktonic bacteria67,68 and may play a major role in perpetuating and spreading an infection.
With its versatile physiology, S. aureus can also survive by invading professional and non-professional phagocytic cells.69–73 This may lead to treatment failure74 and even the spread of infection through haematogenous transport inside host cells.75 Intracellular S. aureus has been documented in skin and soft tissue, bone and upper airway infections72,76,77 and is probably involved in an even wider spectrum of chronic and recurrent infections.58,75,78 The intracellular compartment is a protected niche since it shields S. aureus from the immune system and from the high extracellular antibiotic drug concentrations.75,79,80 However, the specific metabolic state of intracellular S. aureus further reduces the lethality of cell-penetrating antibiotics.81,82,S. aureus from intracellular locations can manifest on the laboratory growth plate as an SCV due to its slowed or delayed growth and hence eponymous reduced colony size.83–85 Compared with normal phenotypes, SCVs not only show reduced metabolism but also reduced expression of regulator and virulence factors.84 SCVs from clinical samples often present as so-called ‘non-stable’ or ‘dynamic’ SCVs, since they rapidly revert to normal colony sizes and metabolism after reculture on rich media.86,87 SCVs have been found to be tolerant to aminoglycosides and cell-wall active antibiotics88,89 and are associated with recalcitrant and relapsing infections.90,91 Nutrient starvation, oxidative stress and low pH are characteristics of the encountered intracellular milieu and all induce the formation of SCVs.85,92,93 Concordantly, alkalinization of the phagolysosome by the antimalarial drug chloroquine reduced survival in antibiotic treatment of intracellular S. aureus.93 Finally, exposure to host defence mechanisms can stimulate SCVs and other tolerant phenotypes in S. aureus during an infection.85,94
Taken together, S. aureus has developed various life forms to persist in different niches of the human body and thereby encounters or produces environmental triggers of increased survival, rendering an antibiotic treatment ineffective.
In vitro evidence for mechanisms of increased antibiotic survival of S. aureus
Understanding the mechanisms of antibiotic survival is essential in order to improve therapeutic strategies against S. aureus infections. Reduced cellular metabolism leading to decreased growth rate or dormancy seems to be a shared feature of most tolerant bacteria.18,30,95,96 This may not just be a passive state but may involve active metabolic changes focusing on adaptation to a hostile environment.24,97–99 Various molecular targets have been found to change the level of antibiotic survival in S. aureus (Table 2). Individual targets may only induce survival to some antibiotics tested whereas other antibiotics may stay effective (Table 2). However, separate studies have found some conflicting results, which likely reflect the complexity of mechanisms and the sensitivity of its regulation. Results may differ greatly depending on the genetic background of the individual strains and on the experimental setup.17 The growth phase strongly influences the level of antibiotic survival since slow- or non-growing S. aureus from high-density stationary-phase cultures are considerably more tolerant to antibiotics than when in exponential growth.30,37,100–104 A low intracellular level of ATP may represent a mechanism of antibiotic survival for S. aureus30 as seen in other bacteria.105 The reduced cellular fuel in the form of ATP slows down or halts cellular processes targeted by antibiotics and thereby protects the bacterial cell from their bactericidal action. Nevertheless, low ATP levels seem not to be a prerequisite, since increased antibiotic survival has been found in S. aureus with normal ATP levels.106,107 Moreover, antibiotic survival may not always be associated with decreased growth according to a study that found a higher rate of cells tolerant to vancomycin in a more rapidly growing WT S. aureus compared with its mutant with defects in purine synthesis.108
Targets and mechanisms associated with increased antibiotic survival of S. aureus
| Target associated with increased antibiotic survival and main known function | Presumable or associated mechanism(s) of increased antibiotic survival | Change of antibiotic survival shown for these antibiotics | No change of antibiotic survival found for these antibiotics | Comments |
|---|---|---|---|---|
| Increased antibiotic survival if amount or function of target decreased | ||||
| dysfunction of agr
|
| ||
| low level of ATP decreases activity of antibiotic targets and downstream bactericidal mechanism | not reported |
| |
| inactivation of GdpP
| |||
| dysfunction of ileS
|
| not reported | |
| decreased activity of hydrolases
| not reported |
| |
| point mutation in pitA leads to
| not reported | ||
| inactivation by mutations of selected TCA enzymes
|
| ||
| deletion mutation of dsp1 or asp1
| not reported | ||
| Increased antibiotic survival if amount or function of target increased | ||||
| ppGpp-activated stringent response
|
| ||
| increased amount of saturated fatty acid
|
| not reported |
|
| Reduced antibiotic survival if amount or function of target decreased | ||||
| lack of Clp
|
| ||
| presumable mechanism not known |
| ||
| presumable mechanism not known | planktonic exponential232
| planktonic exponential232
|
|
planktonic stationary232
| planktonic stationary232
| |||
biofilm232
| biofilm232
| |||
| lack of superoxide dismutase
|
| not reported | |
|
|
| not reported |
|
| Target associated with increased antibiotic survival and main known function | Presumable or associated mechanism(s) of increased antibiotic survival | Change of antibiotic survival shown for these antibiotics | No change of antibiotic survival found for these antibiotics | Comments |
|---|---|---|---|---|
| Increased antibiotic survival if amount or function of target decreased | ||||
| dysfunction of agr
|
| ||
| low level of ATP decreases activity of antibiotic targets and downstream bactericidal mechanism | not reported |
| |
| inactivation of GdpP
| |||
| dysfunction of ileS
|
| not reported | |
| decreased activity of hydrolases
| not reported |
| |
| point mutation in pitA leads to
| not reported | ||
| inactivation by mutations of selected TCA enzymes
|
| ||
| deletion mutation of dsp1 or asp1
| not reported | ||
| Increased antibiotic survival if amount or function of target increased | ||||
| ppGpp-activated stringent response
|
| ||
| increased amount of saturated fatty acid
|
| not reported |
|
| Reduced antibiotic survival if amount or function of target decreased | ||||
| lack of Clp
|
| ||
| presumable mechanism not known |
| ||
| presumable mechanism not known | planktonic exponential232
| planktonic exponential232
|
|
planktonic stationary232
| planktonic stationary232
| |||
biofilm232
| biofilm232
| |||
| lack of superoxide dismutase
|
| not reported | |
|
|
| not reported |
|
ROS, reactive oxygen species.
Targets and mechanisms associated with increased antibiotic survival of S. aureus
| Target associated with increased antibiotic survival and main known function | Presumable or associated mechanism(s) of increased antibiotic survival | Change of antibiotic survival shown for these antibiotics | No change of antibiotic survival found for these antibiotics | Comments |
|---|---|---|---|---|
| Increased antibiotic survival if amount or function of target decreased | ||||
| dysfunction of agr
|
| ||
| low level of ATP decreases activity of antibiotic targets and downstream bactericidal mechanism | not reported |
| |
| inactivation of GdpP
| |||
| dysfunction of ileS
|
| not reported | |
| decreased activity of hydrolases
| not reported |
| |
| point mutation in pitA leads to
| not reported | ||
| inactivation by mutations of selected TCA enzymes
|
| ||
| deletion mutation of dsp1 or asp1
| not reported | ||
| Increased antibiotic survival if amount or function of target increased | ||||
| ppGpp-activated stringent response
|
| ||
| increased amount of saturated fatty acid
|
| not reported |
|
| Reduced antibiotic survival if amount or function of target decreased | ||||
| lack of Clp
|
| ||
| presumable mechanism not known |
| ||
| presumable mechanism not known | planktonic exponential232
| planktonic exponential232
|
|
planktonic stationary232
| planktonic stationary232
| |||
biofilm232
| biofilm232
| |||
| lack of superoxide dismutase
|
| not reported | |
|
|
| not reported |
|
| Target associated with increased antibiotic survival and main known function | Presumable or associated mechanism(s) of increased antibiotic survival | Change of antibiotic survival shown for these antibiotics | No change of antibiotic survival found for these antibiotics | Comments |
|---|---|---|---|---|
| Increased antibiotic survival if amount or function of target decreased | ||||
| dysfunction of agr
|
| ||
| low level of ATP decreases activity of antibiotic targets and downstream bactericidal mechanism | not reported |
| |
| inactivation of GdpP
| |||
| dysfunction of ileS
|
| not reported | |
| decreased activity of hydrolases
| not reported |
| |
| point mutation in pitA leads to
| not reported | ||
| inactivation by mutations of selected TCA enzymes
|
| ||
| deletion mutation of dsp1 or asp1
| not reported | ||
| Increased antibiotic survival if amount or function of target increased | ||||
| ppGpp-activated stringent response
|
| ||
| increased amount of saturated fatty acid
|
| not reported |
|
| Reduced antibiotic survival if amount or function of target decreased | ||||
| lack of Clp
|
| ||
| presumable mechanism not known |
| ||
| presumable mechanism not known | planktonic exponential232
| planktonic exponential232
|
|
planktonic stationary232
| planktonic stationary232
| |||
biofilm232
| biofilm232
| |||
| lack of superoxide dismutase
|
| not reported | |
|
|
| not reported |
|
ROS, reactive oxygen species.
Deletion of the Clp proteolytic system [involved in degradation of several antitoxins in S. aureus toxin/antitoxin systems109 and in regulation of the stress response110,111 as well as the tricarboxylic acid (TCA) cycle112] was associated with increased rates of tolerant S. aureus.113 Similarly, deletion mutants of the S. aureus toxin/antitoxin system mazF/mazE exhibited decreased antibiotic survival after exposure to β-lactam antibiotics or vancomycin in planktonic as well as biofilm growth.114,115 It should be cautioned that this effect on antibiotic survival could not be confirmed after simultaneously knocking out three major toxin–antitoxin systems including MazF/MazE.30 Nevertheless, dysregulation of the MazF/MazE system can phenotypically lead to growth inhibition116 and SCVs85 as an indirect clue to its association with antibiotic survival in S. aureus. Further, several studies found increased antibiotic survival of S. aureus associated with activation of the guanosine tetraphosphate ppGpp-dependent stringent response and vice versa.117–119 This may not be true for all strains or other experimental setups according to one conflicting study.30 Nonetheless, parallels to phenotypes of tolerant life forms again exist since higher levels of ppGpp have been associated with SCVs120 and intraphagosomal survival.121 More than 40 years ago, the lack of autolysis was found to lead to increased survival with β-lactam antibiotics in S. aureus.122 More recent studies were able to associate deficiencies in CidABC and LrgA, both regulators of extracellular murein hydrolases, to increased survival through lack of autolysis.123,124 Concordantly, WGS associated mutation in the murein hydrolase atl with in vitro-generated phenotypes tolerant to oxacillin.125 Addition of glucose increased the activity of CidABC and LrgAB, thereby simultaneously decreasing the level of antibiotic survival.126
Other studies have specifically investigated mechanisms of increased survival of S. aureus with daptomycin, a membrane-targeting bactericidal antibiotic known for its growth-independent killing capacity.101 Changes in the membrane-lipid composition in the stationary phase increased antibiotic survival to daptomycin treatment. This observation could be influenced by the lipids available in the growth medium.104 Similarly, up-regulation of the dlt operon, encoding proteins responsible for d-alanylation of teichoic acids in the Gram-positive cell wall, increased daptomycin-specific antibiotic survival.127,128 However, changes in available glucose can diminish the rate of daptomycin-tolerant S. aureus.129 Recently, a genome-wide analysis identified several new genomic regions associated with daptomycin-tolerant S. aureus, again a strong hint for the plasticity and versatility of mechanisms of antibiotic survival in S. aureus.29
Studies have found a higher level of antibiotic survival in mutants with defects in the quorum sensing accessory gene regulator (agr).107,130 Dysfunctional agr may promote tolerant phenotypes through increased biofilm formation131 or through excreted factors interfering extracellularly with antibiotics such as gentamicin130 or daptomycin.132 Such increased antibiotic survival is supported by clinical data linking agr mutants to persistent infections.133–136
Antibiotics themselves may trigger antibiotic survival by ‘toxifying’ the environment of S. aureus. Indeed, evidence suggests that antibiotics can not only induce biofilm137 and SCV85,138 formation, but also trigger other mechanisms of antibiotic survival.139,140 In the case of S. aureus, repeated in vitro exposure to antibiotics can lead to increased antibiotic survival, as shown for β-lactams,25,118,141 aminoglycosides,142 vancomycin118 or daptomycin.127 For vancomycin, the proposed mechanisms include the induction of the ppGpp-dependent stringent response118 and direct blocking of the access of murein hydrolases to their cell-wall substrates.134,143 Moreover, pre-treatment with an antibiotic can result in increased survival not only for the applied antibiotic but also for other antibiotic classes.144–146 This cross-effect can vary greatly depending on which antibiotics are chosen97,103,147,148 and even which sequence of antibiotics is selected.144 It stretches from full cross-effect (as shown for tobramycin followed by ciprofloxacin) to no cross-effect (as shown for tobramycin and daptomycin).144 Finally, the widely used topical antimicrobial triclosan can increase antibiotic survival up to 10 000-fold, a mechanism again requiring ppGpp synthesis.149
Taken together, multiple mechanisms may mediate increased antibiotic survival of S. aureus. It remains to be seen whether they act in parallel or as redundant or upstream/downstream pathways. Nevertheless, the selective effect on individual antibiotics is a strong clue that different mechanisms of antibiotic survival are present. Clearly, a better understanding of the mechanisms of cellular death mediated by individual antibiotics should help decipher the various mechanisms underlying antibiotic survival.
In vivo experimental evidence for increased antibiotic survival of S. aureus
Although knowledge about in vitro phenomena of increased antibiotic survival is extensive, how this translates in vivo has not been ascertained. Moreover, there is a paucity of in vivo data on the outcome of infections with tolerant S. aureus since most available studies were performed more than 20 years ago. An early animal study by Chuard et al.150 gave proof of the impact of the in vivo environment on antibiotic survival. Measuring the S. aureus MIC and MBC of various antibiotics before and after a 6 week untreated foreign-body infection revealed such a drastic rise of the MBC that even the highest antibiotic concentrations could no longer kill the recovered S. aureus, notably without changes to the MIC. Various other animal studies have shown both the failure of antibiotics to eradicate in vivo infections, possibly due to tolerant phenotypes,55,151,152 and the increased in vivo survival of S. aureus already determined in vitro to be tolerant.108,153,154 However, not all studies found a correlation of in vitro with in vivo antibiotic survival.155,156 In summary, in vivo studies show the relevance of the host environment on antibiotic survival but also reveal the difficulty of translating in vitro measurements into in vivo differences of outcome.
Evidence of the clinical impact of increased antibiotic survival of S. aureus
Although bedside experience and the growing in vitro evidence argue for a major role of antibiotic-tolerant phenotypes of S. aureus in the clinical setting, the number of studies analysing antibiotic survival of S. aureus from patients is limited. The awareness of the clinical relevance of the antibiotic killing capacity, however, is not new and earlier studies have analysed the correlation between in vitro antibiotic survival and clinical outcome (Table 3). The MBC/MIC ratio, the most commonly used clinical measurement of tolerance, is fraught with considerable variability.15,16 Nevertheless, up to now it has been the best available marker of its clinical relevance. Most studies focus on vancomycin and describe a rate of tolerant S. aureus from 6%–43% in clinical strains;157–164 however, up to 63% oxacillin-tolerant S. aureus have been reported in patients with endocarditis.165 Single case reports describe treatment failures with tolerant strains.166–168 Larger series present a wide spectrum of clinical outcomes associated with ‘in vitro tolerant’ S. aureus. It ranges from no clinical difference165 to longer duration of fever,169 longer duration of bacteraemia170,171 and increased treatment failure in composite endpoints172–175 to an increased mortality rate.176–178 The relevance and clinical impact of increased antibiotic survival may depend on the site of infection: one study showed, for example, a worse outcome with tolerant strains only in bacteraemic patients with endocarditis but not in non-endocarditis patients.177 Most studies focus on increased survival with vancomycin, which may be a marker for treatment failure even when no vancomycin is used in the treatment.175 Remarkably, Miyazaki et al.178 were able to demonstrate a correlation of the level of bacterial glycopeptide survival (measured as reduction of cfu in kill curves) with the probability of survival of patients with MRSA bacteraemia. In conclusion, several studies over the last few decades found a correlation between increased antibiotic survival and clinical failure, but the differences in patient population and the lack of standardized methods of measuring antibiotic survival make a comparison between them difficult. Hence, there is an urgent clinical need to reliably measure and understand the dynamics and rate of increased antibiotic survival of S. aureus in patients.179
Clinical studies analysing the outcome of patients infected with tolerant S. aureus
| Year | Study type | Included patients | No. of patients | Tolerance measurement | Antibiotic tested | Outcome/observation if tolerant | Ref |
|---|---|---|---|---|---|---|---|
| 1977 | case report | pneumonia | 1 | MBC vs MIC | penicillin, methicillin | Several recurrences with tolerant S. aureus | 163 |
| 1978 | case report | endocarditis/ bacteraemia | 2 | MBC vs MIC | vancomycin | Treatment failure under vancomycin monotherapy | 168 |
| 1979 | retrospective case series | endocarditis and soft tissue abscess | 20 | MIC and MBC | various | Higher mortality if receiving non-bactericidal antibiotics according to MBC | 176 |
| 1980 | prospective cohort | endocarditis and SAB | 104 | MBC/MIC ≥16 | various |
| 177 |
| 1982 | retrospective cohort | SAB | 20 | MBC/MIC ≥32 | vancomycin | More treatment failure | 172 |
| 1982 | case report | pneumonia | 1 | MBC/MIC ≥32 | oxacillin | Initial treatment failure with oxacillin | 164 |
| 1986 | prospective cohort | SAB | 84 | MBC/MIC ≥32 | nafcillin | Longer fever with tolerant strains (but all cured) | 169 |
| 1987 | prospective cohort | osteomyelitis | 27 | serum bactericidal titre (trough <1:2) | various | More therapeutic failure if trough bactericidal titre <1:2 | 173 |
| 2004 | prospective cohort | SAB | 30 | killing (<4.71, 4. 71–6.26, >6.27 log cfu/72 h) | vancomycin | Lower clinical success rate if decreased killing | 174 |
| 2006 | retrospective cohort | SAB | 3 | MBC/MIC ≥32 | vancomycin | Persistent bacteraemia >72 h | 167 |
| 2007 | prospective cohort | SAB | 34 | killing (>/<2.5 log cfu/24 h) | vancomycin | Longer duration of bacteraemia (<6.5 days vs >10.5 days) | 168 |
| 2011 | retrospective cohort | endocarditis | 62 | MBC/MIC ≥32 |
|
| 165 |
| 2011 | retrospective cohort | SAB | 66 | killing (log cfu/72 h) | vancomycin | Quantitative correlation between probability of survival and amount of killing | 178 |
| 2017 | retrospective cohort | SAB | 225 | MBC/MIC ≥32 | vancomycin | More clinical failure with vancomycin tolerance | 175 |
| Year | Study type | Included patients | No. of patients | Tolerance measurement | Antibiotic tested | Outcome/observation if tolerant | Ref |
|---|---|---|---|---|---|---|---|
| 1977 | case report | pneumonia | 1 | MBC vs MIC | penicillin, methicillin | Several recurrences with tolerant S. aureus | 163 |
| 1978 | case report | endocarditis/ bacteraemia | 2 | MBC vs MIC | vancomycin | Treatment failure under vancomycin monotherapy | 168 |
| 1979 | retrospective case series | endocarditis and soft tissue abscess | 20 | MIC and MBC | various | Higher mortality if receiving non-bactericidal antibiotics according to MBC | 176 |
| 1980 | prospective cohort | endocarditis and SAB | 104 | MBC/MIC ≥16 | various |
| 177 |
| 1982 | retrospective cohort | SAB | 20 | MBC/MIC ≥32 | vancomycin | More treatment failure | 172 |
| 1982 | case report | pneumonia | 1 | MBC/MIC ≥32 | oxacillin | Initial treatment failure with oxacillin | 164 |
| 1986 | prospective cohort | SAB | 84 | MBC/MIC ≥32 | nafcillin | Longer fever with tolerant strains (but all cured) | 169 |
| 1987 | prospective cohort | osteomyelitis | 27 | serum bactericidal titre (trough <1:2) | various | More therapeutic failure if trough bactericidal titre <1:2 | 173 |
| 2004 | prospective cohort | SAB | 30 | killing (<4.71, 4. 71–6.26, >6.27 log cfu/72 h) | vancomycin | Lower clinical success rate if decreased killing | 174 |
| 2006 | retrospective cohort | SAB | 3 | MBC/MIC ≥32 | vancomycin | Persistent bacteraemia >72 h | 167 |
| 2007 | prospective cohort | SAB | 34 | killing (>/<2.5 log cfu/24 h) | vancomycin | Longer duration of bacteraemia (<6.5 days vs >10.5 days) | 168 |
| 2011 | retrospective cohort | endocarditis | 62 | MBC/MIC ≥32 |
|
| 165 |
| 2011 | retrospective cohort | SAB | 66 | killing (log cfu/72 h) | vancomycin | Quantitative correlation between probability of survival and amount of killing | 178 |
| 2017 | retrospective cohort | SAB | 225 | MBC/MIC ≥32 | vancomycin | More clinical failure with vancomycin tolerance | 175 |
SAB, S. aureus bacteraemia.
Clinical studies analysing the outcome of patients infected with tolerant S. aureus
| Year | Study type | Included patients | No. of patients | Tolerance measurement | Antibiotic tested | Outcome/observation if tolerant | Ref |
|---|---|---|---|---|---|---|---|
| 1977 | case report | pneumonia | 1 | MBC vs MIC | penicillin, methicillin | Several recurrences with tolerant S. aureus | 163 |
| 1978 | case report | endocarditis/ bacteraemia | 2 | MBC vs MIC | vancomycin | Treatment failure under vancomycin monotherapy | 168 |
| 1979 | retrospective case series | endocarditis and soft tissue abscess | 20 | MIC and MBC | various | Higher mortality if receiving non-bactericidal antibiotics according to MBC | 176 |
| 1980 | prospective cohort | endocarditis and SAB | 104 | MBC/MIC ≥16 | various |
| 177 |
| 1982 | retrospective cohort | SAB | 20 | MBC/MIC ≥32 | vancomycin | More treatment failure | 172 |
| 1982 | case report | pneumonia | 1 | MBC/MIC ≥32 | oxacillin | Initial treatment failure with oxacillin | 164 |
| 1986 | prospective cohort | SAB | 84 | MBC/MIC ≥32 | nafcillin | Longer fever with tolerant strains (but all cured) | 169 |
| 1987 | prospective cohort | osteomyelitis | 27 | serum bactericidal titre (trough <1:2) | various | More therapeutic failure if trough bactericidal titre <1:2 | 173 |
| 2004 | prospective cohort | SAB | 30 | killing (<4.71, 4. 71–6.26, >6.27 log cfu/72 h) | vancomycin | Lower clinical success rate if decreased killing | 174 |
| 2006 | retrospective cohort | SAB | 3 | MBC/MIC ≥32 | vancomycin | Persistent bacteraemia >72 h | 167 |
| 2007 | prospective cohort | SAB | 34 | killing (>/<2.5 log cfu/24 h) | vancomycin | Longer duration of bacteraemia (<6.5 days vs >10.5 days) | 168 |
| 2011 | retrospective cohort | endocarditis | 62 | MBC/MIC ≥32 |
|
| 165 |
| 2011 | retrospective cohort | SAB | 66 | killing (log cfu/72 h) | vancomycin | Quantitative correlation between probability of survival and amount of killing | 178 |
| 2017 | retrospective cohort | SAB | 225 | MBC/MIC ≥32 | vancomycin | More clinical failure with vancomycin tolerance | 175 |
| Year | Study type | Included patients | No. of patients | Tolerance measurement | Antibiotic tested | Outcome/observation if tolerant | Ref |
|---|---|---|---|---|---|---|---|
| 1977 | case report | pneumonia | 1 | MBC vs MIC | penicillin, methicillin | Several recurrences with tolerant S. aureus | 163 |
| 1978 | case report | endocarditis/ bacteraemia | 2 | MBC vs MIC | vancomycin | Treatment failure under vancomycin monotherapy | 168 |
| 1979 | retrospective case series | endocarditis and soft tissue abscess | 20 | MIC and MBC | various | Higher mortality if receiving non-bactericidal antibiotics according to MBC | 176 |
| 1980 | prospective cohort | endocarditis and SAB | 104 | MBC/MIC ≥16 | various |
| 177 |
| 1982 | retrospective cohort | SAB | 20 | MBC/MIC ≥32 | vancomycin | More treatment failure | 172 |
| 1982 | case report | pneumonia | 1 | MBC/MIC ≥32 | oxacillin | Initial treatment failure with oxacillin | 164 |
| 1986 | prospective cohort | SAB | 84 | MBC/MIC ≥32 | nafcillin | Longer fever with tolerant strains (but all cured) | 169 |
| 1987 | prospective cohort | osteomyelitis | 27 | serum bactericidal titre (trough <1:2) | various | More therapeutic failure if trough bactericidal titre <1:2 | 173 |
| 2004 | prospective cohort | SAB | 30 | killing (<4.71, 4. 71–6.26, >6.27 log cfu/72 h) | vancomycin | Lower clinical success rate if decreased killing | 174 |
| 2006 | retrospective cohort | SAB | 3 | MBC/MIC ≥32 | vancomycin | Persistent bacteraemia >72 h | 167 |
| 2007 | prospective cohort | SAB | 34 | killing (>/<2.5 log cfu/24 h) | vancomycin | Longer duration of bacteraemia (<6.5 days vs >10.5 days) | 168 |
| 2011 | retrospective cohort | endocarditis | 62 | MBC/MIC ≥32 |
|
| 165 |
| 2011 | retrospective cohort | SAB | 66 | killing (log cfu/72 h) | vancomycin | Quantitative correlation between probability of survival and amount of killing | 178 |
| 2017 | retrospective cohort | SAB | 225 | MBC/MIC ≥32 | vancomycin | More clinical failure with vancomycin tolerance | 175 |
SAB, S. aureus bacteraemia.
Although expert consensus on the definition of tolerance and persistence in general has been achieved,17 there is so far no validated and reliable tool to quantify and most of all classify tolerance or persistence for clinical application and decision-making. An optimal test for measuring tolerance or persistence would need to be highly standardized, reproducible within and between laboratories and strains, rapid, not too labour-intensive and correlated with clinical outcome. First promising steps in this direction have been taken with proposed new metrics and methodological approaches.117,180–184 For example, the metric MDK99 (minimal duration necessary for killing 99% of a bacterial population) could be used to quantify tolerance.17,180 However, standardization of pre-test growth conditions, starting inoculum, test media and conditions as well as the applicable antibiotic concentrations still need to be established and clinically relevant cut-offs determined. Moreover, the antibiotic concentration needs to reach saturation for killing in the MDK99 assay, which does not work for all antibiotics.180,185 Finally, the proposed setup to measure MDK99 probably needs a robotic system to fit into the workflow and manpower of a routine diagnostic microbiology laboratory.180
Approaches to treating antibiotic-tolerant S. aureus
With the increasing awareness of increased antibiotic survival, research has turned its focus on new ways of treating and eradicating tolerant S. aureus. Three main approaches have been explored so far to combat tolerant S. aureus (Figure 2): (i) disrupting growth- or metabolism-independent targets of increased antibiotic survival with anti-tolerance compounds; (ii) improving the activity of existing antibiotics (by changing methods and duration of administration); and (iii) resensitizing tolerant cells to existing antibiotics.186
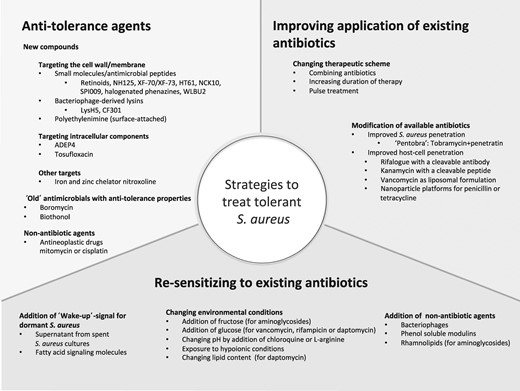
Published strategies for the possible treatment of tolerant S. aureus infections.
Strategies targeting mechanisms of increased antibiotic survival have produced compounds capable of killing tolerant S. aureus, the majority of which target the cell membrane, an optimal target in metabolically quiescent cells. Retinoids, a new class of antibiotics, disrupt the lipid bilayer of tolerant S. aureus.187 Various membrane-active small molecules, such as NH125,188–190 XF-73,191 HT-61192 or NCK10193 have shown good activity against tolerant S. aureus. A broad clinical application may be limited by specific properties such as induction of significant haemolysis through NH125 or low bioavailability of XF-73 and HT-61 due to strong hydrophobicity. Nevertheless, XF-73 has successfully completed Phase I/II tests and HT-61 has passed Phase II trials and is currently undergoing Phase III trials.186 Engineered cationic amphipathic peptides,194 bacteriophage-derived or synthetic lysins,195–197 the potassium ionophore boromycin,198 surface-attached polyethylenimine199 or even the anthelminthic biothionol200 can kill tolerant S. aureus through cell wall/membrane interaction.
Another promising new synthetic antibiotic is ADEP4, which has the unique feature of not blocking but activating its target. It binds intracellularly to the protease ClpP resulting in an ATP-independent uncontrolled self-digestion and death of the cell.55,201,202 However, this substance must be used in combination with other antibiotics as its use is fraught with rapid development of escape mutants.55 Similarly, a clinical drug-library screen identified the quinolone tosufloxacin as highly active against tolerant S. aureus—even more active than ADEP4 in one in vitro study.203 Novel compounds such as SPI009,204 halogenated indoles,205 phenazine-inspired small molecules206 or chelators of iron and zinc207 have an effect on tolerant S. aureus cells. Even the anticancer drugs mitomycin and cisplatin demonstrate bactericidal action on tolerant S. aureus through crosslinking DNA.208,209 However, their clinical application in S. aureus infections seems limited due to their inherently unfavourable toxicity.
Enhancing existing antibiotics may be the most straightforward way of tackling tolerant S. aureus. For example, combinations of antibiotics may achieve a synergistic effect on tolerant S. aureus as shown in vitro for daptomycin and tobramycin.103,144 However, that study applied very high doses of 100× MIC, which is not achievable systemically in patients due to toxicity issues, especially considering protein binding of 92%. Localized administration could, in theory, circumvent systemic toxicity in implant-associated infection.210 Prolonging the duration of antibiotic treatment represents another approach. Concordantly, in vitro long-term antibiotic exposures of up to 24 days led to complete eradication of tolerant S. aureus populations.103 Recently, a mathematical model successfully predicted the duration of treatment for eradication of infections with tolerant bacteria.211 These results foster the discussion of treatment duration in patients and may also explain why short-course antibiotic treatment (<14 days) is associated with more relapse even in uncomplicated S. aureus bacteraemia.212 As early as 1944, Bigger suggested an intermittent antibiotic treatment to let the dormant tolerant subpopulation awaken in antibiotic-free intervals, rendering them amenable to the next antibiotic killing.35 The potential efficacy of this pulsed treatment was recently validated in vitro.213 However, whether this effect can be applied clinically to improve the outcome in treatment of tolerant infections remains to be proven. Another way of improving the available antibiotics is to synthetically fuse them to cleavable moieties with cell-penetrating properties79,214–217 or to change the carrier formulation (Figure 2).218–220 This either increases the penetration into S. aureus per se, as shown for the fusion antibiotic pentobra, or ameliorates the penetration into mammalian cells to access the intracellular S. aureus population.
Finally, resensitizing tolerant S. aureus to existing antibiotics has become a focus of research. Changing bacterial metabolism by varying the composition of environmental metabolites can influence antibiotic killing of tolerant S. aureus. As an example, addition of fructose to biofilm cultures enabled growth-independent killing through aminoglycosides;97 addition of glucose increased the effect of daptomycin,129 vancomycin and rifampicin,126 or of quinolones when combined with oxygen.38 Mechanistically, this has been linked to metabolite-enabled generation of proton-motive force,97 activation of cell respiration38 or increased transcription of murein hydrolase.126 It is worth noting that potentiation is generally antibiotic and metabolite specific,97 supportive of the notion that various tolerance mechanisms are at play. Changing the environment is not limited to metabolic stimuli, as pH modification by addition of the basic amino acid l-arginine or exposing tolerant S. aureus to alkaline conditions increased the killing effect of aminoglycosides.221,222 Further, interfering with chemical signalling by using supernatant from S. aureus medium223,224 or fatty acid signalling molecules225 succeeded in resuscitating dormant S. aureus. These results of sensitizing by ‘environmental reprogramming’ are intriguing; however, the complex nutritional and metabolic interactions in an infected tissue could make it challenging to specifically trigger the desired response for synergistic therapeutic reasons. Finally, the effect of existing antibiotics on tolerant S. aureus may be augmented by adding phenol-soluble modulins,226 bacteriophages227 or rhamnolipids228 to the treatment.
Open questions and conclusions
Our knowledge of the determinants and significance of antibiotic-tolerant S. aureus in patients remains patchy at best. Does the same mechanism of increased antibiotic survival account for a late relapse after stopping antibiotic treatment62 and continuous infection as found in persistent bacteraemia?7 Do the various microenvironments in the infected milieu determine antibiotic survival more than the cellular stochasticity that is thought to underlie the persister phenotype? How relevant is increased antibiotic survival in non-bloodstream infections such as in implant infections? What role does the antibiotic selected have on development of tolerant phenotypes during treatment? What is the therapeutic role of so-called bacteriostatic antibiotics in infections with tolerant S. aureus? What influence does the immune system exert on S. aureus to become tolerant? Can we identify bacterial or host markers for tolerant infections? How long can tolerant S. aureus stay in the metabolically quiescent state in the human body? Do they revert stochastically or only by induction through environmental triggers? Do tolerant subpopulations actually harbour mutations, which rapidly revert back upon subculturing? Would it be an advantage to regularly screen all patients for tolerant S. aureus or only in those with treatment failure? Could we adapt treatment duration to the level of antibiotic survival or should we aim to individualize an effective combination therapy? To answer these and other questions, a reliable and reproducible tool of measuring antibiotic survival is critical.
In conclusion, S. aureus may display increased survival to antibiotic treatment through a plethora of inducers and mechanisms, reflecting its physiological versatility and infectious context. Increased antibiotic survival should be considered as a potential factor for the unacceptably high rate of treatment failure. New insights and promising approaches are appearing on the horizon.
However, clinical studies are needed to better understand the dynamics, mechanisms and relevance of tolerant S. aureus at the site of infection and to step ahead in the fight against one of mankind’s enduring foes.
Funding
R. K. was supported by the research funds of the University of Basel.
Transparency declarations
None to declare.

{kind=link}
{kind=link}