





Abstract
We reported tet(S/M) in Streptococcus pneumoniae and investigated its temporal spread in relation to nationwide clinical interventions.
We whole-genome sequenced 12 254 pneumococcal isolates from 29 countries on an Illumina HiSeq sequencer. Serotype, multilocus ST and antibiotic resistance were inferred from genomes. An SNP tree was built using Gubbins. Temporal spread was reconstructed using a birth–death model.
We identified tet(S/M) in 131 pneumococcal isolates and none carried other known tet genes. Tetracycline susceptibility testing results were available for 121 tet(S/M)-positive isolates and all were resistant. A majority (74%) of tet(S/M)-positive isolates were from South Africa and caused invasive diseases among young children (59% HIV positive, where HIV status was available). All but two tet(S/M)-positive isolates belonged to clonal complex (CC) 230. A global phylogeny of CC230 (n=389) revealed that tet(S/M)-positive isolates formed a sublineage predicted to exhibit resistance to penicillin, co-trimoxazole, erythromycin and tetracycline. The birth–death model detected an unrecognized outbreak of this sublineage in South Africa between 2000 and 2004 with expected secondary infections (effective reproductive number, R) of ∼2.5. R declined to ∼1.0 in 2005 and <1.0 in 2012. The declining epidemic could be related to improved access to ART in 2004 and introduction of pneumococcal conjugate vaccine (PCV) in 2009. Capsular switching from vaccine serotype 14 to non-vaccine serotype 23A was observed within the sublineage.
The prevalence of tet(S/M) in pneumococci was low and its dissemination was due to an unrecognized outbreak of CC230 in South Africa. Capsular switching in this MDR sublineage highlighted its potential to continue to cause disease in the post-PCV13 era.
Introduction
Streptococcus pneumoniae is a major bacterial cause of disease in young children. Despite the success of pneumococcal conjugate vaccines (PCVs), invasive pneumococcal disease (IPD) remains an important health priority owing to increasing disease incidence caused by pneumococci expressing non-vaccine serotypes, a phenomenon known as serotype replacement.1 Serotype replacement could be mediated by capsular switching, in which a cps locus encoding vaccine-type (VT) capsule is replaced by a cps locus encoding non-vaccine-type (NVT) capsule through homologous recombination.2 Capsular switching within MDR lineages, especially those recognized by the Pneumococcal Molecular Epidemiology Network (PMEN; http://spneumoniae.mlst.net/pmen/pmen.asp), is of increasing concern, as these expansions can reduce overall vaccine effectiveness in preventing IPD and temper the reduction in antimicrobial-resistant pneumococcal infections associated with introduction of PCVs.3 The persistence of the MDR lineage ST156 (Spain9V-3, PMEN3) in the USA following the introduction of PCV13 provides a clear example of a historically successful lineage that underwent a capsular switch from VT (serotype 9V, 14 and 19A) to NVT (serotype 35B) and continued to cause IPD in the post-vaccine era.3–5
Resistance to tetracycline has been frequently observed in S. pneumoniae.6 The genetic basis was shown to be tet(M), and less commonly tet(O), which encode for a ribosomal protection protein that prevents tetracycline binding to the bacterial 30S ribosome subunit.6,7 Eleven other classes of ribosomal protection proteins such as tet(S) and 12 mosaic structures of tet genes such as tet(S/M) have not been found previously in pneumococci (http://faculty.washington.edu/marilynr/). tet(S), originally discovered in Listeria monocytogenes strain BM4210,8 has occasionally been found in a variety of streptococci, including Streptococcus suis (NCBI accession number KX077886),9,Streptococcus infantis (NCBI accession number JX275965) and Streptococcus dysgalactiae (NCBI accession number EF682210)10 and is associated with a transposase-containing element IS1216, which potentially mediates chromosomal rearrangement. The mosaic tet(S/M) has been observed on a Tn916 element in Streptococcus intermedius11 and an IS1216 composite in Streptococcus bovis.12 Using a dataset of 12 254 pneumococcal genomes from the Global Pneumococcal Sequencing (GPS) project (https://www.pneumogen.net/gps/), we identified a novel genetic basis for tetracycline resistance in S. pneumoniae, tet(S/M), and characterized its genetic background in relation to nationwide clinical interventions.
Materials and methods
Isolate collection
In the GPS project, each participating country retrospectively and randomly selected pneumococcal disease isolates collected via laboratory-based surveillance and carriage isolates via cohort studies using the following criteria: ∼50% of isolates from children ≤2 years old, 25% from children 3–5 years old and 25% from individuals >5 years old. By May 2017 (last access to the GPS database for this study), 12 254 isolates, representing 29 countries, in Africa (65%), North America (14%), Asia (9%), South America (8%) and Europe (4%), were sequenced, passed quality control and included in this study. The collection spanned 26 years between 1991 and 2016 and included both pneumococcal carriage (n=4863) and disease isolates (n=7391). We compiled the metadata including age, year of collection, sample source, HIV status and phenotypic antimicrobial susceptibility testing results, where available, from each participating site. In children <18 months of age, HIV status was confirmed by PCR assay. Tetracycline resistance phenotype of tet(S/M)-positive isolates was confirmed by either microbroth dilution or Etest. MIC results were interpreted according to CLSI M100-S24.13 When MIC was analysed as ‘>X’, MIC was approximated as value 2X for median and IQR calculations.
Genome sequencing and analyses
The pneumococcal isolates were whole-genome sequenced on an Illumina HiSeq platform and raw data were deposited in the European Nucleotide Archive (ENA) (Supplementary metadata, available as Supplementary data at JAC Online). We inferred serotype, multilocus ST and resistance profile for penicillin, chloramphenicol, co-trimoxazole, erythromycin and tetracycline from the genomic data, as previously described.14 The tet(S/M) gene was identified with a tet(S/M) reference sequence (NCBI accession number AY534326) using ARIBA15 and the promoter region was examined manually by comparing with the reference sequences of tet(M) (NCBI accession number M85225) and tet(S) (NCBI accession number FN555436). We detected the presence of other tet genes in tet(S/M)-positive isolates by BLASTing their assemblies against a list of 13 tet genes encoding ribosomal protection proteins (Table 1).
List of tet genes encoding ribosomal protection proteins for BLAST analysis
| Gene(s) | NCBI accession number |
|---|---|
| tet(M) | MH283017 |
| tet(O) | Y07780 |
| tetA(P) and tetB(P) | L20800 |
| tet(Q) | Z21523 |
| tet(S) | FN555436 |
| tet(T) | L42544 |
| tet(W) | AJ222769 |
| tet(32) | AJ295238 |
| tet(36) | AJ514254 |
| tet(44) | FN594949 |
| tet(61) | KY887560 |
| otr(A) | X53401 |
| tet | M74049 |
| Gene(s) | NCBI accession number |
|---|---|
| tet(M) | MH283017 |
| tet(O) | Y07780 |
| tetA(P) and tetB(P) | L20800 |
| tet(Q) | Z21523 |
| tet(S) | FN555436 |
| tet(T) | L42544 |
| tet(W) | AJ222769 |
| tet(32) | AJ295238 |
| tet(36) | AJ514254 |
| tet(44) | FN594949 |
| tet(61) | KY887560 |
| otr(A) | X53401 |
| tet | M74049 |
The list is adapted from http://faculty.washington.edu/marilynr/tetweb4.pdf.
List of tet genes encoding ribosomal protection proteins for BLAST analysis
| Gene(s) | NCBI accession number |
|---|---|
| tet(M) | MH283017 |
| tet(O) | Y07780 |
| tetA(P) and tetB(P) | L20800 |
| tet(Q) | Z21523 |
| tet(S) | FN555436 |
| tet(T) | L42544 |
| tet(W) | AJ222769 |
| tet(32) | AJ295238 |
| tet(36) | AJ514254 |
| tet(44) | FN594949 |
| tet(61) | KY887560 |
| otr(A) | X53401 |
| tet | M74049 |
| Gene(s) | NCBI accession number |
|---|---|
| tet(M) | MH283017 |
| tet(O) | Y07780 |
| tetA(P) and tetB(P) | L20800 |
| tet(Q) | Z21523 |
| tet(S) | FN555436 |
| tet(T) | L42544 |
| tet(W) | AJ222769 |
| tet(32) | AJ295238 |
| tet(36) | AJ514254 |
| tet(44) | FN594949 |
| tet(61) | KY887560 |
| otr(A) | X53401 |
| tet | M74049 |
The list is adapted from http://faculty.washington.edu/marilynr/tetweb4.pdf.
To reconstruct a global phylogeny, an additional collection of clonal complex (CC) 230 isolates (n=130) from previous studies,16–20 together with the CC230 collection (n=259) in the GPS dataset were included. The phylogeny was built as previously described.14 Based on the international genomic definition of pneumococcal lineages, all CC230 isolates in this study belong to GPS Cluster (GPSC)10.14 The metadata and analysis results of CC230 can be interactively visualized online using the Microreact tool at https://microreact.org/project/GPS_tetSM.
Temporal changes of tet(S/M) CC230 sublineage
Coalescent analysis was performed on tet(S/M) CC230 sublineage (n=129) to date the most recent common ancestor (MRCA) and reconstruct the population demographic history. First, we tested the presence of temporal signal by a linear regression of root-to-tip distances against year of collection using TempEst v1.5.21 Next, a timed phylogeny was constructed using BEAST v2.4.1.22 The Markov chain Monte Carlo (MCMC) chain was run for 100 million generations, sampled every 1000 states using the general time-reversible (GTR) model of nucleotide substitution and the discrete gamma model of heterogeneity among sites. Finally, the population demographic history was reconstructed using a birth–death model23 to examine the temporal changes with the tet(S/M) CC230 sublineage invasive isolates (n=105) but not carriage isolates (n=24), because the model assumes that once an individual is diagnosed with IPD, the individual is no longer transmitting due to treatment and recovery, death or being socially removed from susceptible individuals. Thus, it appeared to be logical to apply this model to the disease but not carriage isolates. This model overcomes the limitations of the coalescent-based skyline plot and is able to examine whether introduction of an intervention had an impact on the epidemiological dynamics in a bacterial population.24 The birth–death skyline plot shows the effective reproductive number (R) over time. R is defined as the number of expected secondary infections from an infected individual. R>1 indicates a growing epidemic, whereas R<1 indicates a declining epidemic. Notably, R ≥ 1 can be reflected in the coalescent-based skyline plot analysis, whereas R<1 cannot. Therefore, we expected the birth–death skyline model would be a better fit for our data. Other Bayesian population-size models (coalescent constant, coalescent exponential and Bayesian skyline), in combination with strict and lognormal-relaxed molecular clocks, were also applied for comparisons using BEAST.
Integrative and conjugative element (ICE)
The ICE was extracted from the de novo assemblies of CC230 isolates and compared using EasyFig version 2.2.2. The NCBI accession numbers for the representative ICE sequences in Figure 5 are FM211187 (ICESp23FST81), MH283017 [ICESp14ST230 with tet(M)], MH283012 [ICESp14ST230 with tet(S/M) and omega cassettes], MH283013 [ICESp14ST230 with tet(M) and omega], MH283012 [ICESp14ST230 with tet(M) and Tn917], MH283016 (ICESp19AST2013), MH283015 (ICESp17FST8812) and MH283014 (ICESp14ST156).
Results
Prevalence of tet(S/M) in a global collection of S. pneumoniae
A tetracycline resistance gene, tet(S/M), was identified in 131 pneumococcal isolates (1%, 131/12 254) from South Africa (n=123), Malawi (n=5), Brazil (n=1), Mozambique (n=1) and the USA (n=1). They were isolated from sterile body sites (invasive isolates) [blood (n=73), CSF (n=30) and pleural fluid (n=4)] and from the nasopharynx (carriage isolates) (n=24). In South Africa, tet(S/M) was found in 3.5% (103/2920) of the invasive isolates that were submitted to the GPS project from 2005 to 2014 and 1.2% (20/1701) of the carriage isolates that were collected in Agincourt and Soweto between 2009 and 2013. Of the 103 invasive isolates, 94% (97/103) were from children with IPD aged ≤5 years (Figure S1). HIV status was known in only 44% (54/123) of individuals with tet(S/M)-positive pneumococci; 59% (32/54) were HIV positive, of which 94% (30/32) were children aged ≤5 years.
MIC of tetracycline was determined for 121 tet(S/M)-positive isolates by either Etest (n=73) or broth dilution (n=48). The remaining 10 isolates were either non-viable (n=5) or unable to be tested due to inadequate resources (n=5). The median MIC was 8 mg/L (IQR 6–8) by Etest and 16 mg/L by broth dilution. Based on the CLSI guideline, 99% (120/121) and 1% (1/121) were fully (≥4 mg/L) and intermediately (2–3 mg/L) resistant to tetracycline, respectively. The tet(S/M) in this study showed 100% nucleotide identity, except for one isolate (GPS_ZA_1982) from South Africa, which varied from the others (G1769A) and resulted in the substitution R590Q. This isolate remained resistant to tetracycline with an MIC of >8 mg/L when measured by broth dilution. Unlike the two previously reported tet(S/M) alleles from S. intermedius11 and S. bovis,12 the amino acid sequence of Tet(S/M) in this study showed 100% identity to Tet(S) (NCBI accession number FN555436) across the first 613 amino acids, with the final 32 amino acids at the C-terminus end being identical to Tet(M) (NCBI accession number M85225) (Figure 1). The promoter region was intact in all tet(S/M)-positive isolates and was of tet(M) origin, rather than tet(S) origin. Between the promoter region and the start codon of tet(S/M), a 38 bp stem loop, which is potentially involved in transcriptional regulation,25 was found in all tet(S/M) genes (Figure 1), apart from one disease isolate (GPS_ZA_1926) from South Africa. The deletion did not affect the tetracycline resistance level, as the MIC remained at >8 mg/L when measured by the broth dilution method. No other tet genes (Table 1) were detected in any tet(S/M)-positive isolates, strongly indicating that Tet(S/M) conferred resistance to tetracycline in S. pneumoniae.
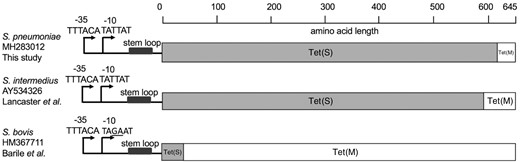
Schematic representation of the mosaic structure of tet(S/M) alleles of the current and previous studies. The bars in grey and in white indicate amino acid sequences with high identity to Tet(S) and Tet(M), respectively. The reference sequences for tet(M) and tet(S) were retrieved from NCBI GenBank using accession numbers M85225 and FN555436, respectively.
Phylogeny and characteristics of the tet(S/M) CC230 sublineage
Of the 131 tet(S/M)-positive isolates, 129 belonged to CC230; one Brazilian and one Malawian isolate belonged to CC156 and ST5359 (a singleton not belonging to any CC), respectively. The global CC230 phylogeny showed that all tet(S/M)-positive isolates formed a sublineage predicted to be resistant to penicillin, erythromycin, tetracycline and co-trimoxazole (Figure 2). The tet(S/M) sublineage was associated predominantly with VT 14 (98%, 127/129) but was also found in two NVT 23A isolates. The two serotype 23A isolates, which both belonged to ST11106 (a single-locus variant of ST230), were recovered from infants after the introduction of PCV13. One was isolated from a nasopharyngeal sample in Soweto in 2012 and the other from blood culture in Johannesburg in 2014. The serotype 23A cps locus sequences of these two isolates were identical and their cps-flanking pbp loci (pbp1a and pbp2x) were also identical to the majority of the serotype 14 isolates within the tet(S/M) sublineage, exhibiting resistance to penicillin with an MIC of 2 mg/L. To identify the potential donor of the serotype 23A cps locus, a phylogenetic tree was built using the cps sequences from all serotype 23A (n=130, belong to eight lineages) pneumococci in the GPS database. This analysis showed that the serotype 23A cps loci of these two CC230 isolates clustered with those originating from a serogroup 23 lineage GPSC7, which is predominantly (99%, 145/146) represented by CC439 (Figure S2), with pairwise nucleotide similarity of 99.97% (24 818/24 825) and 100% coverage. The seven nucleotide variations were found within the IS630 transposase downstream of dexB.
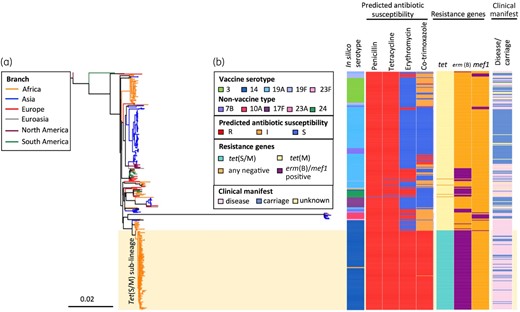
An SNP tree constructed with CC230 tet(S/M)-positive isolates (n=129) and tet(S/M)-negative carriage/disease isolates (n=260) collected from 20 countries. The tree was built based on 13 405 SNPs extracted from an alignment outside recombination regions, created by mapping reads of each isolate to the sequence of an ST230 reference strain, PMEN global clone Denmark14-32, PMEN32 (ENA accession number ERS1706837). Penicillin resistance was predicted based on the pbp1a, pbp2x and pbp2b sequences;38,39 tetracycline and erythromycin resistance were predicted based on the presence of tet(M), tet(O) and tet(S/M), and erm(B) and mef(A), respectively. Co-trimoxazole resistance was predicted based on the presence of mutation I100L in folA and any indel within amino acid residues 56–67 in folP, while the presence of either mutation was predicted to confer a co-trimoxazole-intermediate phenotype.
Temporal spread of the tet(S/M) CC230 sublineage
The sublineage showed a temporal signal in terms of SNP accumulation against time (R2 = 0.4094, P=0.001; Figure S3). Using a birth–death model in BEAST, the tet(S/M) sublineage was estimated to emerge around 1994 [95% highest posterior density (HPD): 1991–96]; the MRCA for the African clade was 1998 (95% HPD: 1996–2000) and for the two serotype 23A isolates was 2009 (95% HPD: 2007–11) (Figure 3). The temporal changes of spread were reconstructed based on a birth–death skyline plot and a coalescent-based skyline plot (Figure 4). Both skyline plots showed that the tet(S/M) sublineage expanded at the beginning of the year 2000 and growth continued until around 2004. The decline of the tet(S/M) sublineage was only captured by the birth–death skyline plot in or around 2005, from expected secondary infections (R) of ∼2.5 to ∼1, and steadily declined until 2012 when the median and HPD of R were <1, indicating a declining epidemic. The coalescent-based skyline plot failed to detect the impact of the epidemic decline, as described in a previous study.23
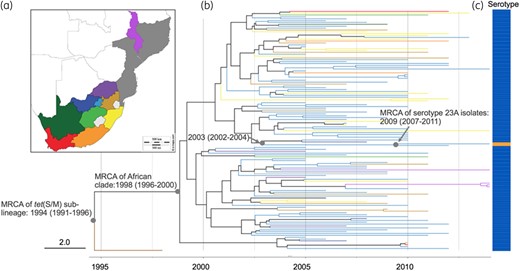
(a) Malawi, Mozambique and administration regions of South Africa. (b) Timed phylogeny for S. pneumoniae tet(S/M) CC230 sublineage (n=129) reconstructed using BEAST. Tree branches are coloured according to the geographical locations in (a), except for the branch for an isolate collected from the USA, coloured in brown. (c) Vaccine serotype 14 is indicated in blue, whereas non-vaccine serotype 23A is indicated in orange.
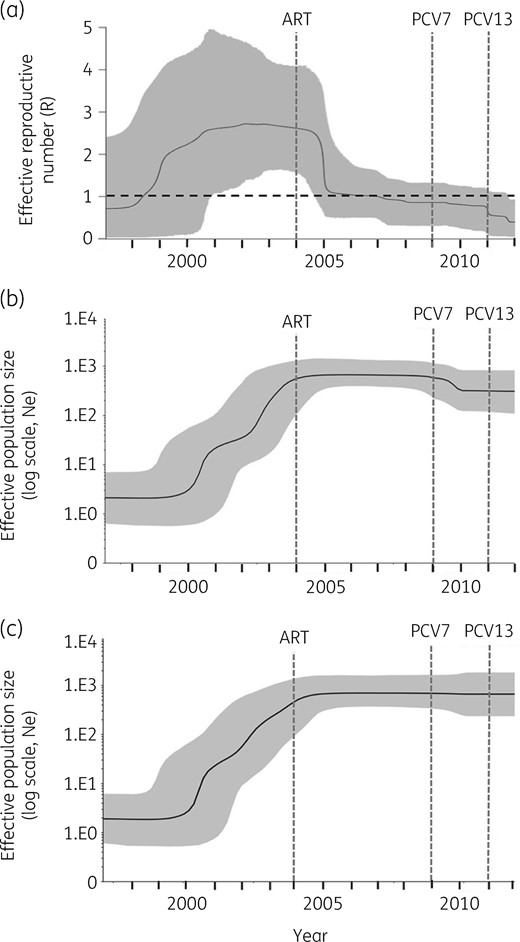
(a) Birth–death skyline plot of inferred changes in R of S. pneumoniae tet(S/M) CC230 sublineage using IPD isolates (n=105). (b) Coalescent-based skyline plot of inferred changes in the effective population size (Ne) of the S. pneumoniae tet(S/M) CC230 sublineage using both IPD and carriage isolates (n=129) and (c) using only IPD isolates (n=105). The black continuous line shows the median of R in (a) and Ne in (b) and (c). The background area represents the 95% HPD intervals. R>1 indicates a growing epidemic, whereas R<1 indicates a declining epidemic.
ICE carrying tet(S/M)
The acquisition of tetracycline and erythromycin resistance determinants by CC230 was the result of the insertion of a Tn5253-type ICE, which shared a similar structure to ICESp23FST81 identified in PMEN1 (Figure 5). Both the tet(M) (n=255) and tet(S/M) (n=131) genes detected in this study were carried on a conserved conjugative Tn916 transposon (Figure S4). Of the 172 macrolide-resistant isolates, insertions of either the ‘omega’ element (n=165) or Tn917 (n=7) harbouring erm(B) were found upstream or downstream of the tet gene, respectively (Figure 5). The insertion of the ‘omega’ element truncated the gene encoding the replication initiation factor, creating an 8 bp DR, CAAAAAAA. The insertion of Tn917 disrupted the gene orf9, which encodes a putative conjugative transposon regulator; no DRs were found.
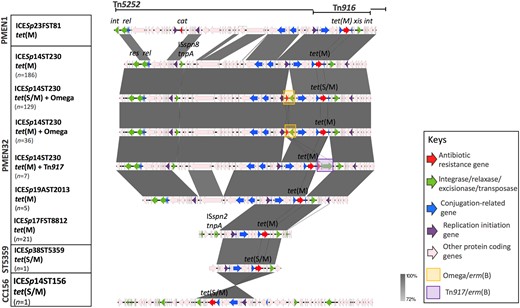
Comparison of ICE identified in CC230 with Spain23F-1 (PMEN1). Grey bands between the sequences indicate BLASTN matches.
Discussion
We used WGS to identify a novel mosaic structure of tet(S/M) in S. pneumoniae. This approach overcame the limitation of PCR that requires specific primers to detect known antibiotic resistance genes. Compared with tet(M), the prevalence of tet(S/M) was low. They were mainly found in a CC230 sublineage that predominantly expressed VT 14 and exhibited MDR in South Africa. Together with its conserved nucleotide sequence and genomic location, our finding strongly suggested a clonal expansion of tet(S/M)-positive CC230 isolates within South Africa prior to the introduction of PCVs.
Unlike what was observed in other countries, CC230 in South Africa predominantly expressed a highly invasive serotype 14 capsule14 and was the clone that represented most of the serotype 14 isolates (43%) in the pre-vaccine era, when serotype 14 was the most prevalent serotype causing IPD in South Africa.26 Any controlling measure to decrease this lineage would not only result in a reduction in the IPD burden but also MDR IPD incidence.
The birth–death model estimated that the decline of the tet(S/M) CC230 sublineage started around 2005, one year after the national ART programme was launched to treat HIV-infected individuals in South Africa.27 The prediction was consistent with a 41% reduction of IPD incidence among HIV-infected children after the introduction of ART.27 Among the IPD caused by tet(S/M) CC230 isolates, almost 60% occurred in HIV-positive children. This observational evidence strengthened the suggestion that ART was likely to contribute to the decline before PCV introduction. In contrast, the large-scale use of co-trimoxazole as prophylaxis to prevent bacterial infections among HIV-positive individuals was unlikely to be responsible for the decline, as the sublineage was resistant to co-trimoxazole. The further decline in 2012 predicted by the model echoed the epidemiological finding that IPD caused by VT pneumococci significantly decreased among children in 2012.28 Our finding demonstrated that we could effectively reconstruct the temporal spread of an epidemic using genomic data and highlighted the possible use of routine genomic surveillance to model outbreaks as they occur.
The MRCA of two CC230 isolates expressing NVT 23A was dated to emerge around 2009, the year when PCV7 was introduced. However, the long branch leading to the MRCA from the internal node that was shared with the closely related serotype 14 isolates indicated that the window of time for capsular switching could be between 2002 and 2011. Although the invasive disease potential for serotype 23A is low,14 a significant increase of this serotype in IPD cases was reported from England,29 Stockholm30 and Taiwan31 after the implementation of PCV13. Serotype 23A is primarily associated with CC338 (GPSC5, PMEN26) and CC439 (GPSC7) and is thus rarely found in a CC230 genetic background. Such serotype and genotype combinations were only identified in two ST9396 isolates (single-locus variant of ST230) from China in 2013 and one ST10921 isolate (double-locus variant of ST230) from Poland in 2013 in the MLST database. In South Africa, CC439 accounted for 62% of serotype 23A (both carriage and disease) isolates14 and is the potential donor of the serotype 23A cps to the tet(S/M) CC230 sublineage, highlighting that capsular switching with the prevalent NVT lineage could enable a VT lineage to evade the vaccine. Capsular switching is usually a result of homologous recombination. When compared with 620 other GPSCs, GPSC10, including 98% (258/262) of CC230 isolates, is a very recombinogenic lineage, which had a significantly high recombination rate [ratio of base substitutions predicted to have been imported through recombination to those occurring through point mutation for GPSC10: 10.9 versus median of 35 dominant GPSCs: 8.3 (IQR, 5.7–10.7), P<0.0001, Wilcoxon signed-rank test].14 Given this recombinogenic nature, together with the established MDR genotypes, it is of concern that any further capsular switching may increase the chance of this MDR lineage surviving and continuing to cause invasive disease.
Like tet(M), tet(S/M) was also carried by Tn916, which was reported as a mobile conjugative transposon with a broad host range.11 The conserved genetic environment of tet(M) and tet(S/M) indicates that the recombination resulting in the mosaic structure of tet(S/M) probably occurred after the acquisition of the gene by Tn916. Comparison of tet(M) sequences in the current collection also revealed a high degree of allelic variations that were probably due to homologous recombination.32 This finding is consistent with previous studies that suggested that tet evolved separately from Tn916.6,32 However, the driving force behind the evolution of tet genes remains unclear, given that tetracycline was not used as a first-line antibiotic to treat pneumococcal disease and was seldom used in young children.33 The allelic diversity of tet may be maintained by: (i) frequent recombination among S. pneumoniae and with closely related species such as normal nasopharyngeal resident Streptococcus mitis34 and the zoonotic pathogen S. suis;35 and (ii) antibiotic-selective pressure via the food chain, as tetracycline is widely used in agriculture36 and its residue is detected in milk.37 Future studies that investigate the driving force behind the evolution of tet will improve our understanding to develop preventive measures to reduce tetracycline resistance in S. pneumoniae.
In conclusion, we identified the tetracycline-resistance determinant tet(S/M) in S. pneumoniae and showed that its dissemination is due to a clonal expansion of the MDR lineage CC230 in South Africa, where the HIV burden is high. With genomic data, we successfully detected the decline in transmission of this MDR lineage using a birth–death model and the fall of this lineage may correlate with the improved treatment of HIV-infected individuals and the implementation of PCVs. Capsular switching within this lineage is potentially of public health importance and may erode the beneficial effect brought about by the implementation of PCVs. The capacity for continuous genomic surveillance in the post-vaccine era provides critical opportunities for monitoring and forecasting the rise of MDR pneumococcal lineages that may also undergo vaccine evasion through capsular switching events.
Acknowledgements
We would like to acknowledge the Bill & Melinda Gates Foundation (grant code OPP1034556) and the Wellcome Sanger Institute (core Wellcome grants 098051 and 206194) for funding this study, as part of the GPS project. We deeply appreciate all members of the Global Pneumococcal Sequencing Consortium for their collaborative spirit and determination in the monumental task of sampling, extracting data and for their intellectual input to this manuscript. We extend our special thanks to Linda De Gouveia from the National Institute for Communicable Diseases of the National Health Laboratory Service, South Africa who helped us with the antimicrobial susceptibility testing. We appreciated critiques and suggestions from all team members in the Genomics of Pneumonia and Meningitis (and neonatal sepsis) group and technical support from the Pathogen Informatic Team in the Parasites and Microbe Programme at the Wellcome Sanger Institute.
Members of the Global Pneumococcal Sequencing Consortium
Abdullah W. Brooks, Alejandra Corso, Alexander Davydov, Alison Maguire, Andrew J. Pollard, Anmol Kiran, Anna Skoczynska, Benild Moiane, Betuel Sigauque, David Aanensen, Deborah Lehmann, Diego Faccone, Ebenezer Foster-Nyarko, Ebrima Bojang, Elena Voropaeva, Eric Sampane-Donkor, Ewa Sadowy, Geetha Nagaraj, Godfrey Bigogo, Helio Mucavele, Houria Belabbès, Idrissa Diawara, Jennifer Moïsi, Jennifer Verani, Jeremy Keenan, Jyothish N. Nair Thulasee Bhai, Kedibone M. Ndlangisa, Khalid Zerouali, Leon J. Bentley, Linda De Gouveia, Maaike Alaerts, Maria-Cristina de Cunto Brandileone, Margaret Ip, Md Hasanuzzaman, Metka Paragi, Mushal Ali, Nicholas Croucher, Nicole Wolter, Noga Givon-Lavi, Özgen Köseoglu Eser, Pak Leung Ho, Patrick E. Akpaka, Paul Turner, Paula Gagetti, Peggy-Estelle Tientcheu, Philip E. Carter, Pierra Law, Rachel Benisty, Rafal Mostowy, Rebecca Ford, Rebecca Henderson, Roly Malaker, Ron Dagan, Sadia Shakoor, Sanjay Doiphode, Shamala Devi Sekaran, Somporn Srifuengfung, Shamala Devi Sekaran, Somporn Srifuengfung, Stephen Obaro, Stuart C. Clarke, Tamara Kastrin, Theresa J. Ochoa, Waleria Hryniewicz, Veeraraghavan Balaji and Yulia Urban.
Funding
This work was co-funded by the Bill & Melinda Gates Foundation (grant code OPP1034556), the Wellcome Sanger Institute (core Wellcome grants 098051 and 206194) and the US Centers for Disease Control and Prevention.
Transparency declarations
R. A. Gladstone reports a PhD studentship from Pfizer, outside the submitted work. A. Skoczynska reports grants from Pfizer, assistance to attend scientific meetings and honoraria for lecturing from GlaxoSmithKline and Pfizer, and participation in Advisory Boards of GlaxoSmithKline and Pfizer, outside the submitted work. S. D. Bentley reports personal fees from Pfizer and personal fees from Merck, outside the submitted work. A. von Gottberg reports grants and other from Pfizer, during the conduct of the study, and grants and other from Sanofi, outside the submitted work. All other authors: none to declare.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
CLSI. Performance Standards for Antimicrobial Susceptibility Testing—Twenty-Fourth Edition: M100.
WHO. Scientific basis of WHO recommendations for treatment of pneumonia. In: Revised WHO Classification and Treatment of Pneumonia in Children at Health Facilities: Evidence Summaries.
Author notes
Members are listed in the Acknowledgements section.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}