





Abstract
The HIV-1 reverse transcriptase (RT) natural polymorphism E138A is included among the mutations with a minor impact on response to etravirine. However, the interpretation of E138A on etravirine susceptibility is not consistent across different genotypic resistance algorithms. The aim of the study was to investigate the effect of E138A on the genetic barrier to resistance to etravirine in vitro.
A panel of 20 clinically derived recombinant viruses (10 with WT 138E and 10 with 138A, all without any other resistance mutation) were cultured in the presence of increasing etravirine concentrations and analysed for genotypic changes at virus breakthrough. Parallel experiments were conducted with 138E/A/G/K/Q NL4-3-based clones.
In the NL4-3 background, codon 138 changes increased etravirine resistance in the following order: Q > K > A > G > E. The 138A viruses were less susceptible to etravirine compared with the 138E viruses [median (IQR) fold change, 1.8 (1.5–2.8) versus 1.3 (0.8–1.8); P = 0.026], overcame etravirine pressure earlier [HR (95% CI) for viral outgrowth with 138A, 5.48 (2.95–28.24); P < 0.001] and grew at higher drug concentrations [median (IQR), 1350 (1350–1350) versus 0 (0–1350) nM; P = 0.005]. A variety of etravirine resistance-related mutations and changes in the RT connection and RNase H domains accumulated without any consistent pattern depending on baseline codon 138.
E138A can contribute to reduced response to etravirine through a decreased genetic barrier to resistance. In vitro drug resistance selection is a valuable complement to define the full potential of low-level resistance mutations.
Introduction
Etravirine is a second-generation NNRTI indicated for the treatment of HIV-1 infection in treatment-experienced patients, both in Europe and the USA.1,2,In vitro experiments demonstrated that etravirine has a higher genetic barrier to resistance and a different resistance profile compared with the first-generation NNRTIs nevirapine and efavirenz.3,4 The efficacy and safety of etravirine were originally investigated in treatment-experienced patients in the DUET-1 and DUET-2 clinical trials, where the addition of etravirine to the background regimen showed significantly higher rates of viral suppression compared with the placebo plus background regimen up to week 96.5 Accordingly, etravirine has since been used in the clinical setting primarily in heavily treatment-experienced patients.6,7 Based on favourable data on durability, tolerability and genetic barriers, etravirine has also been evaluated as a component of two-drug regimens in several studies in patients with either viral suppression or virological failure.8–10 These studies concluded that etravirine-based dual regimens were effective both in maintaining undetectable viraemia in suppressed patients and in contributing to achievement of virological control in highly treated patients with limited therapy options.
Analysis of the correlation between baseline HIV-1 genotype and virological response in the DUET studies led to the identification of 17 etravirine resistance-associated mutations (RAMs) in HIV-1 reverse transcriptase (RT) including V90I, A98G, L100I, K101E/H/P, V106I, E138A, V179D/F/T, Y181C/I/V, G190A/S and M230L,11 while treatment-emergent mutations were most often detected at codons 101, 138, 179 and 181.12 Variants emerging at codon 138 were highly heterogeneous and most often accompanied by other etravirine RAMs, prompting further investigation through site-directed mutagenesis and resulting in the addition of E138G, K and Q to the existing etravirine weighted genotypic score.13 Of note, the etravirine RAM E138A occurs as a natural polymorphism and is more prevalent in subtype C than B in different databases with frequencies ranging from 5.9% to 7.5% versus 0% to 2.3% in treatment-naive patients and 5.9% to 6.1% versus 2.0% to 2.5% in treatment-experienced individuals, respectively.14 However, subtype specificity is subject to geographical variation. For example, E138A is found in 5.2% (270/5209) of RT sequences from treatment-naive patients stored in the Italian Antiviral Response Cohort Analysis (ARCA) database (www.dbarca.net), with comparable frequency in subtype B (6.0%) and C (5.3%). In addition, E138A is interpreted differently by the most widely used genotype interpretation systems, with Stanford HIVdb, REGA and ANRS scoring this variant as potential low-level resistance, full susceptibility and possible resistance to etravirine, respectively. Based on these data, we were interested in assessing the impact of E138A on the genetic barrier to resistance to etravirine. In this study, we evaluated the development of resistance to etravirine by in vitro selection experiments in two panels of clinically derived recombinant strains carrying the WT 138E or the 138A variant in the absence of any other etravirine and NNRTI RAMs. Site-directed mutant infectious plasmids were also used to evaluate the effect of different amino acids at codon 138 in a syngeneic background.
Methods
Clinical samples and ethics
Twenty plasma samples were selected from HIV-1-positive untreated patients previously tested for routine drug resistance genotyping at baseline, as recommended by European guidelines (EACS 2017). Sample choice was based on the presence of the 138E WT codon (n = 10) or the 138A polymorphism (n = 10) within the RT coding region, and no other NNRTI RAMs, according to the IAS-USA drug resistance mutations list,15 as well as no RT mutations (except for E138A itself) conferring any level of etravirine resistance in the Stanford HIVdb algorithm, version 8.6 (A98G, L100I/V, K101E/H/P, E138G/K/Q/R, V179D/E/F/L, Y181C/F/G/I/S/V, Y188L, G190A/C/E/Q/S/T/V, H221Y, F227C and M230I/L). The use of residual, anonymized clinical samples for research studies was regulated by informed patient consent, as approved by the South-East Tuscany Ethics Committee. Clinical and laboratory data of the patients were stored in the ARCA database (www.dbarca.net).
Construction of recombinant viruses and phenotypic determination of susceptibility to etravirine
The method used for the creation and titration of recombinant viruses carrying the patient-derived RT-RNase H coding region has been previously described.16 The PCR primers and the protocol for amplification of the RT-RNase H region from viral RNA are included in the Supplementary data available at JAC Online. The HIV-1 subtype B NL4-3 laboratory strain (harbouring the RT 138E codon) was used as the WT virus. In addition, pNL4-3-based plasmids carrying RT 138A, 138G, 138K and 138Q were constructed through site-directed mutagenesis by using the QuikChange® Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer’s instructions. Baseline etravirine susceptibility was evaluated by quantifying luciferase activity after infection of TZM-bl cells with recombinant viruses in the presence of serial dilutions of the drug. To determine the IC50 of each recombinant virus, TZM-bl cells were seeded in a 96-well plate at 15 000 cells per well and infected at an moi of 0.01 in the presence of 5-fold serial dilutions of etravirine (range 10 000–5.12 nM). After 48 h, the cells were lysed by adding 50 μL/well of Glo-Lysis Buffer (Promega, Madison, WI, USA) and the lysates were transferred to a luminescence plate. Fifty microlitres of Bright-Glo Luciferase Reagent (Promega) was added to each well and the luminescence was measured with the GloMax® Discover Multimode Microplate Reader (Promega). Relative Luminescence Units (RLUs) detected in each well were elaborated with GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA) to calculate the IC50 values of recombinant and WT viruses.
In vitro resistance selection (IVRS)
The recombinant viruses, as well as the reference WT and mutant NL4-3 strains, were used to infect 106 MT-2 cells at an moi of 0.1 in the presence of an initial concentration of 50 nM etravirine corresponding to 4-fold the IC50 of WT 138E. Viral replication was monitored every 48–72 h by microscopic inspection of syncytia. When an advanced cytopathic effect was observed, the supernatant was harvested, cleared by centrifugation for 10 min at 300 g and stored at −80°C until genotypic analysis. Subsequent passages were set-up using the harvested virus to infect a new culture of 106 MT-2 cells in the presence of a 3-fold higher concentration of etravirine (150, 450 and 1350 nM). IVRS cultures were interrupted after viral breakthrough at 1350 nM etravirine or after 108 days from the start of the experiment, whichever occurred first.
RT-RNase H sequencing
The whole RT-RNase H coding region sequence (nt 2550–4229 in the HIV-1 HXB2 reference strain) was obtained from each recombinant virus by Sanger sequencing at baseline and at each viral breakthrough to detect emergent mutations. Viral RNA was extracted using the EZ1 Advanced XL system (QIAGEN, Hilden, Germany) with the EZ1 DSP Virus Kit (QIAGEN), reverse-transcribed and amplified as described in the Supplementary data.
Statistical analysis
The probability of virus breakthrough in cell culture under etravirine pressure was computed by survival analysis and the difference between 138E and 138A viruses was examined by Mantel–Cox log rank analysis. The Mann–Whitney U-test was used to analyse the differences in baseline etravirine susceptibility and in the maximum etravirine concentration at which viral growth occurred with 138E and 138A. All tests were done using GraphPad Prism version 6.0.
Results
Based on phylogenetic analysis of the whole RT-RNase H coding region, the clinical variants carrying the WT 138E codon included six subtype Bs, two subtype F1s and two B/F recombinants close to CRF12_BF, while the clinical variants carrying the mutant 138A codon included nine subtype Bs and one CRF02_AG. Following recombination, the chimeric viruses were confirmed to retain the original sequence as determined at the time of routine drug resistance genotyping, without any NNRTI resistance mutation. Also, NRTI and major PI resistance mutations were not present in any isolate. Natural polymorphisms are shown in Table S1 (available as Supplementary data at JAC Online) and the baseline sequences have been made available through GenBank at accession codes MH682065–MH682084.
Changes in RT genotype detected at virus breakthrough with increasing etravirine concentration in the five NL4-3 variants differing at codon 138
| Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | ||||||
| day | mutations | day | mutations | day | mutations | day | mutations | ||
| E | 1.0 (reference) | 28 | L100I | 31 | L100I | 45 | L100I V106A S400T | 49 | L100I V106A F227C A400T |
| A | 2.8 | 33 | no mutation | 47 | M230I Q269H | no viral growth | NT | ||
| G | 2.4 | 31 | Y181C | 40 | Y181C | 73 | Y181C | 76 | V179F 181C |
| K | 9.3 | 14 | no mutation | no viral growth | NT | NT | |||
| Q | 14.1 | 14 | no mutation | 33 | M230I | 47 | M230I | 76 | F227C M230I V241A |
| Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | ||||||
| day | mutations | day | mutations | day | mutations | day | mutations | ||
| E | 1.0 (reference) | 28 | L100I | 31 | L100I | 45 | L100I V106A S400T | 49 | L100I V106A F227C A400T |
| A | 2.8 | 33 | no mutation | 47 | M230I Q269H | no viral growth | NT | ||
| G | 2.4 | 31 | Y181C | 40 | Y181C | 73 | Y181C | 76 | V179F 181C |
| K | 9.3 | 14 | no mutation | no viral growth | NT | NT | |||
| Q | 14.1 | 14 | no mutation | 33 | M230I | 47 | M230I | 76 | F227C M230I V241A |
NT, not tested.
Fold change with respect to the reference 138E WT NL4-3 virus.
The day at which virus breakthrough occurred and the list of mutations with respect to the individual baseline sequence are shown. Mutations included in the etravirine resistance score derived from the DUET studies are underlined (E138A itself is also included in this list).11 Mutations conferring at least low-level resistance to etravirine according to the Stanford HIVdb 8.6 algorithm are in bold.
Changes in RT genotype detected at virus breakthrough with increasing etravirine concentration in the five NL4-3 variants differing at codon 138
| Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | ||||||
| day | mutations | day | mutations | day | mutations | day | mutations | ||
| E | 1.0 (reference) | 28 | L100I | 31 | L100I | 45 | L100I V106A S400T | 49 | L100I V106A F227C A400T |
| A | 2.8 | 33 | no mutation | 47 | M230I Q269H | no viral growth | NT | ||
| G | 2.4 | 31 | Y181C | 40 | Y181C | 73 | Y181C | 76 | V179F 181C |
| K | 9.3 | 14 | no mutation | no viral growth | NT | NT | |||
| Q | 14.1 | 14 | no mutation | 33 | M230I | 47 | M230I | 76 | F227C M230I V241A |
| Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | ||||||
| day | mutations | day | mutations | day | mutations | day | mutations | ||
| E | 1.0 (reference) | 28 | L100I | 31 | L100I | 45 | L100I V106A S400T | 49 | L100I V106A F227C A400T |
| A | 2.8 | 33 | no mutation | 47 | M230I Q269H | no viral growth | NT | ||
| G | 2.4 | 31 | Y181C | 40 | Y181C | 73 | Y181C | 76 | V179F 181C |
| K | 9.3 | 14 | no mutation | no viral growth | NT | NT | |||
| Q | 14.1 | 14 | no mutation | 33 | M230I | 47 | M230I | 76 | F227C M230I V241A |
NT, not tested.
Fold change with respect to the reference 138E WT NL4-3 virus.
The day at which virus breakthrough occurred and the list of mutations with respect to the individual baseline sequence are shown. Mutations included in the etravirine resistance score derived from the DUET studies are underlined (E138A itself is also included in this list).11 Mutations conferring at least low-level resistance to etravirine according to the Stanford HIVdb 8.6 algorithm are in bold.
At baseline, the fold-change etravirine susceptibility with respect to the reference NL4-3 was significantly higher with 138A- compared with 138E-carrying viruses (median and IQR 1.8, 1.5–2.8 versus 1.3, 0.8–1.8; P = 0.026) (Figure 1). All site-directed NL4-3 mutants carrying different amino acids at codon 138 were less susceptible to etravirine compared with the WT 138E reference virus (Table 1). However, the 138K NL4-3 virus replicated poorly in drug resistance selection experiments yielding a cytopathic effect only at 50 nM etravirine in the absence of any RT mutation. Also, the 138A NL4-3 virus was not able to grow at etravirine concentrations higher than 150 nM, despite initial selection of one etravirine resistance mutation (M230I). By contrast, the WT 138E and the mutant 138G and 138Q NL4-3 viruses all grew up to the highest etravirine concentration used but selected different combinations of mutations at codons involved in resistance to etravirine, i.e. 100, 106, 179, 181, 227, 230.
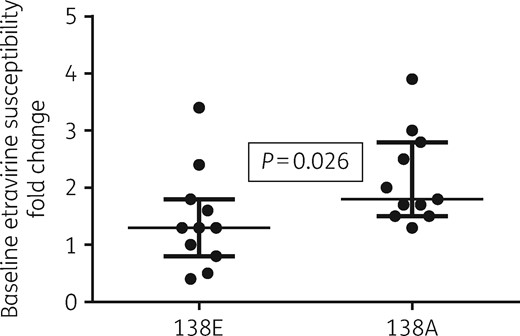
Baseline fold-change etravirine susceptibility with respect to the reference NL4-3 138E virus for the panel of 138E and 138A viruses.
All of the 10 clinically derived 138A viruses replicated up to 450 nM etravirine and all but one replicated up to the final highest 1350 nM etravirine concentration. By contrast, only three of the clinically derived 138E viruses were able to replicate at the highest concentration, one grew up to 450 nM etravirine and the remaining six did not even grow at the lowest 50 nM etravirine concentration used. The difference between the 138E and 138A groups was statistically significant, both in terms of time to first virus breakthrough (HR for viral outgrowth with 138A, 5.48, 95% CI 2.95–28.24; P < 0.001) (Figure 2) and in terms of maximum etravirine concentration overcome by viral growth (median and IQR: 0 and 0–1350 nM for 138E versus 1350 and 1350–1350 for 138A; P = 0.005) (Figure 3).
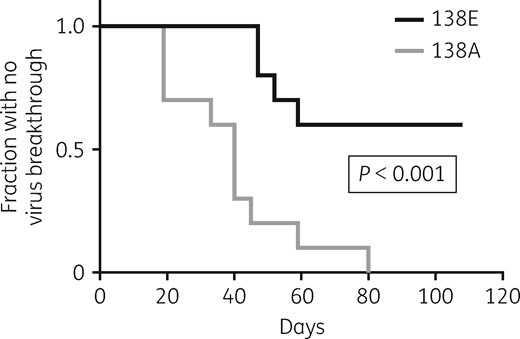
Kaplan–Meier curve showing the probability of cell cultures remaining free from virus breakthrough at the initial etravirine concentration (50 nM) starting with WT 138E (n = 10) and mutant 138A (n = 10) virus.
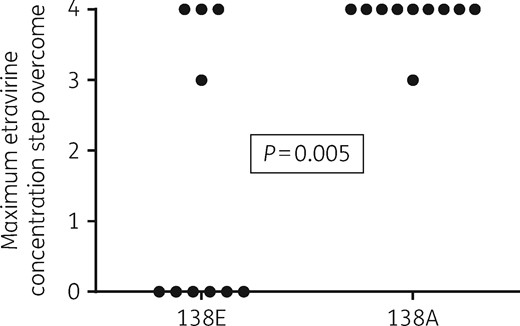
Comparison between the maximum etravirine concentration step overcome by viral growth in the 138E (n = 10) and 138A (n = 10) virus groups. Steps 0 to 4 correspond to 0, 50, 150, 450 and 1350 nM etravirine, with the cultures terminated at day 108.
Table 2 shows the RT amino acid changes detected in the breakthrough virus for all clinical isolates at the different etravirine concentration steps, with respect to the individual baseline sequence. Mutations emerging with at least three isolates at any timepoint included V179D/E/F (7 cases), Y181C (5), M230I (4) and T240I (3). Of these, T240I was the only mutation exclusively emerging with baseline 138A viruses (the three clinically derived recombinants and the 138A NL4-3 reference virus). Of the 14 viruses replicating at least up to the 450 nM etravirine concentration, 12 eventually carried at least one of the mutations included in the etravirine resistance score derived from the DUET studies and/or conferring at least low-level resistance to etravirine according to the Stanford HIVdb algorithm. One of the two cases without any of these mutations showed the V179E change conferring only potential low-level resistance to etravirine in the Stanford HIVdb algorithm (sample 138185) while sample 146102, which was the 138E virus with the largest decrease in baseline etravirine susceptibility, selected mutations not known to be involved in any NNRTI resistance and did not grow at the highest etravirine concentration used.
Changes in RT genotype detected at virus breakthrough with increasing etravirine concentration in the 20 clinically derived recombinant viruses
| Sample code (subtype) | Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | |||||||
| day | mutations | day | mutations | day | mutations | day | mutations | |||
| 52420 (B) | A | 1.5 | 33 | no mutation | 52 | T240I | 59 | T240I | 91 | H221Y L228H M230I K275R T477N A554FS |
| 60154 (B) | A | 2.0 | 19 | R356K A376S K527R | 40 | K102R M230I K527R | 66 | V8I V179DM230I G359S A376S K527R | 83 | V8I V179DM230I G359S A376S K527R |
| 72009 (CRF02_AG) | A | 1.5 | 80 | T107A G333V | 95 | T107A V179E Y181C G333V | 105 | T107A Y181C G333V | no viral growth | |
| 81958 (B) | A | 3.0 | 19 | Q197K E370A | 59 | T240I E370A K558R | 66 | T240I E370A K558R | 80 | L100I T240I E370A K558R |
| 95551 (B) | A | 1.7 | 45 | E6D V35I A554T | 59 | E6D E29K I47N A554T | 66 | E6D G190E A554T | 73 | E6D G190E A554T |
| 100442 (B) | A | 2.5 | 40 | P294A H483Y | 52 | P294A H483Y | 59 | G190E P294A H483Y | 66 | G190E P294A N348I E413D T477I H483Y |
| 127757 (B) | A | 1.7 | 40 | M230I K451R Q464R L517Q V531I | 52 | M230I R461K L517Q | 73 | M230I P420L K451R Q464R L517Q V531I V536G | 83 | V179DM230I K451R Q464R L517Q V531I |
| 138185 (B) | A | 3.9 | 19 | V179E R461K S468F | 52 | V179E V381I K451R Q464R V531I | 59 | V179E T240I R461K | 66 | L120F V179E T240I R461K |
| 138992 (B) | A | 1.3 | 40 | V10I | 59 | M230I | 73 | F227C M230I | 108 | Y181C F227C M230I |
| 141163 (B) | A | 1.8 | 59 | I135T V179D A360T | 66 | I135T Y144F V179D Q336H R358I A360T | 80 | I135T V179D A360T | 83 | I135T V179DY181CNS A360T A400T |
| 146473 (F1) | E | 1.3 | 52 | L100I E169G I178L K281R R358S E370G K530R | 59 | L100I E169G I178L K281R R358S E370G K530R | 73 | L100I E169G I178L K281R R358S E370G K530R | 76 | L100I E169G I178L K281R R358S E370G K530R |
| 146446 (CRF12_BF) | E | 0.8 | no viral growth | NT | NT | NT | ||||
| 146440 (CRF12_BF) | E | 1.6 | no viral growth | NT | NT | NT | ||||
| 146356 (B) | E | 0.4 | no viral growth | NT | NT | NT | ||||
| 146352 (B) | E | 0.5 | no viral growth | NT | NT | NT | ||||
| 146213 (B) | E | 2.4 | no viral growth | NT | NT | NT | ||||
| 146150 (F1) | E | 1.3 | no viral growth | NT | NT | NT | ||||
| 143035 (B) | E | 1.3 | 47 | V35I K64R Y181C K527R | 52 | V35I K64R Y181C K527R | 59 | V35I K64R Y181C K527R | 73 | V35I K64R V179F Y181C K527R |
| 143026 (B) | E | 1.8 | 59 | Y181C | 66 | Y181C V372L | 76 | V179F Y181C | 108 | V179F Y181C |
| 146102 (B) | E | 3.4 | 47 | T165K | 66 | R78K T165K G273E | 80 | T165K G273E | no viral growth | |
| Sample code (subtype) | Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | |||||||
| day | mutations | day | mutations | day | mutations | day | mutations | |||
| 52420 (B) | A | 1.5 | 33 | no mutation | 52 | T240I | 59 | T240I | 91 | H221Y L228H M230I K275R T477N A554FS |
| 60154 (B) | A | 2.0 | 19 | R356K A376S K527R | 40 | K102R M230I K527R | 66 | V8I V179DM230I G359S A376S K527R | 83 | V8I V179DM230I G359S A376S K527R |
| 72009 (CRF02_AG) | A | 1.5 | 80 | T107A G333V | 95 | T107A V179E Y181C G333V | 105 | T107A Y181C G333V | no viral growth | |
| 81958 (B) | A | 3.0 | 19 | Q197K E370A | 59 | T240I E370A K558R | 66 | T240I E370A K558R | 80 | L100I T240I E370A K558R |
| 95551 (B) | A | 1.7 | 45 | E6D V35I A554T | 59 | E6D E29K I47N A554T | 66 | E6D G190E A554T | 73 | E6D G190E A554T |
| 100442 (B) | A | 2.5 | 40 | P294A H483Y | 52 | P294A H483Y | 59 | G190E P294A H483Y | 66 | G190E P294A N348I E413D T477I H483Y |
| 127757 (B) | A | 1.7 | 40 | M230I K451R Q464R L517Q V531I | 52 | M230I R461K L517Q | 73 | M230I P420L K451R Q464R L517Q V531I V536G | 83 | V179DM230I K451R Q464R L517Q V531I |
| 138185 (B) | A | 3.9 | 19 | V179E R461K S468F | 52 | V179E V381I K451R Q464R V531I | 59 | V179E T240I R461K | 66 | L120F V179E T240I R461K |
| 138992 (B) | A | 1.3 | 40 | V10I | 59 | M230I | 73 | F227C M230I | 108 | Y181C F227C M230I |
| 141163 (B) | A | 1.8 | 59 | I135T V179D A360T | 66 | I135T Y144F V179D Q336H R358I A360T | 80 | I135T V179D A360T | 83 | I135T V179DY181CNS A360T A400T |
| 146473 (F1) | E | 1.3 | 52 | L100I E169G I178L K281R R358S E370G K530R | 59 | L100I E169G I178L K281R R358S E370G K530R | 73 | L100I E169G I178L K281R R358S E370G K530R | 76 | L100I E169G I178L K281R R358S E370G K530R |
| 146446 (CRF12_BF) | E | 0.8 | no viral growth | NT | NT | NT | ||||
| 146440 (CRF12_BF) | E | 1.6 | no viral growth | NT | NT | NT | ||||
| 146356 (B) | E | 0.4 | no viral growth | NT | NT | NT | ||||
| 146352 (B) | E | 0.5 | no viral growth | NT | NT | NT | ||||
| 146213 (B) | E | 2.4 | no viral growth | NT | NT | NT | ||||
| 146150 (F1) | E | 1.3 | no viral growth | NT | NT | NT | ||||
| 143035 (B) | E | 1.3 | 47 | V35I K64R Y181C K527R | 52 | V35I K64R Y181C K527R | 59 | V35I K64R Y181C K527R | 73 | V35I K64R V179F Y181C K527R |
| 143026 (B) | E | 1.8 | 59 | Y181C | 66 | Y181C V372L | 76 | V179F Y181C | 108 | V179F Y181C |
| 146102 (B) | E | 3.4 | 47 | T165K | 66 | R78K T165K G273E | 80 | T165K G273E | no viral growth | |
NT, not tested.
Fold change with respect to the reference 138E WT NL4-3 virus.
The day at which virus breakthrough occurred and the list of mutations with respect to the individual baseline sequence are shown. Mutations included in the etravirine resistance score derived from the DUET studies are underlined (E138A itself is also included in this list).11 Mutations conferring at least low-level resistance to etravirine according to the Stanford HIVdb 8.6 algorithm are in bold.
Changes in RT genotype detected at virus breakthrough with increasing etravirine concentration in the 20 clinically derived recombinant viruses
| Sample code (subtype) | Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | |||||||
| day | mutations | day | mutations | day | mutations | day | mutations | |||
| 52420 (B) | A | 1.5 | 33 | no mutation | 52 | T240I | 59 | T240I | 91 | H221Y L228H M230I K275R T477N A554FS |
| 60154 (B) | A | 2.0 | 19 | R356K A376S K527R | 40 | K102R M230I K527R | 66 | V8I V179DM230I G359S A376S K527R | 83 | V8I V179DM230I G359S A376S K527R |
| 72009 (CRF02_AG) | A | 1.5 | 80 | T107A G333V | 95 | T107A V179E Y181C G333V | 105 | T107A Y181C G333V | no viral growth | |
| 81958 (B) | A | 3.0 | 19 | Q197K E370A | 59 | T240I E370A K558R | 66 | T240I E370A K558R | 80 | L100I T240I E370A K558R |
| 95551 (B) | A | 1.7 | 45 | E6D V35I A554T | 59 | E6D E29K I47N A554T | 66 | E6D G190E A554T | 73 | E6D G190E A554T |
| 100442 (B) | A | 2.5 | 40 | P294A H483Y | 52 | P294A H483Y | 59 | G190E P294A H483Y | 66 | G190E P294A N348I E413D T477I H483Y |
| 127757 (B) | A | 1.7 | 40 | M230I K451R Q464R L517Q V531I | 52 | M230I R461K L517Q | 73 | M230I P420L K451R Q464R L517Q V531I V536G | 83 | V179DM230I K451R Q464R L517Q V531I |
| 138185 (B) | A | 3.9 | 19 | V179E R461K S468F | 52 | V179E V381I K451R Q464R V531I | 59 | V179E T240I R461K | 66 | L120F V179E T240I R461K |
| 138992 (B) | A | 1.3 | 40 | V10I | 59 | M230I | 73 | F227C M230I | 108 | Y181C F227C M230I |
| 141163 (B) | A | 1.8 | 59 | I135T V179D A360T | 66 | I135T Y144F V179D Q336H R358I A360T | 80 | I135T V179D A360T | 83 | I135T V179DY181CNS A360T A400T |
| 146473 (F1) | E | 1.3 | 52 | L100I E169G I178L K281R R358S E370G K530R | 59 | L100I E169G I178L K281R R358S E370G K530R | 73 | L100I E169G I178L K281R R358S E370G K530R | 76 | L100I E169G I178L K281R R358S E370G K530R |
| 146446 (CRF12_BF) | E | 0.8 | no viral growth | NT | NT | NT | ||||
| 146440 (CRF12_BF) | E | 1.6 | no viral growth | NT | NT | NT | ||||
| 146356 (B) | E | 0.4 | no viral growth | NT | NT | NT | ||||
| 146352 (B) | E | 0.5 | no viral growth | NT | NT | NT | ||||
| 146213 (B) | E | 2.4 | no viral growth | NT | NT | NT | ||||
| 146150 (F1) | E | 1.3 | no viral growth | NT | NT | NT | ||||
| 143035 (B) | E | 1.3 | 47 | V35I K64R Y181C K527R | 52 | V35I K64R Y181C K527R | 59 | V35I K64R Y181C K527R | 73 | V35I K64R V179F Y181C K527R |
| 143026 (B) | E | 1.8 | 59 | Y181C | 66 | Y181C V372L | 76 | V179F Y181C | 108 | V179F Y181C |
| 146102 (B) | E | 3.4 | 47 | T165K | 66 | R78K T165K G273E | 80 | T165K G273E | no viral growth | |
| Sample code (subtype) | Codon 138 | Etravirine susceptibility fold changea | Breakthrough at increasing etravirine concentrationb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50 nM | 150 nM | 450 nM | 1350 nM | |||||||
| day | mutations | day | mutations | day | mutations | day | mutations | |||
| 52420 (B) | A | 1.5 | 33 | no mutation | 52 | T240I | 59 | T240I | 91 | H221Y L228H M230I K275R T477N A554FS |
| 60154 (B) | A | 2.0 | 19 | R356K A376S K527R | 40 | K102R M230I K527R | 66 | V8I V179DM230I G359S A376S K527R | 83 | V8I V179DM230I G359S A376S K527R |
| 72009 (CRF02_AG) | A | 1.5 | 80 | T107A G333V | 95 | T107A V179E Y181C G333V | 105 | T107A Y181C G333V | no viral growth | |
| 81958 (B) | A | 3.0 | 19 | Q197K E370A | 59 | T240I E370A K558R | 66 | T240I E370A K558R | 80 | L100I T240I E370A K558R |
| 95551 (B) | A | 1.7 | 45 | E6D V35I A554T | 59 | E6D E29K I47N A554T | 66 | E6D G190E A554T | 73 | E6D G190E A554T |
| 100442 (B) | A | 2.5 | 40 | P294A H483Y | 52 | P294A H483Y | 59 | G190E P294A H483Y | 66 | G190E P294A N348I E413D T477I H483Y |
| 127757 (B) | A | 1.7 | 40 | M230I K451R Q464R L517Q V531I | 52 | M230I R461K L517Q | 73 | M230I P420L K451R Q464R L517Q V531I V536G | 83 | V179DM230I K451R Q464R L517Q V531I |
| 138185 (B) | A | 3.9 | 19 | V179E R461K S468F | 52 | V179E V381I K451R Q464R V531I | 59 | V179E T240I R461K | 66 | L120F V179E T240I R461K |
| 138992 (B) | A | 1.3 | 40 | V10I | 59 | M230I | 73 | F227C M230I | 108 | Y181C F227C M230I |
| 141163 (B) | A | 1.8 | 59 | I135T V179D A360T | 66 | I135T Y144F V179D Q336H R358I A360T | 80 | I135T V179D A360T | 83 | I135T V179DY181CNS A360T A400T |
| 146473 (F1) | E | 1.3 | 52 | L100I E169G I178L K281R R358S E370G K530R | 59 | L100I E169G I178L K281R R358S E370G K530R | 73 | L100I E169G I178L K281R R358S E370G K530R | 76 | L100I E169G I178L K281R R358S E370G K530R |
| 146446 (CRF12_BF) | E | 0.8 | no viral growth | NT | NT | NT | ||||
| 146440 (CRF12_BF) | E | 1.6 | no viral growth | NT | NT | NT | ||||
| 146356 (B) | E | 0.4 | no viral growth | NT | NT | NT | ||||
| 146352 (B) | E | 0.5 | no viral growth | NT | NT | NT | ||||
| 146213 (B) | E | 2.4 | no viral growth | NT | NT | NT | ||||
| 146150 (F1) | E | 1.3 | no viral growth | NT | NT | NT | ||||
| 143035 (B) | E | 1.3 | 47 | V35I K64R Y181C K527R | 52 | V35I K64R Y181C K527R | 59 | V35I K64R Y181C K527R | 73 | V35I K64R V179F Y181C K527R |
| 143026 (B) | E | 1.8 | 59 | Y181C | 66 | Y181C V372L | 76 | V179F Y181C | 108 | V179F Y181C |
| 146102 (B) | E | 3.4 | 47 | T165K | 66 | R78K T165K G273E | 80 | T165K G273E | no viral growth | |
NT, not tested.
Fold change with respect to the reference 138E WT NL4-3 virus.
The day at which virus breakthrough occurred and the list of mutations with respect to the individual baseline sequence are shown. Mutations included in the etravirine resistance score derived from the DUET studies are underlined (E138A itself is also included in this list).11 Mutations conferring at least low-level resistance to etravirine according to the Stanford HIVdb 8.6 algorithm are in bold.
Interestingly, mutations in the RT connection domain (amino acids 320 to 440) emerged in two 138E (143026 and 146473) and seven 138A (60154, 72009, 81958, 100442, 127757, 138185 and 141163) clinically derived viruses. Likewise, mutations in the RNase H domain (amino acids 441 to 560) also emerged in two 138E (143035 and 146473) and seven 138A (52420, 60154, 81958, 95551, 100442, 127757 and 138185) clinically derived viruses. Overall, as many as 31 distinct connection or RNase H domain mutations emerged in 12 clinically derived viruses, appearing before or concomitantly with etravirine resistance mutations in 5 and 5 cases, respectively. However, mutational patterns were highly diversified, with only a few mutations selected with two different viruses (K451R, R461K, Q464R, K527R and V531I) and no mutation selected in more than two. By contrast, the only mutation selected outside the RT domain with the NL4-3 viruses was A400T (also detected with sample 141163) emerging in the WT 138E virus following selection of the etravirine resistance mutation L100I (Table 1).
Discussion
Etravirine was originally approved as a component of salvage regimens in patients harbouring MDR virus. Indeed, etravirine retains activity against several mutants selected at failure of first-generation NNRTIs and can be a key drug even in deep salvage.6 Both in vitro and in vivo data soon revealed that resistance to etravirine is much more complex than resistance to first-generation NNRTIs and genotypic etravirine susceptibility scoring systems have included up to 49 mutations.17 Although different systems mostly agree on mutations conferring high resistance, there is much less consensus on the role of minor mutations. Among these, E138A is particularly interesting because it occurs in nature in up to 1.8%–7.8% of isolates, depending on HIV-1 subtype (Stanford HIVdb, https://hivdb.stanford.edu/cgi-bin/MutPrevBySubtypeRx.cgi), and it is interpreted differently by the most common genotype algorithms. In addition, codon 138 can accommodate different polymorphisms in nature (mainly A and Q) and selects for different variants following etravirine pressure.13
The 2.8-fold decrease in etravirine susceptibility measured in our study for the 138A clone is remarkably similar to the 2.9-fold and the 2.5-fold previously obtained with subtype B and C clones, respectively.13,18 The same applies to the 138G clone, while in our experiments the 138K and 138Q constructs showed a fold change closer to the data obtained in a subtype C, rather than B, background.13,18 The lower clinical cut-off used for etravirine in the PhenoSense assay from Monogram is 2.9-fold,19 confirming that the impact of E138A on etravirine susceptibility is difficult to define. Interestingly, codon 138 appeared to dictate different resistance pathways within the same NL4-3 backbone, with L100I, Y181C and M230I as the initial etravirine resistance mutation emerging from 138E, 138G and 138A/Q, respectively. However, the choice of different major etravirine mutations may have been partly stochastic as different changes have been documented in multiple IVRS studies, all starting from the WT 138E virus.4,20–22 The 138A, 138K and 138Q variants replicated at the lowest etravirine concentration used without selecting for any mutation, a finding compatible with their lower baseline susceptibility to etravirine. Accordingly, the most resistant 138K and 138Q clones generated a cytopathic effect significantly earlier than the other variants. However, the 138K clone was lost at the first increase in etravirine concentration (150 nM), despite baseline resistance, suggesting reduced fitness as previously documented.23 Similarly, the 138A clone did not grow at 450 nM etravirine, irrespective of previous acquisition of M230I. On the other hand, the WT 138E and the 138G and 138Q variants accumulated further etravirine resistance mutations at higher drug concentration. This indicates a delicate balance between resistance and fitness for different variants at codon 138.
With the panel of 20 clinically derived recombinant viruses, E138A was confirmed to confer a low but significant level of resistance to etravirine. The median fold change in etravirine susceptibility (1.8) was lower than that of the 138A NL4-3 clone (2.8), possibly reflecting better adaptation of the laboratory NL4-3 virus to cell culture. However, two patient-derived 138A viruses had fold-change values larger than the 138A NL4-3, suggesting a role for background polymorphisms. Accordingly, among isolates without any other NNRTI mutation included in the Monogram database, E138A was present in 71.4% of 49 clinically derived viruses with >2.9 (median 3.5) etravirine susceptibility fold change as compared with 3.3% of another 9409 samples with fold change <2.9.24 The most interesting and novel data shown in this study are that E138A significantly lowers the HIV-1 genetic barrier to resistance to etravirine as indicated by both a shorter time to virus breakthrough and the ability to grow at higher etravirine concentration compared with the WT 138E. Of note, 6 of the 10 138E viruses did not even grow at the lowest etravirine concentration used indicating that the experimental condition was challenging in the absence of the E138A mutation. The resistance pathway was highly diversified in the other 14 viruses and did not allow for definition of any preferential pattern for 138A versus 138E at baseline. This highlights that etravirine has a complex resistance pattern but also that there are multiple options for resistance to emerge, likely guided by natural polymorphisms. One 138A and one 138E accumulated only non-etravirine-related mutations and grew up to the last and penultimate drug concentration used, respectively, suggesting that alternative resistance patterns may go undetected by currently used genotypic interpretation systems.
RT evolution also involved the RT connection domain and/or RNase H domain in 12 of the 14 clinically derived viruses overcoming drug pressure. Again, the pattern of emergent mutations was highly heterogeneous and no specific mutation was significantly associated with either 138E or 138A. A number of RT connection and RNase H domain mutations have been reported to enhance NRTI resistance and, in a few cases, to contribute to NNRTI resistance likely through decreased RNase activity leading to increased time for NRTI excision or NNRTI dissociation from RT.25 C-terminal RT mutations of this kind emerging in this etravirine resistance selection study include N348I, G359S, A360T, A376S, A400T, A554T and K558R. Among these, a possible role in NNRTI resistance has been suggested for N348I,26–31 A376S28,30,32 and A400T,33 although the impact on etravirine activity could be modest.30 A negative interaction between N348I and E138K has also been reported.34 It must be noted that under our experimental conditions several other C-terminal mutations emerged, possibly reflecting adaptation of the different virus backbones to cell culture. Indeed, with the laboratory-adapted NL4-3 viruses only one C-terminal RT mutation emerged, A400T in the WT 138E virus, as also reported in one previous etravirine resistance selection experiment.22
In summary, this study suggests that the main contribution of E138A to reduced response to etravirine may result from an impact on the genetic barrier to resistance rather than from, or in addition to, low-level resistance. These data support inclusion of E138A as an etravirine resistance mutation in the REGA genotype interpretation algorithm, similar to HIVdb and ANRS. However, the naturally occurring E138A variant may still have a higher genetic barrier to resistance to etravirine compared with first-generation low-barrier NNRTIs such as efavirenz and nevirapine. A role for E138A in response to etravirine-based therapy in treatment-experienced viraemic patients has been shown both in clinical trials and observational cohorts.13,35 By contrast, limited data suggest that naturally occurring E138A does not impact virological response to rilpivirine.36 It remains to be established whether such a role remains when using etravirine as a component of treatment switch in patients under suppressive therapy.37 As a more general model, it may be advisable to examine the potential of low-level resistance mutations to decrease the genetic barrier to resistance to specific antiretrovirals, particularly those being used in dual regimens.
Acknowledgements
The pNL4-3 vector, etravirine and MT-2 and TZM-bl cell lines were obtained through the NIH AIDS Reagent Program. We thank Stefano Rusconi for helpful discussions regarding the design of the experiments.
Funding
This work was supported by internal funding.
Transparency declarations
M. Z. reports consultancy for ViiV Healthcare, Gilead Sciences and Janssen-Cilag, and grants for his institution from ViiV Healthcare and Gilead. All other authors: none to declare.
References
European AIDS Clinical Society (EACS). Guidelines for Treatment of HIV-Positive Adults in Europe, Version 9.0, October 2017. http://www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html.
Department of Health and Human Services - Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Last Updated 30 May 2018. https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv/0.

{kind=link}
{kind=link}
{kind=link}