





Sir,
We read with interest the work of Soge et al.1 describing two unusual MRSA clones found in macaques at a US primate research centre in 2015. The first, ST188-MRSA-IV, had previously been reported in humans, with ST188-MSSA found in primates, whereas the second, ST3268-MRSA-V, had initially been discovered in quarantined primates originating outside Washington in the same year, but had subsequently also been identified in their primate colony.1 We had also identified ST3268-MRSA-V and its single-locus variant (SLV) ST2817-MRSA-V among long-tailed macaques (Macaca fascicularis) used in experimental surgery in 2014, and describe our experience here, comparing our isolates to the two US ST3268 isolates via genomic analysis.1
The SingHealth Experimental Medicine Centre (SEMC) has facilities at three separate locations in Singapore, with limited movement of animals between them. The macaques in these facilities originated primarily from Vietnam and had been imported at various times starting from 2009. One macaque developed an MRSA wound infection in January 2014 following head implant surgery; Staphylococcus aureus was identified via MALDI-TOF MS and the isolate was resistant to cefoxitin. This led to mass screening of all remaining 51 macaques (surgical site, nasal and perianal swabs) as well as all 28 of their human contacts (nasal swabs) across the three facilities, using MRSA plates (bioMérieux SA, Marcy-l’Étoile, France).
All macaques and two humans were colonized. The macaques were treated with either sulfamethoxazole/trimethoprim or vancomycin, and the MRSA cluster was eventually cleared by isolating the colonized macaques and decolonizing both humans and macaques by bathing with 0.05% chlorhexidine solution and applying mupirocin to the nares and any wound sites.
All MRSA isolates underwent antimicrobial susceptibility testing according to the CLSI standards2 and multi-locus variable number tandem repeat analysis (MLVA).3 The results are available in Table S1 (available as Supplementary data at JAC Online). Both human isolates and randomly selected representative macaque isolates from each of six MLVA profiles were subject to WGS, which was performed as previously described.4 The MLST STs of these isolates were inferred from the WGS output.5 WGS data for the TXA and TXB isolates described by Soge et al.1 were obtained from the National Center for Biotechnology Information Sequence Read Archive database (accession number SRP067697). Paired-end sequence reads were mapped to the chromosome of reference strain CA-347,6 with SNPs identified as previously described.4 Regions of homologous recombination were predicted by using Gubbins.7 A phylogenetic tree was constructed from core genome SNPs, minus recombination regions, using RAxML.8
All but one of the macaque MRSA isolates belonged to MLVA clusters where representative sequenced isolates were assigned to either ST3268-MRSA-V or ST2817-MRSA-V. The final macaque MRSA was found to be ST22-MRSA-IV, the major human healthcare-associated MRSA clone in Singapore.4 One human who performed animal husbandry, including blood collection from macaques, was colonized with ST3268-MRSA-V. The second human performed surgical procedures on macaques and carried ST2817-MRSA-V. WGS analysis demonstrated that the ST22-MRSA-IV isolated from the macaque was phylogenetically related to Singaporean human isolates (data not shown), suggesting an anthroponotic source.4
Comparative genomic analysis revealed evidence of homologous recombination in the chromosomes of the ST2817 and ST3268 isolates. Phylogenetic analysis, removing SNP variation associated with recombination, revealed the clonal relationship of the ST2817 and ST3268 isolates (Figure 1) and also evidence of close genetic relationships between isolates belonging to each of the STs, supportive of local transmission. Moreover, inclusion of the ST3268 isolates from the Washington primate facility1 revealed they belong to the same clone as the Singapore isolates, with comparable genetic relatedness. Phylogenomic analysis of the ST2817 isolates revealed that they emerged from the ST3268 population and that there were numerous recombination events that accompanied the emergence of the ST; 21 recombination regions encompassing 7362 SNPs and 40 core SNPs (Figure 1).
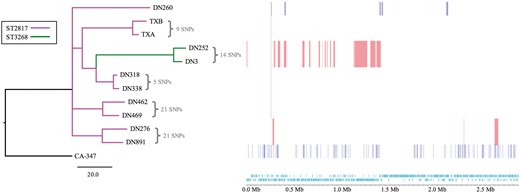
Prediction of recombination in the S. aureus isolate chromosomes. Regions of variation in the genomes of the 11 clinical S. aureus and the reference strain CA-347, which are predicted to have arisen by homologous recombination, are shown on the right. Red blocks indicate recombination predicted to have occurred on internal nodes and blue indicates taxon-specific recombination. Isolates are ordered according to the phylogenetic tree displayed on the left. The tree is a maximum likelihood tree constructed with core chromosome SNPs, with SNPs in recombination regions removed, and rooted with the ST45 CA-347 reference. The branches are colour coded according to the ST with the key indicated in the figure. The track along the bottom of the figure displays the CA-347 chromosome and annotation, in which protein-coding sequences are indicated in light blue.
The phylogenetic relationships and genetic diversity indicate that this was not a single point source outbreak originating at the SEMC primate facilities, although some of the SNP distances between the isolates suggest that there could have been localized transmission of some parts of the population. Interestingly, TXA and TXB from the Washington facility are both placed within the larger cluster of ST3268 isolates from Singapore and are predicted to be within 36 SNPs of the nearest SEMC isolate. Unfortunately, we were unable to ascertain the country of origin of the US primates or whether they had shared a common transit facility with any of the Singaporean macaques.
Collectively, our experience and results suggest that ST3268-MRSA-V and its SLV ST2817-MRSA-V is probably a macaque-specific MRSA clone that is capable of zoonotic transmission, in much the same way as the porcine- and bovine-specific MRSA clones.9,10 The genomic diversity of the macaque isolates coupled with the paucity of ST3268-MRSA-V and its SLV in humans argue against a human-to-macaque transmission with subsequent spread in the caged macaques. More studies need to be done to determine if this clone is endemic in macaques or represents an exceptional expansion of uncommon S. aureus types in defined monkey populations.
Funding
Bioinformatics and Computational Biology analyses were supported by the University of St Andrews Bioinformatics Unit that is funded by a Wellcome Trust Institutional Strategic Support Fund (ISSF) award (grant 097831/Z/11/Z). M. T. G. H. and K. P. were supported by the Scottish Infection Research Network and Chief Scientist Office through the Scottish Healthcare Associated Infection Prevention Institute consortium funding (CSO Reference: SIRN10). The work in Singapore was funded via internal funding from the respective institutions, including intramural support from the Singapore Infectious Diseases Initiative.
Transparency declarations
None to declare.
Supplementary data
Table S1 is available as Supplementary data at JAC Online.

{kind=link}