





Abstract
Beyond plasmid-encoded resistance (mcr genes) prevalence in strain collections, large epidemiological studies to estimate the human burden of colistin-resistant Escherichia coli gut carriage are lacking.
To evaluate the prevalence of colistin-resistant E. coli carriage in inpatients and decipher the molecular support of resistance and the genetic background of the strains.
During a 3 month period in 2017, we prospectively screened patients in six Parisian hospitals for rectal carriage of colistin-resistant E. coli using a selective medium, a biochemical confirmatory test and MIC determination. WGS of the resistant strains and their corresponding plasmids was performed.
Among the 1217 screened patients, 153 colistin-resistant E. coli strains were isolated from 152 patients (12.5%). The mcr-1 gene was identified in only seven isolates (4.6%) on different plasmid scaffolds. The genetic background of these MCR-1 producers argued for an animal origin. Conversely, the remaining 146 colistin-resistant E. coli exhibited a phylogenetic distribution corresponding to human gut commensal/clinical population structure (B2 and D phylogroup predominance); 72.6% of those isolates harboured convergent mutations in the PmrA and PmrB proteins, constituting a two-component system shown to be associated with colistin resistance.
We showed that the occurrence at a high rate of colistin resistance in human faecal E. coli is the result of two distinct evolutionary pathways, i.e. the occurrence of chromosomal mutations in an endogenous E. coli population and the rare acquisition of exogenous mcr-1-bearing strains probably of animal origin. The involved selective pressures need to be identified in order to develop preventative strategies.
Introduction
Colistin is among the very few last-resort antibiotics in the armamentarium for treating infections caused by MDR Gram-negative bacteria. The emergence and spread of plasmid-encoded colistin resistance among Enterobacteriaceae is therefore of great concern, threatening the clinical relevance of the polymyxin antibiotic family.1 The occurrence of colistin resistance, and more particularly transferable colistin resistance, in Escherichia coli is of paramount importance. Currently, E. coli ranks first as the bacterial species responsible for community and hospital infections worldwide. Beyond the report of infection cases, the extent of the human reservoir for colistin-resistant Enterobacteriaceae remains unclear. Most of the epidemiological data originated from strain collections that were selected either according to their resistance to another antimicrobial class (mainly isolates producing ESBLs or carbapenemases) or according to their clinical origin (e.g. bloodstream isolates).2–4 Consequently the prevalence of intestinal carriage of acquired colistin resistance in humans in Europe remains unknown, mostly because of the lack of an adequate screening medium. In the present work we performed a prospective multicentre study using a screening medium to: (i) measure the prevalence of colistin-resistant human faecal E. coli; (ii) decipher the molecular mechanisms mediating colistin resistance; and (iii) identify the genetic background of colistin-resistant E. coli isolates.
Methods
Study design and material
We performed a prospective multicentre non-interventional study among six University hospitals (‘Assistance Publique-Hôpitaux de Paris’) geographically distributed in the Paris area [two in Paris intramuros and four in the suburbs; see Figure S1 (available as Supplementary data at JAC Online) for details], France, namely the ColiRED study. During a 3 month period in 2017 (January–March), each adult patient screened at hospital admission because of risk factors for carriage of XDR bacteria (such as carbapenemase-producing Enterobacteriaceae) or at an ICU admission for MDR bacteria (i.e. ESBL-producing Enterobacteriaceae) was additionally screened for carriage of colistin-resistant E. coli using Superpolymyxin® agar plates (ELITECH Microbiology, Signes, France) according to the recommendations of the manufacturer. Regarding the French guidelines, patients at risk for carriage of eXDR bacteria (emerging extensively drug-resistant bacteria, i.e. VRE or carbapenemase-producing Enterobacteriaceae) were direct repatriates as inpatients from an overseas health institution for a hospital stay, patients with a history of hospitalization abroad within the previous year, patients with past medical history of eXDR carriage or patients with a history of contact with eXDR patients.5 Only one single rectal swab (Eswab®, Copan, Italia) per patient was included. In order to prevent the emergence of adaptive resistance, we limited the use of selective medium to the initial screening step; picking and streaking of subsequent colonies were performed on non-selective medium. Basic clinical data such as gender, age, dates of hospital admission and of specimen collection, and history of admission in the preceding year in overseas hospitals were recorded.
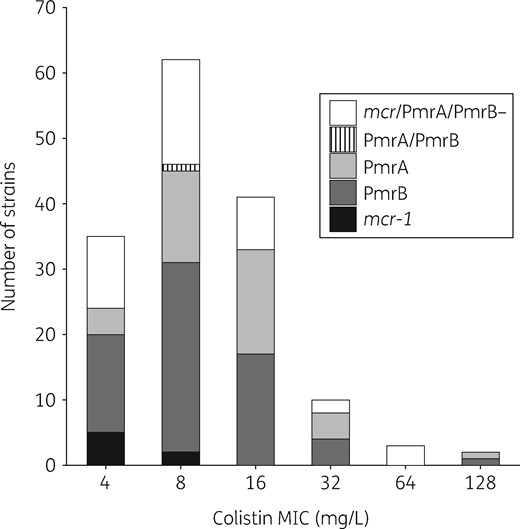
Colistin MIC distribution for the 153 colistin-resistant E. coli strains. MICs were determined using the broth microdilution method. The MICs for the seven mcr-1-positive colistin-resistant E. coli were 4 mg/L (n = 5) and 8 mg/L (n = 2) (indicated in black). The MIC distribution of the PmrA and PmrB mutations is indicated in different shades of grey. The strain mutated in both PmrA and PmrB is indicated by the hatched lines. The strain without additional mcr gene and no mutation in PmrA or in PmrB is indicated in white.
Ethics
Informed consent was obtained from each participant; this study was approved by the Ethics Committee – Institutional Review Board from Henri Mondor Hospital (IRB Mondor 20180607 Decousser).
Antimicrobial susceptibility testing
After collection, the swabs were immediately grown on the screening medium. Each different morphotype of colonies was identified at the species level using MALDI-TOF MS (Andromas®, Beckman Coulter, Villepinte, France). The colistin resistance of E. coli isolates was confirmed by using the Rapid Polymyxin NP® test (Elitech Microbiology, Signes, France) according to the recommendations of the manufacturer. MICs of colistin were determined for each strain by the broth microdilution method (BMD) according to the EUCAST recommendations (http://www.eucast.org/ast_of_bacteria/mic_determination/? no_cache=1). Antimicrobial susceptibility testing of the other antimicrobial compounds was performed using the disc diffusion method according to the EUCAST recommendations (http://www.eucast.org/ast_of_bacteria/disk_diffusion_methodology/). The list of antimicrobial compounds that were tested is reported in the Supplementary data. Carbapenemase production was tested using the Rapidec Carba NP test® (bioMérieux, Craponne, France).
WGS and analysis
The whole genomes of all E. coli isolates exhibiting an MIC >2 mg/L were sequenced using the Illumina technology and analysed with an in-house bioinformatics pipeline (see Table S1 and Table S2 for details). Briefly, after SPAdes assembly, the chromosome sequences were ordered against reference genomes with Ragout to enhance the assembly quality.6,7 The phylogroup was determined in silico using the ClermonTyping method, and read-based determination of MLST through the Warwick scheme and serotype were performed with SRST2.8,9 The resistome, the virulome and the plasmid type were defined by BlastN approach with Abricate (https://github.com/tseemann/abricate) using the ResFinder database, a custom database, and PlasmidFinder, respectively.10,11 The plasmid sequences of each isolate were also predicted by PlaScope.12 The contigs carrying mcr genes were annotated using Prokka (v1.13.3) to explore the genetic environment.13 Phylogenetic history of the strains was reconstructed using SNPs of the core genome. Briefly, SNP calling was performed for each strain against E. coli K-12 MG1655 with Snippy (v4.0) (https://github.com/tseemann/snippy). Then, only core SNPs were conserved and used to construct a maximum likelihood phylogenetic tree using FastTree (v2.1.9) with the general time reversible evolution model and a gamma distribution of rates across sites.14 All the genomic data are publicly available through the bioproject PRJEB28020.
Main characteristics of the MCR-1-producing colistin-resistant E. coli isolates
| Isolate | Hospital (name) | Hospitalization abroad historya | ST Warwick | ST Pasteur | Phylogenetic group | Serotype | Colistin resistance genotype | Incompatibility group | ESBL | Ampicillin resistance genotype | Tetracycline resistance genotype | Sulphonamide/ trimethoprim resistance genotype | Phenicol resistance genotype | Intestinal pathogenic E. coli virulence gene | Extra-intestinal E. coli virulence gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 370D | Avicenne | none | 7056 | 869 | clade I | Onovel27: H45 | mcr-1 | IncX4 | – | blaTEM-1b | tet(B), tet(M) | sul2, sul3, dfrA12 | cmlA1 | – | ompT |
| 436A | Lariboisière | none | 8154 | 842 | B1 | O54: H8 | mcr-1 | IncX4 | – | – | tet(A) | sul2, dfrA17 | – | – | iha, ireA, ompT |
| 635A | Avicenne | Thailand | 189 | 809 | A | O177: H26 | mcr-1.9 | IncX4 | – | blaTEM-1b | tet(A), tet(M) | sul1, sul3, dfrA12 | catA2 | eae | traT |
| 925B | Louis Mourrier | none | 10 | 2 | A | O89: H10 | mcr-1 | IncI2 | – | – | – | – | – | – | – |
| 933A | Avicenne | none | 48 | 843 | A | O6: H16 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A) | sul2, dfrA14 | catA1, floR | – | iha, iucC, iroN, traT |
| 1057A | Henri Mondor | Cambodia | 219 | 870 | E | O114: H34 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A), tet(M) | sul2, sul3 | cmlA1, floR | – | iroN, traT |
| 1263A | Henri Mondor | South Korea | 7329 | 881 | B1 | Onovel9: H21 | mcr-1 | IncI2 | – | blaTEM-1b | tet(A) | sul1, sul2, dfrA1 | – | – | hra, iroN, traT, ompT |
| Isolate | Hospital (name) | Hospitalization abroad historya | ST Warwick | ST Pasteur | Phylogenetic group | Serotype | Colistin resistance genotype | Incompatibility group | ESBL | Ampicillin resistance genotype | Tetracycline resistance genotype | Sulphonamide/ trimethoprim resistance genotype | Phenicol resistance genotype | Intestinal pathogenic E. coli virulence gene | Extra-intestinal E. coli virulence gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 370D | Avicenne | none | 7056 | 869 | clade I | Onovel27: H45 | mcr-1 | IncX4 | – | blaTEM-1b | tet(B), tet(M) | sul2, sul3, dfrA12 | cmlA1 | – | ompT |
| 436A | Lariboisière | none | 8154 | 842 | B1 | O54: H8 | mcr-1 | IncX4 | – | – | tet(A) | sul2, dfrA17 | – | – | iha, ireA, ompT |
| 635A | Avicenne | Thailand | 189 | 809 | A | O177: H26 | mcr-1.9 | IncX4 | – | blaTEM-1b | tet(A), tet(M) | sul1, sul3, dfrA12 | catA2 | eae | traT |
| 925B | Louis Mourrier | none | 10 | 2 | A | O89: H10 | mcr-1 | IncI2 | – | – | – | – | – | – | – |
| 933A | Avicenne | none | 48 | 843 | A | O6: H16 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A) | sul2, dfrA14 | catA1, floR | – | iha, iucC, iroN, traT |
| 1057A | Henri Mondor | Cambodia | 219 | 870 | E | O114: H34 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A), tet(M) | sul2, sul3 | cmlA1, floR | – | iroN, traT |
| 1263A | Henri Mondor | South Korea | 7329 | 881 | B1 | Onovel9: H21 | mcr-1 | IncI2 | – | blaTEM-1b | tet(A) | sul1, sul2, dfrA1 | – | – | hra, iroN, traT, ompT |
During the previous year.
Main characteristics of the MCR-1-producing colistin-resistant E. coli isolates
| Isolate | Hospital (name) | Hospitalization abroad historya | ST Warwick | ST Pasteur | Phylogenetic group | Serotype | Colistin resistance genotype | Incompatibility group | ESBL | Ampicillin resistance genotype | Tetracycline resistance genotype | Sulphonamide/ trimethoprim resistance genotype | Phenicol resistance genotype | Intestinal pathogenic E. coli virulence gene | Extra-intestinal E. coli virulence gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 370D | Avicenne | none | 7056 | 869 | clade I | Onovel27: H45 | mcr-1 | IncX4 | – | blaTEM-1b | tet(B), tet(M) | sul2, sul3, dfrA12 | cmlA1 | – | ompT |
| 436A | Lariboisière | none | 8154 | 842 | B1 | O54: H8 | mcr-1 | IncX4 | – | – | tet(A) | sul2, dfrA17 | – | – | iha, ireA, ompT |
| 635A | Avicenne | Thailand | 189 | 809 | A | O177: H26 | mcr-1.9 | IncX4 | – | blaTEM-1b | tet(A), tet(M) | sul1, sul3, dfrA12 | catA2 | eae | traT |
| 925B | Louis Mourrier | none | 10 | 2 | A | O89: H10 | mcr-1 | IncI2 | – | – | – | – | – | – | – |
| 933A | Avicenne | none | 48 | 843 | A | O6: H16 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A) | sul2, dfrA14 | catA1, floR | – | iha, iucC, iroN, traT |
| 1057A | Henri Mondor | Cambodia | 219 | 870 | E | O114: H34 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A), tet(M) | sul2, sul3 | cmlA1, floR | – | iroN, traT |
| 1263A | Henri Mondor | South Korea | 7329 | 881 | B1 | Onovel9: H21 | mcr-1 | IncI2 | – | blaTEM-1b | tet(A) | sul1, sul2, dfrA1 | – | – | hra, iroN, traT, ompT |
| Isolate | Hospital (name) | Hospitalization abroad historya | ST Warwick | ST Pasteur | Phylogenetic group | Serotype | Colistin resistance genotype | Incompatibility group | ESBL | Ampicillin resistance genotype | Tetracycline resistance genotype | Sulphonamide/ trimethoprim resistance genotype | Phenicol resistance genotype | Intestinal pathogenic E. coli virulence gene | Extra-intestinal E. coli virulence gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 370D | Avicenne | none | 7056 | 869 | clade I | Onovel27: H45 | mcr-1 | IncX4 | – | blaTEM-1b | tet(B), tet(M) | sul2, sul3, dfrA12 | cmlA1 | – | ompT |
| 436A | Lariboisière | none | 8154 | 842 | B1 | O54: H8 | mcr-1 | IncX4 | – | – | tet(A) | sul2, dfrA17 | – | – | iha, ireA, ompT |
| 635A | Avicenne | Thailand | 189 | 809 | A | O177: H26 | mcr-1.9 | IncX4 | – | blaTEM-1b | tet(A), tet(M) | sul1, sul3, dfrA12 | catA2 | eae | traT |
| 925B | Louis Mourrier | none | 10 | 2 | A | O89: H10 | mcr-1 | IncI2 | – | – | – | – | – | – | – |
| 933A | Avicenne | none | 48 | 843 | A | O6: H16 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A) | sul2, dfrA14 | catA1, floR | – | iha, iucC, iroN, traT |
| 1057A | Henri Mondor | Cambodia | 219 | 870 | E | O114: H34 | mcr-1 | IncHI2 | – | blaTEM-1b | tet(A), tet(M) | sul2, sul3 | cmlA1, floR | – | iroN, traT |
| 1263A | Henri Mondor | South Korea | 7329 | 881 | B1 | Onovel9: H21 | mcr-1 | IncI2 | – | blaTEM-1b | tet(A) | sul1, sul2, dfrA1 | – | – | hra, iroN, traT, ompT |
During the previous year.
Phylogenetic group/subgroup distribution of the 146 mcr-negative colistin-resistant E. coli with their resistance and virulence characteristics
| Phylogenetic group/subgroup | Strains, n (%) | PmrA/PmrB mutationa, n (%) | ESBL, n (%) | Ampicillin resistance, n (%) | Tetracycline resistance, n (%) | Sulphonamide and/or trimethoprim resistance, n (%) | Phenicol resistance, n (%) | Intestinal pathogenic E. coli virulence gene score, M (min–max) | Extra-intestinal E. coli virulence gene score, M (min–max) |
|---|---|---|---|---|---|---|---|---|---|
| A | 17 (11.6) | 10 (58.8) | 2 (11.8) | 9 (52.9) | 5 (29.4) | 5 (29.4) | 2 (11.8) | 0.1 (0–1) | 2.8 (0–6) |
| B1 | 12 (8.2) | 10 (83.3) | 1 (8.3) | 6 (50.0) | 4 (33.3) | 6 (50.0) | 1 (8.3) | 0.3 (0–2) | 2.6 (0–9) |
| CC87 | 3 (25.0) | 3 (100.0) | 0 | 3 (100.0) | 3 (100.0) | 3 (100.0) | 1 (33.3) | 0.3 (0–1) | 7.3 (6–9) |
| B2 | 68 (46.6) | 48 (70.6) | 11 (16.2) | 36 (52.9) | 20 (29.4) | 26 (38.2) | 4 (5.9) | 0.0 (0–1) | 10.4 (3–17) |
| I ST131 | 14 (20.6) | 12 (85.7) | 9 (64.3) | 14 (100.0) | 9 (64.3) | 8 (57.1) | 2 (14.3) | 0.1 (0–1) | 8.8 (5–12) |
| I non-ST131 | 3 (4.4) | 3 (100.0) | 0 | 2 (66.7) | 2 (66.7) | 1 (33.3) | 0 | 0.0 (0–0) | 7.3 (6–9) |
| II | 17 (25) | 9 (52.9) | 0 | 8 (47.1) | 2 (11.8) | 7 (41.2) | 1 (5.9) | 0.0 (0–0) | 13.2 (7–17) |
| III | 1 (1.5) | 1 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 12.0 (12–12) |
| IV | 2 (2.9) | 2 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 9.0 (6–12) |
| V | 1 (1.5) | 1 (100.0) | 0 | 1 (100.0) | 0 | 1 (100.0) | 0 | 0.0 (0–0) | 12 (12–12) |
| VI | 4 (5.9) | 3 (75.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 11.8 (11–12) |
| VII | 11 (16.2) | 4 (36.4) | 0 | 9 (81.8) | 4 (36.4) | 5 (45.5) | 0 | 0.1 (0–1) | 9.1 (6–13) |
| IX | 7 (10.3) | 6 (85.7) | 1 (14.3) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 1 (14.3) | 0.0 (0–0) | 11.9 (10–14) |
| UA | 8 (11.8) | 7 (87.5) | 1 (12.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 0 | 0.0 (0–0) | 8.6 (3–13) |
| C | 4 (2.7) | 4 (100.0) | 0 | 3 (75.0) | 4 (100.0) | 3 (75.0) | 3 (75.0) | 0.3 (0–1) | 8.3 (7–10) |
| D | 29 (19.9) | 24 (82.8) | 4 (13.8) | 23 (79.3) | 12 (41.4) | 18 (62.1) | 4 (13.8) | 0.2 (0–2) | 5.9 (0–11) |
| CGA | 20 (69.0) | 16 (80.0) | 2 (10.0) | 17 (85.0) | 11 (55.0) | 13 (65.0) | 2 (10.0) | 0.0 (0–0) | 7.2 (1–11) |
| non-CGA | 9 (31.0) | 8 (88.9) | 2 (22.2) | 6 (66.7) | 1 (11.1) | 5 (55.6) | 2 (22.2) | 0.6 (0–2) | 3.1 (0–6) |
| E | 3 (2.1) | 3 (100.0) | 1 (33.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 0 | 0.0 (0–0) | 3.3 (1–5) |
| F | 12 (8.2) | 7 (58.3) | 1 (8.3) | 5 (41.7) | 2 (16.7) | 6 (50.0) | 1 (8.3) | 0.1 (0–1) | 7.3 (4–12) |
| Clade V | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 0.0 (0–0) |
| Total | 146 (100.0) | 106 (72.6) | 20 (13.7) | 83 (56.8) | 47 (32.2) | 65 (44.5) | 15 (10.3) | 0.1 (0–2) | 7.5 (0–17) |
| Phylogenetic group/subgroup | Strains, n (%) | PmrA/PmrB mutationa, n (%) | ESBL, n (%) | Ampicillin resistance, n (%) | Tetracycline resistance, n (%) | Sulphonamide and/or trimethoprim resistance, n (%) | Phenicol resistance, n (%) | Intestinal pathogenic E. coli virulence gene score, M (min–max) | Extra-intestinal E. coli virulence gene score, M (min–max) |
|---|---|---|---|---|---|---|---|---|---|
| A | 17 (11.6) | 10 (58.8) | 2 (11.8) | 9 (52.9) | 5 (29.4) | 5 (29.4) | 2 (11.8) | 0.1 (0–1) | 2.8 (0–6) |
| B1 | 12 (8.2) | 10 (83.3) | 1 (8.3) | 6 (50.0) | 4 (33.3) | 6 (50.0) | 1 (8.3) | 0.3 (0–2) | 2.6 (0–9) |
| CC87 | 3 (25.0) | 3 (100.0) | 0 | 3 (100.0) | 3 (100.0) | 3 (100.0) | 1 (33.3) | 0.3 (0–1) | 7.3 (6–9) |
| B2 | 68 (46.6) | 48 (70.6) | 11 (16.2) | 36 (52.9) | 20 (29.4) | 26 (38.2) | 4 (5.9) | 0.0 (0–1) | 10.4 (3–17) |
| I ST131 | 14 (20.6) | 12 (85.7) | 9 (64.3) | 14 (100.0) | 9 (64.3) | 8 (57.1) | 2 (14.3) | 0.1 (0–1) | 8.8 (5–12) |
| I non-ST131 | 3 (4.4) | 3 (100.0) | 0 | 2 (66.7) | 2 (66.7) | 1 (33.3) | 0 | 0.0 (0–0) | 7.3 (6–9) |
| II | 17 (25) | 9 (52.9) | 0 | 8 (47.1) | 2 (11.8) | 7 (41.2) | 1 (5.9) | 0.0 (0–0) | 13.2 (7–17) |
| III | 1 (1.5) | 1 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 12.0 (12–12) |
| IV | 2 (2.9) | 2 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 9.0 (6–12) |
| V | 1 (1.5) | 1 (100.0) | 0 | 1 (100.0) | 0 | 1 (100.0) | 0 | 0.0 (0–0) | 12 (12–12) |
| VI | 4 (5.9) | 3 (75.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 11.8 (11–12) |
| VII | 11 (16.2) | 4 (36.4) | 0 | 9 (81.8) | 4 (36.4) | 5 (45.5) | 0 | 0.1 (0–1) | 9.1 (6–13) |
| IX | 7 (10.3) | 6 (85.7) | 1 (14.3) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 1 (14.3) | 0.0 (0–0) | 11.9 (10–14) |
| UA | 8 (11.8) | 7 (87.5) | 1 (12.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 0 | 0.0 (0–0) | 8.6 (3–13) |
| C | 4 (2.7) | 4 (100.0) | 0 | 3 (75.0) | 4 (100.0) | 3 (75.0) | 3 (75.0) | 0.3 (0–1) | 8.3 (7–10) |
| D | 29 (19.9) | 24 (82.8) | 4 (13.8) | 23 (79.3) | 12 (41.4) | 18 (62.1) | 4 (13.8) | 0.2 (0–2) | 5.9 (0–11) |
| CGA | 20 (69.0) | 16 (80.0) | 2 (10.0) | 17 (85.0) | 11 (55.0) | 13 (65.0) | 2 (10.0) | 0.0 (0–0) | 7.2 (1–11) |
| non-CGA | 9 (31.0) | 8 (88.9) | 2 (22.2) | 6 (66.7) | 1 (11.1) | 5 (55.6) | 2 (22.2) | 0.6 (0–2) | 3.1 (0–6) |
| E | 3 (2.1) | 3 (100.0) | 1 (33.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 0 | 0.0 (0–0) | 3.3 (1–5) |
| F | 12 (8.2) | 7 (58.3) | 1 (8.3) | 5 (41.7) | 2 (16.7) | 6 (50.0) | 1 (8.3) | 0.1 (0–1) | 7.3 (4–12) |
| Clade V | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 0.0 (0–0) |
| Total | 146 (100.0) | 106 (72.6) | 20 (13.7) | 83 (56.8) | 47 (32.2) | 65 (44.5) | 15 (10.3) | 0.1 (0–2) | 7.5 (0–17) |
Missense mutations previously reported to be associated with colistin resistance as well as mutations modifying the length of the protein were considered. For the sake of clarity, strain 130A belonging to ST131 and exhibiting two mutations (one in PmrA and one in PmrB) was scored with a unique mutation.
Phylogenetic group/subgroup distribution of the 146 mcr-negative colistin-resistant E. coli with their resistance and virulence characteristics
| Phylogenetic group/subgroup | Strains, n (%) | PmrA/PmrB mutationa, n (%) | ESBL, n (%) | Ampicillin resistance, n (%) | Tetracycline resistance, n (%) | Sulphonamide and/or trimethoprim resistance, n (%) | Phenicol resistance, n (%) | Intestinal pathogenic E. coli virulence gene score, M (min–max) | Extra-intestinal E. coli virulence gene score, M (min–max) |
|---|---|---|---|---|---|---|---|---|---|
| A | 17 (11.6) | 10 (58.8) | 2 (11.8) | 9 (52.9) | 5 (29.4) | 5 (29.4) | 2 (11.8) | 0.1 (0–1) | 2.8 (0–6) |
| B1 | 12 (8.2) | 10 (83.3) | 1 (8.3) | 6 (50.0) | 4 (33.3) | 6 (50.0) | 1 (8.3) | 0.3 (0–2) | 2.6 (0–9) |
| CC87 | 3 (25.0) | 3 (100.0) | 0 | 3 (100.0) | 3 (100.0) | 3 (100.0) | 1 (33.3) | 0.3 (0–1) | 7.3 (6–9) |
| B2 | 68 (46.6) | 48 (70.6) | 11 (16.2) | 36 (52.9) | 20 (29.4) | 26 (38.2) | 4 (5.9) | 0.0 (0–1) | 10.4 (3–17) |
| I ST131 | 14 (20.6) | 12 (85.7) | 9 (64.3) | 14 (100.0) | 9 (64.3) | 8 (57.1) | 2 (14.3) | 0.1 (0–1) | 8.8 (5–12) |
| I non-ST131 | 3 (4.4) | 3 (100.0) | 0 | 2 (66.7) | 2 (66.7) | 1 (33.3) | 0 | 0.0 (0–0) | 7.3 (6–9) |
| II | 17 (25) | 9 (52.9) | 0 | 8 (47.1) | 2 (11.8) | 7 (41.2) | 1 (5.9) | 0.0 (0–0) | 13.2 (7–17) |
| III | 1 (1.5) | 1 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 12.0 (12–12) |
| IV | 2 (2.9) | 2 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 9.0 (6–12) |
| V | 1 (1.5) | 1 (100.0) | 0 | 1 (100.0) | 0 | 1 (100.0) | 0 | 0.0 (0–0) | 12 (12–12) |
| VI | 4 (5.9) | 3 (75.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 11.8 (11–12) |
| VII | 11 (16.2) | 4 (36.4) | 0 | 9 (81.8) | 4 (36.4) | 5 (45.5) | 0 | 0.1 (0–1) | 9.1 (6–13) |
| IX | 7 (10.3) | 6 (85.7) | 1 (14.3) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 1 (14.3) | 0.0 (0–0) | 11.9 (10–14) |
| UA | 8 (11.8) | 7 (87.5) | 1 (12.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 0 | 0.0 (0–0) | 8.6 (3–13) |
| C | 4 (2.7) | 4 (100.0) | 0 | 3 (75.0) | 4 (100.0) | 3 (75.0) | 3 (75.0) | 0.3 (0–1) | 8.3 (7–10) |
| D | 29 (19.9) | 24 (82.8) | 4 (13.8) | 23 (79.3) | 12 (41.4) | 18 (62.1) | 4 (13.8) | 0.2 (0–2) | 5.9 (0–11) |
| CGA | 20 (69.0) | 16 (80.0) | 2 (10.0) | 17 (85.0) | 11 (55.0) | 13 (65.0) | 2 (10.0) | 0.0 (0–0) | 7.2 (1–11) |
| non-CGA | 9 (31.0) | 8 (88.9) | 2 (22.2) | 6 (66.7) | 1 (11.1) | 5 (55.6) | 2 (22.2) | 0.6 (0–2) | 3.1 (0–6) |
| E | 3 (2.1) | 3 (100.0) | 1 (33.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 0 | 0.0 (0–0) | 3.3 (1–5) |
| F | 12 (8.2) | 7 (58.3) | 1 (8.3) | 5 (41.7) | 2 (16.7) | 6 (50.0) | 1 (8.3) | 0.1 (0–1) | 7.3 (4–12) |
| Clade V | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 0.0 (0–0) |
| Total | 146 (100.0) | 106 (72.6) | 20 (13.7) | 83 (56.8) | 47 (32.2) | 65 (44.5) | 15 (10.3) | 0.1 (0–2) | 7.5 (0–17) |
| Phylogenetic group/subgroup | Strains, n (%) | PmrA/PmrB mutationa, n (%) | ESBL, n (%) | Ampicillin resistance, n (%) | Tetracycline resistance, n (%) | Sulphonamide and/or trimethoprim resistance, n (%) | Phenicol resistance, n (%) | Intestinal pathogenic E. coli virulence gene score, M (min–max) | Extra-intestinal E. coli virulence gene score, M (min–max) |
|---|---|---|---|---|---|---|---|---|---|
| A | 17 (11.6) | 10 (58.8) | 2 (11.8) | 9 (52.9) | 5 (29.4) | 5 (29.4) | 2 (11.8) | 0.1 (0–1) | 2.8 (0–6) |
| B1 | 12 (8.2) | 10 (83.3) | 1 (8.3) | 6 (50.0) | 4 (33.3) | 6 (50.0) | 1 (8.3) | 0.3 (0–2) | 2.6 (0–9) |
| CC87 | 3 (25.0) | 3 (100.0) | 0 | 3 (100.0) | 3 (100.0) | 3 (100.0) | 1 (33.3) | 0.3 (0–1) | 7.3 (6–9) |
| B2 | 68 (46.6) | 48 (70.6) | 11 (16.2) | 36 (52.9) | 20 (29.4) | 26 (38.2) | 4 (5.9) | 0.0 (0–1) | 10.4 (3–17) |
| I ST131 | 14 (20.6) | 12 (85.7) | 9 (64.3) | 14 (100.0) | 9 (64.3) | 8 (57.1) | 2 (14.3) | 0.1 (0–1) | 8.8 (5–12) |
| I non-ST131 | 3 (4.4) | 3 (100.0) | 0 | 2 (66.7) | 2 (66.7) | 1 (33.3) | 0 | 0.0 (0–0) | 7.3 (6–9) |
| II | 17 (25) | 9 (52.9) | 0 | 8 (47.1) | 2 (11.8) | 7 (41.2) | 1 (5.9) | 0.0 (0–0) | 13.2 (7–17) |
| III | 1 (1.5) | 1 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 12.0 (12–12) |
| IV | 2 (2.9) | 2 (100.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 9.0 (6–12) |
| V | 1 (1.5) | 1 (100.0) | 0 | 1 (100.0) | 0 | 1 (100.0) | 0 | 0.0 (0–0) | 12 (12–12) |
| VI | 4 (5.9) | 3 (75.0) | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 11.8 (11–12) |
| VII | 11 (16.2) | 4 (36.4) | 0 | 9 (81.8) | 4 (36.4) | 5 (45.5) | 0 | 0.1 (0–1) | 9.1 (6–13) |
| IX | 7 (10.3) | 6 (85.7) | 1 (14.3) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 1 (14.3) | 0.0 (0–0) | 11.9 (10–14) |
| UA | 8 (11.8) | 7 (87.5) | 1 (12.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 0 | 0.0 (0–0) | 8.6 (3–13) |
| C | 4 (2.7) | 4 (100.0) | 0 | 3 (75.0) | 4 (100.0) | 3 (75.0) | 3 (75.0) | 0.3 (0–1) | 8.3 (7–10) |
| D | 29 (19.9) | 24 (82.8) | 4 (13.8) | 23 (79.3) | 12 (41.4) | 18 (62.1) | 4 (13.8) | 0.2 (0–2) | 5.9 (0–11) |
| CGA | 20 (69.0) | 16 (80.0) | 2 (10.0) | 17 (85.0) | 11 (55.0) | 13 (65.0) | 2 (10.0) | 0.0 (0–0) | 7.2 (1–11) |
| non-CGA | 9 (31.0) | 8 (88.9) | 2 (22.2) | 6 (66.7) | 1 (11.1) | 5 (55.6) | 2 (22.2) | 0.6 (0–2) | 3.1 (0–6) |
| E | 3 (2.1) | 3 (100.0) | 1 (33.3) | 1 (33.3) | 0 (0.0) | 1 (33.3) | 0 | 0.0 (0–0) | 3.3 (1–5) |
| F | 12 (8.2) | 7 (58.3) | 1 (8.3) | 5 (41.7) | 2 (16.7) | 6 (50.0) | 1 (8.3) | 0.1 (0–1) | 7.3 (4–12) |
| Clade V | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0.0 (0–0) | 0.0 (0–0) |
| Total | 146 (100.0) | 106 (72.6) | 20 (13.7) | 83 (56.8) | 47 (32.2) | 65 (44.5) | 15 (10.3) | 0.1 (0–2) | 7.5 (0–17) |
Missense mutations previously reported to be associated with colistin resistance as well as mutations modifying the length of the protein were considered. For the sake of clarity, strain 130A belonging to ST131 and exhibiting two mutations (one in PmrA and one in PmrB) was scored with a unique mutation.
Plasmid sequencing and identification of plasmid-mediated colistin resistance determinants
To confirm the data of plasmid sequences obtained with WGS of the natural isolates, we performed conjugation assays for all the mcr-positive strains using E. coli J53 as the recipient strain, followed by PCR-based replicon typing as described to identify the genetic background of the mcr-bearing plasmids.15 The whole genome of each E. coli J53 transconjugant was also sequenced by using the Illumina technology and assembled with SPAdes. Then, E. coli J53 transconjugants were aligned against the E. coli J53 complete reference sequence using Quast 4.5 with standard parameters.16,17 Unaligned contigs (i.e. plasmid sequences) were analysed with ResFinder and PlasmidFinder.
Prediction of the impact of amino acid substitutions of PmrA and PmrB proteins
Functional effects of missense mutations observed in PmrA and PmrB were predicted with SIFT, PolyPhen-2 and PROVEAN software as proposed in Desroches et al.18 Mutations were considered as impacting the protein when they were predicted by at least two of these as damaging/deleterious. Additionally, we looked for mutations in pmrA and pmrB genes that modified the length of the protein.19
Mutations in other genes involved in chromosomal colistin resistance in other bacteria
We looked for mutations in Klebsiella pneumoniae mgrB and crrB orthologous genes in E. coli strains that have been reported in colistin-resistant K. pneumoniae.1 Regarding mgrB, which is also named yobG, we tested its presence and putative associated mutation in the corresponding protein using the online available reference sequence (https://www.uniprot.org/uniprot/P64512). Regarding ccrB, we looked for genes presenting at least 30% similarity in terms of protein counterpart.
Data management and statistical analysis
Clinical data were collected into a Microsoft Excel 2010 database that was password protected. The data management was approved by the Commission Nationale de l’Informatique et des Libertés (#2201069 v 0). The Mann–Whitney test for between-group comparisons with Bonferroni correction was used to compare colistin MICs according to resistance mechanisms. All tests were two-sided and the level of statistical significance was set at 0.05.
Results
Population characteristics
During the study period, 1217 patients originating from the six hospitals were prospectively screened. The median age was 65 years and the male/female ratio was 1.49. The reason for screening was the presence of risk factors for MDR/XDR carriage at ICU admission (1111/1217, 91.3%) or the presence of risk factors for XDR carriage at hospital admission (106/1217, 8.7%).
Prevalence of colistin-resistant strains
Most of the included patients were hospitalized <48 h before screening (817/1217, 67.1%). The screening test was positive for 178 patients from which 184 E. coli isolates identified by MALDI TOF were recovered. The rapid confirmation test for colistin resistance was positive for 165 patients, with a total of 168 positive strains (Figure S2). The determination of MICs identified 153 E. coli strains with an MIC >2 mg/L considered as colistin-resistant E. coli from 152 patients (Figure 1 and Figure S2). According to the BMD results, the specificities of the Superpolymyxin® screening agar and the Rapid Polymyxin NP® test observed in our study were therefore 83.2% and 91.1%, respectively (Figure S2). The 16 isolates for which negative results were obtained with the confirmation test, as well as the 15 isolates with an MIC of colistin ≤2 mg/L were all negative for mcr genes (data not shown). Overall, the prevalence of colistin-resistant E. coli carriage was 12.5% (152/1217).
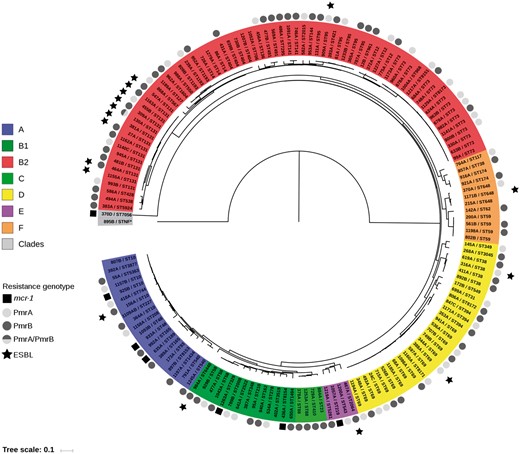
Maximum likelihood tree of the 153 colistin-resistant E. coli strains reconstructed from 422324 core SNPs. The tree was rooted on strain 895B (Escherichia clade V). The strains are indicated by their ID and ST (Warwick scheme). The main E. coli phylogroups (A, B1, B2, C, D, E and F) and the Escherichia clades are represented by colours. The presence of mcr genes, PmrA/PmrB mutations (missense or altering the length of the protein) and production of an ESBL are indicated outside the strain ID circle according to the given code. The scale at the bottom represents genetic distances in nucleotide substitutions per site. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Prevalence and characteristics of mcr-positive strains
Seven out of the 153 colistin-resistant E. coli isolates (4.6%) (Figure 1 and Figure S2) recovered from four out of the six participating centres and from seven distinct patients were positive for the mcr-1 gene. No isolate was found to be positive for other known mcr genes (mcr-2 to mcr-8) (Table 1). Although the number of colistin-resistant mcr-positive E. coli was low (n = 7), these strains seem to be more frequent in the group of patients screened at hospital admission compared with the group of patients screened at ICU admission (data not shown). Three out of the seven patients were hospitalized abroad during the previous year (Thailand, Cambodia and South Korea), and six out of the seven patients had been hospitalized for <48 h. The plasmid support of the mcr-1 gene was determined using the PlaScope pipeline and confirmed in all except one isolate by in vitro and in silico analyses of E. coli transconjugants. The plasmids carrying the mcr-1 gene possessed different backbones, including IncX4, IncI2 and IncHI2 (Table 1 and Table S3). Those plasmid types have been reported in association with the mcr-1 gene in previous studies.1 The observed mcr-1 genetic environments were also typical, with the presence of the pap2 gene downstream, the presence of parA/pir and nikB genes in the IncX4 and IncI2 plasmids, respectively, and the infrequent presence of ISApl1 (Figure S3).20 Among the participating patients, 0.6% (7/1217) carried a colistin-resistant E. coli that harboured mcr-1. With the exception of isolate 925B, all the mcr-1-positive E. coli belonged to different phylogenetic groups (A, B1, E or Escherichia clade I) and STs frequently reported in animals, but rarely in humans (Figure 2).21–25 A single isolate (635A) exhibited the eae gene coding for intimin and can be considered as an atypical enteropathogenic E. coli. All the mcr-1-positive E. coli isolates possessed a very limited number of extra-intestinal virulence genes (Table 1). Furthermore, all mcr-1-positive E. coli except one (isolate 925B) harboured resistance determinants to antibiotics frequently used in veterinary medicine, i.e. tetracycline, sulphonamide/trimethoprim and phenicols (Table 1). All isolates remained susceptible to expanded-spectrum cephalosporins and carbapenems. No mutation previously associated with colistin resistance was identified in the chromosomal genes (pmrA, pmrB, phoP, phoQ and yobG; see below) of those MCR producers.1,26
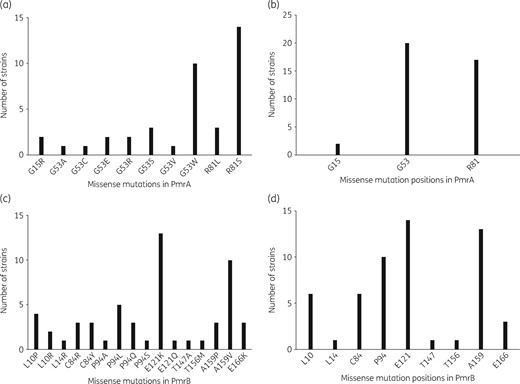
Amino acid sequence missense mutations in PmrA and PmrB with their locations in the 146 colistin-resistant mcr-negative E. coli strains. (a and b) and (c and d) correspond to PmrA and PmrB proteins, respectively. (a and c) correspond to the different mutations identified, whereas (b and d) represent the location of the mutations in the proteins. The one-letter code for the amino acid designation is used.
Prevalence and characteristics of colistin-resistant mcr-negative strains
Among the 146 colistin-resistant E. coli isolates that did not carry any mcr gene, missense mutations in the pmrA or pmrB gene, coding for a two-component system associated with colistin resistance, were identified in 39 (26.7%) and 55 (37.7%) isolates, respectively (Figure 1).27–32 The mutations were mostly localized in two (G53 and R81) and five (L10, C84, P94, E121 and A159) amino acid hot spots in PmrA and PmrB, respectively (Figure 3 and Table S4); these positions were shown to be critical for colistin resistance.1 All except one of these mutations (strain 806A) were predicted as impacting the protein (Table S4). In addition, we identified one and 12 mutations in pmrA and pmrB genes, respectively, modifying the length of the protein: four truncating mutations (nonsense n = 2, frameshift n = 2), three amino acid duplications (2–10 amino acids) and six deletions (1–10 amino acids) (Table S5). Of note, the strain 130A (ST131 O25b: H4) exhibiting the amino acid deletion in PmrA also possessed a missense mutation in PmrB (colistin MIC = 8 mg/L). Most of the mutations occurred in specific domains of the proteins: the REC domain (residues 1–112) for PmrA and the HAMP (residues 92–114) and HisKA (residues 145–205) domains for PmrB (Figure 3). The strains bearing these mutations belonged to various phylogenetic groups and STs (Table 2 and Figure 2). The frequent occurrence of PmrA/PmrB mutations (missense or modified length) targeting specific domains in distinct clones argues for multiple independent mutational events and a convergent evolution. No mutation reported in association with colistin resistance was observed in the PhoP/PhoQ proteins.
Regarding yobG, the orthologue of mgrB in E. coli, we did not identify any mutation or IS insertion that modified the length of the protein. We found only mutations that corresponded to polymorphisms associated with the phylogroup (the V8A mutation in strains from the B2 phylogroup, V8A and V12A mutations in the strain from clade V, and the I41L mutation in the seven strains belonging to STc95). We did not identify a crrB orthologue in E. coli; this must be a K. pneumoniae-specific gene.
The mcr-negative isolates mainly belonged to the phylogenetic group B2. Their distribution within the seven main phylogenetic groups actually mirrored that of the overall commensal/clinical isolates previously identified in the Paris area (Table 2 and Figure 2).14,33 Those isolates possessed a limited number of intestinal pathogenic genes, but a high rate of extra-intestinal pathogenic genes. According to the resistome analysis, a rate of 11.6% of ESBL carriers was found, and 32.2%, 44.5% and 10.3% of the isolates showed resistance to tetracycline, sulphonamide/trimethoprim and phenicols, respectively (Table 2). According to the results of the disc diffusion antimicrobial susceptibility testing, the percentage of expanded-spectrum cephalosporin- or carbapenem-resistant strains among pmrA/B mutants was 11.3% and 0%, respectively. Of note, comparison of colistin MICs according to resistance mechanisms (i.e. mcr-1, PmrA mutation, PmrB mutation, unknown) revealed a significantly lower MIC for mcr-1-bearing strains versus PmrA- or PmrB-mutated strains [Mann–Whitney P = 0.00084 (Bonferroni corrected P = 0.0083) and Mann–Whitney P = 0.0078 (Bonferroni corrected P = 0.0083), respectively]. Regarding the PmrA- and/or PmrB-mutated and PmrA/B non-mutated strains, no difference in terms of phylogenetic groups, MIC50, MIC90 and MIC range was reported (data not shown).
Discussion
The occurrence of colistin-resistant E. coli prevalence in humans is a pivotal issue, as the reasons for their selection remain intriguing in many instances and the extent of their dissemination remains largely unknown. In order possibly to prevent their further selection and spread, their prevalence must be evaluated globally (i.e. the gut carriage) without considering other antibiotic resistance traits, since many of those isolates do not exhibit additional resistance mechanisms. Evaluating the rate of faecal carriage with colistin-resistant Gram-negatives is a challenging process owing to: (i) the interfering effect of intrinsically resistant bacterial species; (ii) some inoculum effect associated with colistin resistance; (iii) the issues of colistin stability and availability in solid agar medium; and (iv) the phenotypic expression of colistin resistance that may be inducible or lead to low-level resistance, especially for mcr-associated resistance.1 To achieve this goal, we used an agar screening medium, corresponding to the commercial product derived from a previously published home-made medium.34 Using such a technical approach, we focused on a well-defined test population.
Surprisingly, we showed that the prevalence of patients colonized by colistin-resistant E. coli was very high (12.5%) compared with published studies.35 The contribution of plasmid-mediated MCR-mediated resistance was, however, quite low (4.6%). In 2017, two single-centre studies based on a selective agar culture failed to identify any plasmid-mediated colistin-resistant E. coli in France and Switzerland among 653 and 1144 specimens, respectively.35,36 When excluding naturally resistant species (such as Hafnia alvei, Proteus spp., Morganella spp., Providencia spp., and Serratia spp.), the prevalence of colistin-resistant Enterobacteriaceae ranged from 0.8% to 1.5%, with no identification of mcr-1-positive strains. A Dutch study focusing on the mcr-1 prevalence among travellers through a metagenomic approach found only a single MCR-producing colistin-resistant E. coli carrier out of 122 healthy humans before travel.37 Here in France, a study focusing on the prevalence of mcr-1 in patients attending a tertiary care hospital through a real-time PCR approach identified a 0.35% prevalence rate (576 tested patients).38 In China, several studies focused on the carriage of MCR-producing strains. Zhong et al.39 reported a 6.2% mcr-1 prevalence from inpatients and outpatients of three hospitals, mainly corresponding to MDR E. coli. This prevalence was 1.6% in another study and rose to 9.8% when hospitalized children were targeted.40,41 In Hong Kong, the prevalence was 2.0% in a mixed population of healthy subjects and inpatients.42
In our study, the prevalence of plasmid-mediated colistin-resistant E. coli was 0.5%. This prevalence could be underestimated by the use of a selective screening medium and the well-known existence of colistin-susceptible mcr-positive strains.1 Among the seven carriers, three were hospitalized in Asia in the previous year. Although we did not know how many mcr non-carriers presented such risk factors, our work therefore could support that travelling to Asia and hospitalization abroad might be considered as risk factors for acquisition of MCR-producing Enterobacteriaceae.43 The genetic supports and environments of the mcr genes identified in our study were very similar to those reported in different parts of the world, with three different plasmid types identified belonging to commonly identified incompatibility groups (Table 1 and Figure S3).4,20
Interestingly, for all except one isolate, the genetic backgrounds of those MCR-1-producing colistin-resistant E. coli were particular since they corresponded to strains usually identified in animals (Table 1). The Escherichia clade I isolate, exhibiting the − − + − genotype using the Clermont method, actually corresponds to STc2715 (Warwick scheme) and is found in 73 strains of the EnteroBase database (https://enterobase.warwick.ac.uk/species/index/ecoli), all from animal (mainly livestock) and environmental origins.44 Similarly, ST219 (isolate 1057A) and ST189 (isolate 635A) were identified in 14 and 51 strains of the EnteroBase database, respectively, almost all originating from poultry and livestock. Isolate 1263A belonged to the ST58/ST155 complex recently reported as being of animal origin and spreading in humans.24 Noticeably, strains belonging to this clonal complex have a high capacity for acquiring and disseminating resistance genes, thus making the identification of the mcr-1 gene in such a clone even more worrisome.24 Isolate 436A belonged to a very rare B1 phylogroup ST and B1 strains are mostly of animal origin, and isolate 933A belonged to the phylogroup A ST48, frequently associated with animals (383 strains identified in the EnteroBase database, mainly from livestock and poultry) and considered as zoonotic.21,25 Finally, the phylogroup A ST10 isolate (925B) might be of either human or animal origin.
Altogether, these observations are in line with a recent study including >300 mcr-1-positive E. coli that showed a predominance of STc10 (including ST48), ST189 and ST155, almost all of animal origin.4 In combination with their genetic background, the resistance gene armamentarium of those strains (except 925B) supports their animal origin. Indeed, a high rate of genes encoding resistance to chloramphenicol (floR, catA and cml), tetracycline (tet family) and/or sulphonamide/trimethoprim (sul and dfr) was found among MCR-1 producers in our study (Table 1) with those resistance genes corresponding to antibiotics widely used in veterinary practice. The subsequent acquisition of an mcr-bearing plasmid by E. coli strains belonging to a highly virulent human clone (i.e. ST131) as reported by Wang et al.41 does not contradict this hypothesis.
Beyond identification of plasmid-mediated colistin resistance, the second relevant finding obtained through the present study was the unexpected high prevalence of mcr-negative colistin-resistant E. coli carriage (12.0%). As opposed to MCR-1-producing colistin-resistant E. coli, the genetic background (phylogenetic group to which it belongs, presence of extra-intestinal genes) as well as the pattern of antibiotic resistance (production of ESBL, low level of resistance to tetracycline, sulphonamide/trimethoprim and phenicols) of the other colistin-resistant E. coli actually point to a human origin.22,33,45 The identification of missense mutations at specific positions in the PmrA/PmrB two-component system, resulting sometimes from different nucleotide substitutions (Figure 3 and Table S4), as well as mutations modifying the length of PmrB (Table S5), in variable genetic backgrounds (Figure 2), indicates a convergent evolution, a strong hallmark of selection.46 Moreover most of the mutations occurred in specific domains of the proteins: the REC domain (residues 1–112) for PmrA and the HAMP (residues 92–141) and HisKA (residues 145–205) domains for PmrB (Figure 3).
Altogether, these findings clearly indicate two evolutionary paths leading to colistin resistance in human faecal E. coli, corresponding to a minority of plasmid-encoded MCR-1-producing isolates of animal origin (probably exposed to colistin use) and a vast majority of isolates of human origin, mostly exhibiting resistance through chromosomally encoded mechanisms.1 The selective pressure for such a high level of non-plasmid-encoded colistin resistance remains to be identified. In France, considering that colistin therapeutic use is limited to a last resort antimicrobial option in hospitalized patients with a history of XDR carriage and very rarely for treating infections due to E. coli, the selective pressure is definitely very low, if present at all. As most of the included patients were hospitalized <48 h before screening (817/1217, 67.1%), the colistin consumption in the French community is a relevant factor: the corresponding values were 0.018 DDDs/1000 inhabitants/day in 2016 and 2017 (https://ecdc.europa.eu/en/antimicrobial-consumption/database/distribution-by-antimicrobial-group). So, we can consider that the selective pressure due to colistin consumption in the community setting was quite low in France compared with other countries such as the UK (0.104 DDDs/1000 inhabitants/day in 2017). The most probable explanation resides in a coincidental evolution as has been shown in E. coli grown at high temperature that became resistant to rifampicin by parallel mutations within rpoB, the gene encoding the beta subunit of RNA polymerase, despite never being in contact with rifampicin.47 An alternative selective pressure leading to colistin resistance could be the exposure in the gut to non-antibiotic human-targeted drugs. It has been recently reported that such drugs can render E. coli resistant to antibiotics by mechanisms involving membrane modifications, among others.48 Also, exposure to chlorhexidine has been shown to select mutations conferring colistin resistance in K. pneumoniae.49
Our study presents several limitations. Firstly, we focused on E. coli although other enterobacterial species were reported as acquiring colistin resistance, particularly K. pneumoniae. Secondly, we did not perform an exhaustive analysis of the clinical history of the 1217 patients, including previous antimicrobial consumption or short periods of travel outside the country. Our study population, namely patients admitted to a healthcare facility and putatively harbouring some risk factors for MDR bacteria colonization, could have artificially increased the prevalence of colistin-resistant E. coli; an additional study focusing on a healthy population would be needed to confirm our findings. Thirdly, the low number of MCR-1-producing colistin-resistant E. coli forced us to balance our conclusions on this population of strains, although consistent with a larger previously published study.4 Lastly we focused on chromosomal mutations that have been previously proved to be responsible for colistin resistance in E. coli, Salmonella spp. and K. pneumoniae or that modified the length of the protein; the screening, identification and confirmation of the role of new mutations is a relevant, but independent, work. This broad and non-targeted prospective investigation of colistin resistance in E. coli constitutes a fully fledged work that is currently underway. Nevertheless, the strength of our work is supported by a robust methodological approach including non-biased multicentre rectal sampling, a marketed culture-based screening method, a reference confirmation test and WGS analysis of all the colistin-resistant isolates.
Conclusions
This is the first multicentre study focusing on the human faecal carriage prevalence of colistin-resistant and mcr-positive E. coli in Europe. Although the prevalence of mcr-positive E. coli carriage was low, the high rate of colistin-resistant, but mcr-negative, E. coli is of concern. The genetic background study of these two populations of colistin-resistant E. coli clearly indicated two distinct evolutionary pathways as the origin of this phenomenon. The selective pressures leading to this pattern of emergence need to be identified to allow the development of preventative strategies.
Acknowledgements
Other members of the IAME Resistance Group
Matthieu LAFAURIE, Béatrice BERCOT (St Louis Hospital), Violaine WALEWSKI, Mathilde LESCAT, Etienne CARBONNELLE (Avicenne-Jean-Verdier Hospitals), Fateh OUSSER, Nadia IDRI, Jean-Damien RICARD, Luce LANDRAUD, (Louis Mourier Hospital), Mathieu LE DORZE, Hervé JACQUIER, Emmanuelle CAMBAU (Lariboisière Hospital), Raphaël LEPEULE and Camille GOMART (Henri-Mondor Hospital).
Funding
This work was partially supported by the Laboratoire Européen Associé INSERM ‘Emerging Antibiotic Resistance in Gram-Negative Bacteria’ and by the Swiss National Science Foundation (project FNS-407240_177381). G. R. was supported by the Assistance Publique - Hôpitaux de Paris [poste d’accueil AP-HP – CEA (Commissariat à l’énergie atomique et aux énergies alternatives) 2016–2018]. E. D. was partially supported by the ‘Fondation pour la Recherche Médicale’ (Equipe FRM 2016, grant number DEQ20161136698).
The funders had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Transparency declarations
Conflicts of interest: none to declare. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
References
Author notes
Anne Sophie Bourrel, Laurent Poirel and Guilhem Royer Contributed equally.
Other members are listed in the Acknowledgements section.

{kind=link}
{kind=link}
{kind=link}