





Abstract
Carbapenemase-producing Enterobacteriaceae (CPE) pose a major global health risk. Mobile genetic elements account for much of the increasing CPE burden.
To investigate CPE colonization and the impact of antibiotic exposure on subsequent resistance gene dissemination within the gut microbiota using a model to simulate the human colon.
Gut models seeded with CPE-negative human faeces [screened with BioMérieux chromID® CARBA-SMART (Carba-Smart), Cepheid Xpert® Carba-R assay (XCR)] were inoculated with distinct carbapenemase-producing Klebsiella pneumoniae strains (KPC, NDM) and challenged with imipenem or piperacillin/tazobactam then meropenem. Resistant populations were enumerated daily on selective agars (Carba-Smart); CPE genes were confirmed by PCR (XCR, Check-Direct CPE Screen for BD MAX™). CPE gene dissemination was tracked using PacBio long-read sequencing.
CPE populations increased during inoculation, plateauing at ∼105 log10 cfu/mL in both models and persisting throughout the experiments (>65 days), with no evidence of CPE ‘washout’. After antibiotic administration, there was evidence of interspecies plasmid transfer of blaKPC-2 (111742 bp IncFII/IncR plasmid, 99% identity to pKpQIL-D2) and blaNDM-1 (∼170 kb IncFIB/IncFII plasmid), and CPE populations rose from <0.01% to >45% of the total lactose-fermenting populations in the KPC model. Isolation of a blaNDM-1K. pneumoniae with one chromosomal single-nucleotide variant compared with the inoculated strain indicated clonal expansion within the model. Antibiotic administration exposed a previously undetected K. pneumoniae encoding blaOXA-232 (KPC model).
CPE exposure can lead to colonization, clonal expansion and resistance gene transfer within intact human colonic microbiota. Furthermore, under antibiotic selective pressure, new resistant populations emerge, emphasizing the need to control exposure to antimicrobials.
Introduction
Carbapenemase producing Enterobacteriaceae (CPE) pose a major global health risk, with reports of outbreaks of CPE throughout Europe1 including in the UK,2,3 most notably in the north-west of England, where a large outbreak has contributed to national spread.4,5 Factors driving CPE acquisition and subsequent gene dissemination include increasing age,4 antibiotic therapy,4,6–8 presence of an intravascular access device/invasive procedure,6,8,9 prolonged hospital stay,6,7,9 multiple co-morbidities8 and proximity to the index case.6,7 Such reports identify potential at-risk individuals, but do not fully explain why some outbreaks are contained with infection control measures,10–12 whereas others are not.13,14 Unlike nosocomial outbreaks of Clostridioides (Clostridium) difficile or MRSA, CPE outbreaks often involve multiple bacterial species and multiple resistance genes carried on mobile genetic elements.12,15,16
The epidemiology of carbapenem resistance and specifically the genes encoding carbapenemases is complex.17,18 For example, while much of the global spread of CPE is due to Klebsiella pneumoniae harbouring KPC resistance genes,3,19,20 clonal populations of K. pneumoniae encoding VIM genes in Greece,21 outbreaks of OXA-48 in Turkey and the UK22,23 and the higher prevalence of IMP resistance genes in southern Europe and Asia24 demonstrate heterogeneous epidemiology. This implies that there are factors beyond bacterial species or single exposure events that potentiate or limit the spread of specific carbapenem resistance genes.
Susceptible bacterial populations gain resistance through mutation or via horizontal gene transfer of mobile genetic elements. The human large intestine is the ideal setting for resistance gene transfer, largely owing to its high biomass. Here we have used an in vitro human gut model that has previously successfully simulated the colon in the context of C. difficile infection25–27 to assess the factors favouring the dissemination of carbapenem resistance genes, and used long-read single-molecule real-time (SMRT) sequencing to further understand CPE resistance gene dissemination.
Methods
In vitro human gut model
Two triple-stage human gut models were used, as previously described.26 Briefly, the model consists of a triple-stage chemostat that is seeded with faecal slurry providing the indigenous gut microbiota. The model vessels are continually purged with nitrogen to maintain an anaerobic environment. The pH of each vessel is controlled to replicate that found in the proximal, medial and distal sections of the human colon, respectively, and the system is top fed with a complex growth medium in keeping with the nutritional profile of the large intestine.
Faecal inoculum
Faeces from healthy volunteers with no preceding antibiotic exposure in the last 3 months and no CPE risk factors (no travel/no hospitalization in a high-risk area), and which were negative on CPE screening [BioMérieux chromID® CARBA-SMART (Carba-Smart), Cepheid Xpert® Carba-R assay (XCR) multiplex real-time PCR assay and Check-Direct CPE Screen for BD MAX™ (CD CPE) multiplex real-time PCR assay], were used in this study. Faecal samples from the volunteers were pooled to produce ∼50 g of faeces, which was then mixed with 500 mL of pre-reduced PBS and filtered through muslin to produce a smooth faecal slurry (10% w/v). The same volunteers were used for both gut model experiments.
CPE strains
Two distinct clinical strains of carbapenemase-producing K. pneumoniae were used in this study, both supplied by Leeds Teaching Hospitals NHS Trust: an NDM-containing strain (MICs: ertapenem ≥32 mg/L, meropenem 8 mg/L and imipenem 1 mg/L); and a KPC-containing strain (MICs: ertapenem 4 mg/L, imipenem 8 mg/L and meropenem 4 mg/L). Each gut model was inoculated with a different strain that had been reconstituted on blood agar from −80°C freezer storage, and sub-cultured onto CPE-selective media (Carba-Smart). The presence of carbapenemase genes was confirmed using XCR and CD CPE multiplex real-time PCR assays.
Experimental design
Two distinct gut models were used in this study to examine the behaviours of the different carbapenemase gene-containing K. pneumoniae strains (Figure 1). Both models were primed with faecal slurry and left without interventions for 2 weeks to allow the bacterial populations to stabilize.
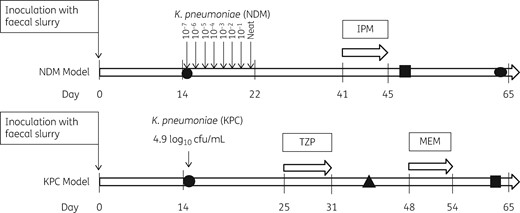
Schematic of experimental design. Shapes indicate the timepoints at which strains were sequenced: a circle denotes the original strain inoculated into the model; a square denotes plasmid uptake; and a triangle denotes a strain with new resistance profile. IPM, imipenem; TZP, piperacillin/tazobactam; MEM, meropenem.
The NDM gut model was challenged with increasing inocula of K. pneumoniae encoding blaNDM resistance genes. Inoculation commenced on day 15 of the experiment and continued for 8 days. The reconstituted strain was diluted in a 10-fold series to 10−7 in peptone water. Each day, 1 mL of an overnight culture (5 mL of nutrient broth) of K. pneumoniae encoding blaNDM was added to the model. The lowest dilution (10−7) was added on day 15 and the inocula were increased daily until a neat solution was added on day 22 of the experiment. Overnight cultures of the diluted K. pneumoniae encoding blaNDM were enumerated on MacConkey agar to ensure the inocula were as expected (Table S1, available as Supplementary data at JAC Online). Following the inoculation period, the model underwent a single antibiotic exposure event with imipenem. The model was dosed to achieve human in vivo gut intraluminal concentrations28 using 11 mg/L imipenem three times daily for 5 days (days 41–45).
The KPC gut model was inoculated with a single inoculum of 4.9 log10 cfu/mL of K. pneumoniae encoding blaKPC genes on day 15 of the experiment. Following inoculation, the KPC model was challenged with two separate antibiotic exposure events. Piperacillin/tazobactam (358 mg/L) was instilled between days 25 and 31, and meropenem (11 mg/L) between days 48 and 54, again dosed to simulate in vivo concentrations. Bacterial populations were monitored throughout (Figure 1) and were sampled daily for CPE and Enterobacteriaceae populations during the inoculation and antibiotic installation periods, and sporadically thereafter. The presence/absence of carbapenemase genes was monitored via the XCR assay.
Population monitoring
Sporadic sampling of the indigenous gut populations took place before CPE inoculation. For the NDM model, twice daily sampling of CPE populations took place during the inoculation phase (days 14–21), and indigenous populations were monitored once a day. Following inoculation, both carbapenemase-producing (CP) and indigenous bacterial populations were monitored daily. Both the CPE and the indigenous populations were sampled daily. Enumeration of the bacterial populations was performed by quantitative culture using Carba-Smart for CPE and MacConkey agar for indigenous populations.
These plates were read at 48 h using the appropriate dilution factor to allow enumeration in log10 cfu/mL. For CPE populations, enumeration took place 1 h after KPC inoculation. Fifty microlitres of the diluted aliquot (up to 10−3) was plated in triplicate on Carba-Smart and ESBL agars, incubated at 37°C and read at 24 h. Again, the plates were read at a factor allowing enumeration in log10 cfu/mL. The percentage of carbapenemase-producing bacteria was calculated from cfu/mL on the MacConkey plate [representing total lactose-fermenting (TLF) population] divided by cfu/mL on the Carba-Smart plate and multiplied by 100 to give a percentage of CP bacteria in the TLF bacteria.
For the molecular platforms (XCR, CD CPE), 50 μL of neat gut model fluid was analysed the same day, in accordance with the manufacturers’ instructions, except that we used gut model fluid instead of a rectal swab. Please see Supplementary Methods for further information.
Selection of CPE isolates for sequencing
Three isolates were chosen from each model for sequencing.
From the NDM model we sequenced the CP K. pneumoniae strain inoculated into the model, designated LCC079, the CP K. pneumoniae strain isolated at the end of the experiment, designated LCC088, and an Escherichia coli strain with carbapenem resistance, designated LCC081.
From the KPC model we sequenced the original CP K. pneumoniae strain inoculated into the model, designated LCC078, an E. coli strain from the end of the experiment with carbapenem resistance, designated LCC096, and a K. pneumoniae strain growing on the OXA side of the Carba-Smart plate (Bi-plate) on day 40, designated LCC093.
Strains were sequenced using Illumina technology as previously described and PacBio long-read sequencing at the Icahn Institute and Department of Genetics and Genomic Sciences, Mount Sinai, New York.
Sequencing and genetic analysis
Long-read SMRT sequencing and initial de novo assembly were performed as previously described29 using the latest P6 enzyme and chemistry and a single SMRTcell on the RSII platform. Chromosomal single-nucleotide variants (SNVs) were determined by mapping Illumina reads to chromosomal references for E. coli CFT073 (AE014075.1) or K. pneumoniae MGH78478 (CP000647.1), as previously described.30 To assemble the long-read sequencing data, we initially performed hybrid assembly using Unicycler v0.4.031 in bold mode with otherwise default parameters. For the KPC and OXA-232 samples, this produced complete, closed assemblies (i.e. all contigs were flagged as circular). For the NDM samples, the assemblies contained multiple un-circularized contigs, and visualization of mapped Illumina reads indicated likely misassemblies. For these samples, we therefore performed a de novo long-read assembly on the PacBio subreads using HGAP3 as previously described.29 Plasmid Inc typing was performed using the February 2018 version of the PlasmidFinder Enterobacteriaceae database,32 with an identity threshold of 95% and minimum length 60%. Copy number in LCC079, LCC081 and LCC088 was calculated using the Unicycler assembly for LCC081, which contained blaNDM-1 on the IncFIB/IncFII plasmid described below, with a single copy of the repeat unit. For each sample, we mapped Illumina reads to a reference consisting of this plasmid structure plus the HGAP3-assembled chromosomal contig for that sample. Mapping was performed using BWA MEM v0.7.12-r1039. Samtools version 1.4.1 was used to filter out supplementary alignments and calculate depth of coverage. blaNDM-1 coverage relative to the plasmid backbone was calculated as the median coverage across blaNDM-1 divided by the median coverage across the entire plasmid sequence excluding the repeat region. Similarly, the same median coverage value for the plasmid backbone was divided by the median coverage across the chromosomal contig to determine plasmid coverage relative to the chromosome. Please see Supplementary Methods for further information.
Results
No CPE were identified during the 2 week stabilization phase in either gut model experiment. In both models, CPE populations increased after antibiotic administration, reaching a plateau at∼105 log10 cfu/mL, and persisted for the duration of the experiments, with no evidence of CPE washout.
Plate enumeration results
NDM model
CP K. pneumoniae were detected after the addition of ∼4.9 log10 cfu/mL (day 18); CP K. pneumoniae increased with increasing inocula, peaking at 5–6 log10 cfu/mL before stabilizing at ∼3.5 log10 cfu/mL. Following antibiotic (imipenem) exposure, the CP K. pneumoniae population reduced, before expanding after administration stopped, finally accounting for ∼3.7% of the TLF population. On day 47 of the experiment (2 days after antibiotic exposure), we began to see the emergence of a resistant CP E. coli population, which appeared sporadically on the Carba-Smart agar until the end of the experiment (Figure 2).
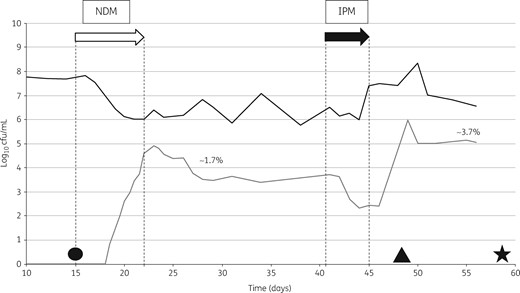
NDM model. Bacterial growth on logarithmic scale. The open arrow indicates the inoculation phase and the filled arrow indicates antibiotic instillation. NDM, K. pneumoniae with blaNDM; IPM, imipenem. The black line shows the total lactose-fermenting population and the grey line shows carbapenemase-producing K. pneumoniae. The percentages shown refer to the proportion of resistant bacteria out of the total Enterobacteriaceae. Circle, strain LCC079; triangle, strain LCC081; star, strain LCC088.
KPC model
Post-inoculation, CP K. pneumoniae counts were ∼1–2 log10 cfu/mL, accounting for <0.01% of the TLF population. Dosing with piperacillin/tazobactam resulted in an increase of >8 log10 cfu/mL in CP K. pneumoniae populations, which comprised ∼25% of the TLF population after dosing stopped. On day 35 of the experiment (20 days after CP K. pneumoniae inoculation and 5 days after the end of antibiotic administration), a CP E. coli emerged, peaking at 5.5 log10 cfu/mL on day 45. Following a second antibiotic exposure to meropenem, there were further increases in CP populations at day 65 (end of the experiment), comprising both the CP K. pneumoniae and CP E. coli populations, which combined accounted for ∼45% of the TLF population (Figure 3).
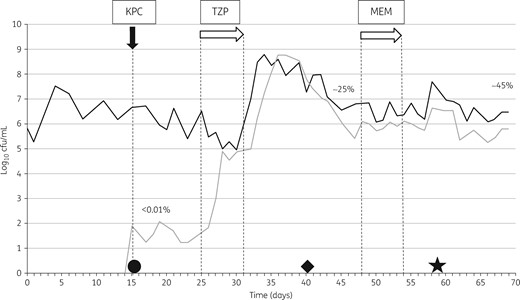
KPC model. Bacterial growth on logarithmic scale. The filled arrow indicates the inoculation phase and the open arrows indicate antibiotic instillation. KPC, K. pneumoniae with blaKPC, TZP, piperacillin/tazobactam; MEM, meropenem. The black line shows the total lactose-fermenting population and the grey line shows carbapenemase-producing K. pneumoniae. The percentages of resistant bacteria are calculated as a proportion of the total Enterobacteriaceae. Circle, strain LCC078; diamond, strain LCC093; star, strain LCC096.
Long read sequencing
Table 1 presents a summary of each bacterial isolate and its characteristics.
Summary of bacterial strain characteristics
| Model and strain number | Day | Isolate | Subtype | Plasmid encoding carbapenemase |
|---|---|---|---|---|
| NDM | ||||
| LCC079 | 15 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC088 | 57 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC081 | 48 | E. coli | ST88 | IncFIB/IncFII |
| KPC | ||||
| LCC078 | 15 | K. pneumoniae | ST661 | pKpQIL-D2 |
| LCC096 | 57 | E. coli | ST357 | pKpQIL-D2 |
| LCC093 | 40 | K. pneumoniae | ST14 | ColKP3 |
| Model and strain number | Day | Isolate | Subtype | Plasmid encoding carbapenemase |
|---|---|---|---|---|
| NDM | ||||
| LCC079 | 15 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC088 | 57 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC081 | 48 | E. coli | ST88 | IncFIB/IncFII |
| KPC | ||||
| LCC078 | 15 | K. pneumoniae | ST661 | pKpQIL-D2 |
| LCC096 | 57 | E. coli | ST357 | pKpQIL-D2 |
| LCC093 | 40 | K. pneumoniae | ST14 | ColKP3 |
Summary of bacterial strain characteristics
| Model and strain number | Day | Isolate | Subtype | Plasmid encoding carbapenemase |
|---|---|---|---|---|
| NDM | ||||
| LCC079 | 15 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC088 | 57 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC081 | 48 | E. coli | ST88 | IncFIB/IncFII |
| KPC | ||||
| LCC078 | 15 | K. pneumoniae | ST661 | pKpQIL-D2 |
| LCC096 | 57 | E. coli | ST357 | pKpQIL-D2 |
| LCC093 | 40 | K. pneumoniae | ST14 | ColKP3 |
| Model and strain number | Day | Isolate | Subtype | Plasmid encoding carbapenemase |
|---|---|---|---|---|
| NDM | ||||
| LCC079 | 15 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC088 | 57 | K. pneumoniae | ST147 | IncFIB/IncFII |
| LCC081 | 48 | E. coli | ST88 | IncFIB/IncFII |
| KPC | ||||
| LCC078 | 15 | K. pneumoniae | ST661 | pKpQIL-D2 |
| LCC096 | 57 | E. coli | ST357 | pKpQIL-D2 |
| LCC093 | 40 | K. pneumoniae | ST14 | ColKP3 |
NDM model
LCC079 (K. pneumoniae, single-locus variant of ST147, original strain) and LCC088 (K. pneumoniae, same single-locus variant of ST147, isolated at the end of the experiment) differed by only one chromosomal SNV, indicating that the original inoculated strain was maintained throughout the experiment. For both these isolates, as well as LCC081 (the CP E. coli ST88 isolated on day 47), the blaNDM-1 gene was located on the same plasmid backbone, indicating blaNDM-1 plasmid transfer from K. pneumoniae to E. coli during the experiment. This plasmid represents a previously undescribed >170 kb IncFIB/IncFII plasmid. Within this plasmid, the blaNDM-1 gene was located within a 6893 bp region flanked by 1695 bp direct repeats. There was evidence that the 6893 + 1695 bp structure was tandemly repeated; however, the exact number of repeat units in each case could not be ascertained from the long-read data, and Illumina coverage suggested variation in the number of repeat units between the three isolates (Table 2).
Estimated genomic coverage of tandem repeat
| Strain | blaNDM-1 coverage relative to plasmid backbone | Plasmid coverage relative to chromosome |
|---|---|---|
| LCC079 | 2.7 | 1.7 |
| LCC081 | 1.6 | 0.9 |
| LCC088 | 2.3 | 1.8 |
| Strain | blaNDM-1 coverage relative to plasmid backbone | Plasmid coverage relative to chromosome |
|---|---|---|
| LCC079 | 2.7 | 1.7 |
| LCC081 | 1.6 | 0.9 |
| LCC088 | 2.3 | 1.8 |
Estimated genomic coverage of tandem repeat
| Strain | blaNDM-1 coverage relative to plasmid backbone | Plasmid coverage relative to chromosome |
|---|---|---|
| LCC079 | 2.7 | 1.7 |
| LCC081 | 1.6 | 0.9 |
| LCC088 | 2.3 | 1.8 |
| Strain | blaNDM-1 coverage relative to plasmid backbone | Plasmid coverage relative to chromosome |
|---|---|---|
| LCC079 | 2.7 | 1.7 |
| LCC081 | 1.6 | 0.9 |
| LCC088 | 2.3 | 1.8 |
The same blaNDM-1-containing repeat structure has previously been described in the unrelated IncHI1B/IncFIB plasmid pPMK1-NDM from a K. pneumoniae isolated in Nepal in 2011.33 There was no evidence for any other shared plasmids between the K. pneumoniae and E. coli isolates.
KPC model
LCC078 (K. pneumoniae ST661, original strain) and LCC096 (E. coli ST357, isolated on day 57) both contained blaKPC-2 on an identical 111742 bp IncFII/IncR plasmid with >99% sequence identity to pKpQIL-D2,5 indicating blaKPC plasmid transfer from K. pneumoniae to E. coli during the experiment. There were no other shared plasmids between these two isolates; LCC078 additionally contained a 249604 bp IncFIB plasmid, a 4350 bp non-typeable plasmid and a 4000 bp Col440I plasmid, and LCC096 additionally contained a 148387 bp IncFIB/IncFIC plasmid and a 3904 bp Col156 plasmid.
LCC093 (K. pneumoniae ST14, taken on day 40) contained blaOXA-232 on a 6141 bp ColKP3 plasmid, which was identical to a previously described blaOXA-232 plasmid from a K. pneumoniae ST14 isolated in the USA in 2013.34 LCC093 additionally contained a 273971 bp IncHI1B/IncFIB plasmid, a 211819 bp IncFII/IncFIB plasmid, a 4167 bp non-typeable plasmid and a 2095 bp non-typeable plasmid.
Discussion
The in vitro human gut model provides a unique insight into CPE colonization dynamics in the colon, allowing biomass enumeration of the microbiota in real time. In the forerunner experiments of our gut model, microbiota composition was validated against the colonic bacteria found in sudden road traffic accident victims.35 Our model has since been used extensively to successfully predict the behaviour of antibiotics in the context of C. difficile infection, which is clearly mediated via alterations to the gut microbiota.36 Utilizing our predictive model, we can follow changes that occur in both resistant and susceptible bacterial populations and count viable bacterial populations with carbapenem resistance genes. The additional long-read sequencing data enabled us to track the ancestral strains/plasmids as they disseminated within the simulated colon.
We chose to study blaKPC and blaNDM as these represent two of the ‘big five’ carbapenemases present worldwide and are frequently encountered in clinical practice and hence freely available for study. In the NDM model, increasing inocula were added over 7 days to determine the threshold at which all screening tests (in triplicate) became positive. We then used our data to determine the inoculum size for the KPC model, and so used a single exposure event to replicate what we believe is most likely to occur in clinical practice. The selected antibiotics represent those commonly used in clinical practice; the regimen of piperacillin/tazobactam followed by meropenem was utilized to represent an often-used escalation of antibiotic treatment in patients who have failed to respond to initial empirical (piperacillin/tazobactam) antibiotic therapy.
Within the NDM model, the use of increasing daily inocula demonstrated that at low levels (<4.9 log10 cfu/mL) commonly used screening methods did not detect CPE. This is a phenomenon that we have observed before and is not unique to the NDM strain (C. M. Rooney, K. Davies, M. H. Wilcox and C. H. Chilton, unpublished data). For the increasing inocula (NDM model) and the single inoculum (KPC model), the resistant populations stabilized before antibiotic exposure, suggesting that antibiotic-induced dysbiosis is not a prerequisite for CPE gut colonization. However, after antibiotic dosing, selective pressure led to a marked increase in both CPE strains. Current understanding4,6–8 suggests that antibiotic exposure is a risk factor for CPE colonization, which is in keeping with our findings. Nonetheless, if antibiotic selective pressure is driving resistant populations that are already present into a detectable range, then true colonization rates could be far higher than reported.
Adding further weight to this argument, in the KPC model we identified on day 40 a K. pneumoniae strain that carried blaOXA232 resistance genes, an isolate we had neither inoculated into the model nor detected on screening. This isolate was only detected after antibiotic exposure and was present at very low levels (sporadic colonies). Interestingly, the plasmid encoding the blaOXA232 has been previously described34 in a US strain carrying dual carbapenem resistance genes (NDM and OXA), although sequencing of our strain did not show both these resistance mechanisms. We hypothesize that this isolate was undetectable until clonal expansion of the population occurred under antibiotic selective pressure. We acknowledge that environmental contamination and the presence of CPE in donor samples are possible alternative explanations for our results; however, these scenarios are unlikely given that no blaOXA232 strains were used in the laboratory at the time of this experiment, and this was not observed in the NDM model (which was primed with faeces from the same donors as used in the KPC model), furthermore we used multiple platforms to screen our donors which failed to demonstrate CPE in any of the volunteers. Metagenomics sequencing would be one way to investigate this in greater detail.
In the NDM model, we have shown that the viable resistant population doubled after antibiotic exposure; combining these results and the long-read sequencing data, we have demonstrated that this was due to an expanding clonal population. This provides clear evidence that the biomass of resistant bacteria and the subsequent transmission risk is greatest after antibiotic selective pressure. We also identified the emergence of an E. coli population with newly acquired carbapenem resistance, a similar finding to that in the KPC model. Again, as in the KPC model, in the NDM model this population shared the same plasmid as the inoculated strain, providing evidence that horizontal gene transfer had taken place, promoted by antibiotic exposure, from the dominant clonal population to susceptible bacterial populations, as is hypothesized to occur in vivo.37 Factors influencing conjugation rate include temperature, substrate, plasmid content, and donor and recipient strain identity.38 Many of these parameters are tightly controlled in our gut model, which simulates the human colonic habitat. Others have shown biofilm formation provides the optimal surroundings for horizontal gene transfer to take place;39 we intend to explore such possibilities in future CPE gut model experiments, but note that biofilm does form on the inner surfaces of vessels (although this was not specifically sampled in these studies).
The KPC model had a much higher CPE biomass than the NDM model, despite greater total inocula in the NDM model. This is true for both the inoculated strain LCC078 and the newly resistant E. coli population LCC096 when compared with LCC079 and LCC081, respectively (Figures 2 and 3), potentially related to the different antibiotics used (piperacillin/tazobactam compared with imipenem, respectively) and their impact on other faecal flora. We hypothesize that at a higher biomass burden within the gut we would observe an increased frequency of horizontal gene transfer events. However, the exact number of such events would be difficult to determine given concomitant vertical transmission. We caution, nevertheless, that the CPE biomass between gut models may not be directly comparable. Although the same donors have been used to increase reproducibility between models, piperacillin/tazobactam dosing in the KPC model resulted in a significant increase in the CP K. pneumoniae population (Figure 3), more so than imipenem exposure in the NDM model (Figure 2) (reflecting the relatively low imipenem MIC for LCC079). Therefore, the higher biomass of CP K. pneumoniae in the KPC model is reliant on both strain characteristics and antibiotic exposure.
This highlights the fact that antimicrobial stewardship is crucial to containing a CPE outbreak as inappropriate antibiotic prescribing may lead to large clonal expansion within the gut, leading to higher biomass, frequent horizontal transfer events and subsequent spread of CP organisms within the ward environment. Given that we were unable to detect low-level CPE colonization within the NDM gut model even when inoculating known quantities, and that extended-spectrum antibiotics are highly selective for carbapenemase producers, we advocate strict antimicrobial stewardship policies for all patients in a ward/unit environment during a CPE outbreak. Slow implementation or lack of such an intervention might explain why some outbreaks expand and persist, and potentially lead to endemicity in some cases.
Conclusions
We have used an in vitro human gut model to investigate the possible outcomes of CPE exposure leading to CPE colonization. Under antibiotic selective pressure, we have demonstrated clonal expansion and, critically, resistance gene transfer between species, within a healthy human gut microbiota. Furthermore, previously unidentified, resistant populations were found. This study provides important data regarding CPE colonization and dissemination within the gut microbiota, which may have key implications for antibiotic prescribing following introduction of CPE into a clinical setting and/or a CPE outbreak.
Funding
The research was funded by the National Institute for Health Research (NIHR) Health Protection Research Unit in Healthcare Associated Infections and Antimicrobial Resistance at University of Oxford in partnership with PHE (grant HPRU-2012–10041). C. M. R. was supported by the NIHR ACF scheme. A. S. W. and D. W. C. are supported by the Oxford NIHR Biomedical Research Centre. The NIHR Health Protection Research Unit Steering Committee consists of J. Coia, N. French, C. Marwick and M. Sharland.
Transparency declarations
M. H. W. has received consulting fees from Abbott Laboratories, Actelion, Antabio, AiCuris, Astellas, Astra-Zeneca, Bayer, BioMérieux, Cambimune, Cerexa, Da Volterra, The European Tissue Symposium, Ferring, The Medicines Company, Menarini, Merck, Meridian, Motif Biosciences, Nabriva, Paratek, Pfizer, Qiagen, Roche, Seres, Spero, Surface Skins, Sanofi-Pasteur, Seres, Summit, Synthetic Biologics and Valneva; lecture fees from Abbott, Alere, Allergan, Astellas, Astra-Zeneca, Merck, Nabriva, Pfizer, Roche and Seres; and grant support from Abbott, Actelion, Astellas, BioMérieux, Cubist, Da Volterra, Merck, MicroPharm, Morphochem AG, Motif Biosciences, Nabriva, Paratek, Pfizer, Sanofi-Pasteur, Seres, Summit and The European Tissue Symposium. C. H. C. has received research funding from Da Volterra, Paratek Plc, Actavis, Teva Pharmaceuticals USA Inc, Astellas Pharma Europe Ltd, Cubist Pharmaceuticals, Seres Therapeutics and has received an honorarium from Astellas Pharma Europe Ltd. The remaining authors have none to declare.
Disclaimer
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health or PHE.

{kind=link}
{kind=link}
{kind=link}