





We evaluated whether maintenance therapy with atazanavir/ritonavir plus lamivudine (ATV/r + 3TC) was non-inferior to ATV/r plus two nucleosides (ATV/r + 2NUCs) at 96 weeks of follow-up.
SALT is a multicentre, open-label, non-inferiority clinical trial in HIV-1-infected virologically suppressed patients. Hepatitis B virus surface antigen-negative subjects with no previous treatment failure/resistance mutations and HIV-1-RNA <50 copies/mL for ≥6 months were randomized (1 : 1) to ATV/r + 3TC or ATV/r + 2NUCs. The primary endpoint was HIV-1-RNA <50 copies/mL in the PP population. Non-inferiority was demonstrated if the lower bound of the 95% CI for the difference was not below −12%.
Some 286 patients were analysed. At week 96, 74.4% had HIV-1-RNA <50 copies/mL in the ATV/r + 3TC arm versus 73.9% in the ATV/r + 2NUCs arm (95% CI for the difference, −9.9%–11.0%). In both groups, similar values were observed for patients with confirmed virological failure in ATV/r + 3TC versus ATV/r + 2NUCs (9 versus 5), death (1 versus 0), discontinuation due to ART-related toxicity (7 versus 11), withdrawal from the study (7 versus 9) and loss to follow-up (6 versus 6). One patient taking ATV/r + 2NUCs developed resistance mutations (M184V and L63P). Similar values were obtained for change in mean CD4 count [19 versus 18 cells/mm3 (95% CI for the difference, −49.3–50.7), grade 3–4 adverse events (70.7% versus 70.2%) and changes in the global deficit score, −0.3 (95% CI, −0.5 to −0.1) for ATV/r + 3TC, versus −0.2 (95% CI, −0.4 to −0.1) for ATV/r + 2NUCs].
The long-term results of switching to ATV/r + 3TC show that this strategy is effective, safe and non-inferior to ATV + 2NUCs in virologically suppressed HIV-infected patients.
Introduction
The goal of combination ART (cART) is to suppress HIV replication and thus improve quality of life and life expectancy. However, besides requiring strict adherence, lifelong cART is associated with a series of disadvantages in the form of adverse events, complex treatment schedules and requirements and cost, all of which affect the long-term effectiveness of treatment. Consequently, simplification strategies to improve the convenience of cART while maintaining effectiveness have been sought.
The combination of boosted atazanavir plus lamivudine has the potential to suppress some of the long-term adverse events associated with nucleos(t)ide reverse transcriptase inhibitors (NUCs), preserve future treatment options and reduce the cost of cART, as anticipated in a small single-arm pilot study of 40 patients.1 SALT [Simplification to Atazanavir and Lamivudine dual Therapy versus atazanavir/ritonavir and two nucleos(t)ides in virologically stable patients; GeSIDA 7011 study] is the first randomized clinical trial to compare atazanavir/ritonavir plus lamivudine (ATV/r + 3TC) with atazanavir/ritonavir plus two NUCs (ATV/r + 2NUCs) in a well-powered study that shows non-inferior efficacy and equivalent safety of dual treatment compared with standard cART.2 This finding is in line with those of the OLE and AtLaS-M clinical trials, in which patients on ritonavir-boosted PI (PI/r) triple therapy were switched to lopinavir/ritonavir plus lamivudine3 or ATV/r + 3TC,4 respectively. As a primary endpoint, the 48 week results of the SALT,2 OLE3 and AtLaS-M4 trials showed that dual treatment based on a PI/r plus lamivudine is a switch option that is effective, safe and well tolerated for patients with virologically suppressed HIV-1 infection.
Data beyond the first 48 weeks of therapy would add value to this simplification strategy, showing the durability of the efficacy and safety results. To date, only the AtLaS pilot single-arm study has provided data on long-term follow-up (144 weeks) for dual therapy with ATV/r + 3TC.1 In the present paper, we report the long-term efficacy and safety results (96 weeks) of dual treatment with ATV/r + 3TC in virologically suppressed patients who switched any standard cART because of intolerance or toxicity or for simplification of their regimen.
Patients and methods
The design of the SALT study has been reported elsewhere.2 SALT is a 96 week, multicentre randomized, open-label, non-inferiority, controlled clinical trial that compares dual therapy with 300 mg of atazanavir + 100 mg of ritonavir + 300 mg of lamivudine administered once daily, with 300 mg of atazanavir + 100 mg of ritonavir administered once daily and two NUCs (2NUCs) (selected at the discretion of the investigator) in HIV-1-infected patients on a stable three-drug regimen who switched therapy for reasons of toxicity, intolerance or simplification. Randomization (1 : 1) was stratified by active hepatitis C virus (HCV) infection (positive HCV-RNA) and previous therapy (NNRTI, PI/r, CCR5 antagonist or integrase inhibitor).
The study population comprised patients aged >18 years with chronic HIV-1 infection and no previous cART failure, no resistance mutations to the study medication, HIV-1-RNA <50 copies/mL for ≥6 months, hepatitis B virus surface antigen-negative status and good general health. Patients assigned to the dual-therapy arm were on triple therapy with ATV/r + 2NUCs (selected at the investigator's discretion) for 1 month before switching to dual therapy with ATV/r + 3TC. This transition phase was made in order to prevent induction of cytochrome P450 by efavirenz or nevirapine, which could lower atazanavir plasma levels and jeopardize the efficacy of dual therapy.
The primary endpoint was virological response rate, defined as the proportion of patients with HIV-1-RNA <50 copies/mL at week 48 in the PP population (which comprises all randomized patients with no major protocol violations). The response rate at 96 weeks was a secondary endpoint. Virological efficacy was evaluated using the time to loss of virological response (TLOVR) and snapshot algorithms in the PP and ITT populations (all randomized patients). Virological failure was defined as two consecutive HIV-1-RNA determinations (≥14 days but not >30 days apart) ≥50 copies/mL at any scheduled visit. Missing patients and changes in any study drug were also considered failures. The primary objective was to evaluate in the PP population whether maintenance therapy with ATV/r + 3TC was non-inferior to ATV/r + 2NUCs at 48 weeks (secondary objective at 96 weeks) with a non-inferiority margin of 12%. Dual therapy was considered non-inferior to standard triple therapy if the lower bound of the 95% CI of the difference between the efficacy of ATV/r + 3TC and that of ATV/r + 2NUCs was >12%.
Adherence was assessed on the basis of pill count and using a simplified medication adherence questionnaire.5 Non-adherence was defined as a rate <90% at any visit during the study period. All laboratory tests were performed at the respective hospital and included determination of HIV-1 resistance mutations.
Safety assessments included adverse event recording, clinical laboratory tests, physical examination, neuropsychological testing, dual-energy X-ray absorptiometry (for bone mineral density and fat distribution), vitamin D levels and anthropometric measurements.
Participants in the neurocognitive substudy were assessed at baseline, week 48 and week 96. The tests applied to assess cognition were the Trail Making Test A, Trail Making Test B, Digit Symbol Substitution Test and Grooved Pegboard Test using both the dominant and non-dominant hand (five neurocognitive domains, one test per domain). Raw test scores were converted to demographically corrected standard scores using the best available normative standards for the Spanish population. Global neurocognitive performance was estimated using the global deficit score (GDS).6
Continuous variables were expressed as the mean and standard deviation when normally distributed or as the median and IQR if their distribution was skewed. Discrete variables were expressed as percentages. We used an independent samples t-test to compare normally distributed continuous variables and the Mann–Whitney test to compare non-normally distributed continuous variables. The association between categorical variables was evaluated using the χ2 test when samples were of sufficient size or with the Fisher exact test when they were not. Neurocognitive change, defined as the difference between GDS at baseline and at week 96, was assessed using analysis of covariance.
Ethics
Ethics committee approval was obtained at all participating centres and all patients gave their written informed consent before undergoing any study procedure. The study was registered at ClinicalTrials.gov (NCT01307488).
Results
![Trial profile [primary endpoint analysis (96 weeks)]. One patient in the ATV/r + 3TC group and two patients in the ATV/r + 2NUCs group showed HIV-1-RNA ≥50 copies/mL at the last study visit, with no further confirmation.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jac/72/1/10.1093_jac_dkw379/3/m_dkw37901.jpeg?Expires=1749057251&Signature=FUg2Go24JEXU4fcZFn6xiyX9FIwEviK84CXQKnqaTsp476oBdTxHtndggbz8a-3uuCidUaC-qjeQ3nj9fnokUMMPLKI0t19~lkQnKFHuHTFSYiSyrintr-zngQejVJrRtyAU6lONX2RXHzDDndCl07kbeoC7g~ph8WDib4FdeurFsuzntSqPoeMjWRPhUWYbF8g4ySUbpgBOqZur07V9D9hV2nUnKdgXX4~C~~Z3sup3zpEM-o31tk7-wZE8VcsG7XAEb2dhQDErpxdPJiQ1Sy-tf~fgIGI8vs2yuyjR1FnUDQ5pADX3tpe-QQD43RMeH-fDqkTCTWTm5zfnLjEzSw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Trial profile [primary endpoint analysis (96 weeks)]. One patient in the ATV/r + 3TC group and two patients in the ATV/r + 2NUCs group showed HIV-1-RNA ≥50 copies/mL at the last study visit, with no further confirmation.
At the baseline visit, 65% of patients were taking a PI/r (39.4% ATV/r) and 32.5% an NNRTI (27% efavirenz). Tenofovir plus emtricitabine and abacavir plus lamivudine were taken at baseline by 83.2% and 15.3% of participants, respectively. After randomization, 75.6% of the patients in the triple-therapy arm were prescribed tenofovir/emtricitabine, 23.7% abacavir/lamivudine and 0.7% zidovudine/lamivudine.
In the efficacy analysis (PP population, TLOVR algorithm), the virological response rate was 74.4% at week 96 in the ATV/r + 3TC arm compared with 73.9% in the ATV/r + 2NUCs arm (Table 1). Non-inferiority was demonstrated at the pre-specified level of −12%; the difference in efficacy between dual and triple therapy was 0.5% (95% CI, −9.9%–11.0%). In comparison, differences at 48 weeks were somewhat higher: the virological response rate was 83.5% and 77.8% in the ATV/r + 3TC and ATV/r + 2NUCs arms, respectively. In the same way, the difference in efficacy between dual and triple therapy was 5.7% (95% CI, −4.5%–15.9%).
Treatment efficacy at 96 weeks (TLOVR; PP population; n = 267)
| Item | ATV/r + 3TC (n = 133) | ATV/r + 2NUCs (n = 134) |
|---|---|---|
| HIV-1-RNA <50 copies/mL | 99 (74.4) | 99 (73.9) |
| HIV-1-RNA ≥50 copies/mL | 10 (7.5) | 7 (5.2) |
| Withdrawal of informed consent | 7 (5.3) | 9 (6.7) |
| Discontinuation because of adverse events | 7 (5.3) | 11 (8.2) |
| Loss to follow-up | 6 (4.5) | 6 (4.5) |
| Death | 1 (0.8) | 0 (0.0) |
| Other | 3 (2.3) | 2 (1.5) |
| Total | 133 (100) | 134 (100) |
| Patients with blips not leading to interruption of cART | 36 (27.1) | 30 (22.4) |
| Item | ATV/r + 3TC (n = 133) | ATV/r + 2NUCs (n = 134) |
|---|---|---|
| HIV-1-RNA <50 copies/mL | 99 (74.4) | 99 (73.9) |
| HIV-1-RNA ≥50 copies/mL | 10 (7.5) | 7 (5.2) |
| Withdrawal of informed consent | 7 (5.3) | 9 (6.7) |
| Discontinuation because of adverse events | 7 (5.3) | 11 (8.2) |
| Loss to follow-up | 6 (4.5) | 6 (4.5) |
| Death | 1 (0.8) | 0 (0.0) |
| Other | 3 (2.3) | 2 (1.5) |
| Total | 133 (100) | 134 (100) |
| Patients with blips not leading to interruption of cART | 36 (27.1) | 30 (22.4) |
All variables are expressed as absolute number and percentage; n (%). ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two nucleos(t)ide analogue inhibitors.
Treatment efficacy at 96 weeks (TLOVR; PP population; n = 267)
| Item | ATV/r + 3TC (n = 133) | ATV/r + 2NUCs (n = 134) |
|---|---|---|
| HIV-1-RNA <50 copies/mL | 99 (74.4) | 99 (73.9) |
| HIV-1-RNA ≥50 copies/mL | 10 (7.5) | 7 (5.2) |
| Withdrawal of informed consent | 7 (5.3) | 9 (6.7) |
| Discontinuation because of adverse events | 7 (5.3) | 11 (8.2) |
| Loss to follow-up | 6 (4.5) | 6 (4.5) |
| Death | 1 (0.8) | 0 (0.0) |
| Other | 3 (2.3) | 2 (1.5) |
| Total | 133 (100) | 134 (100) |
| Patients with blips not leading to interruption of cART | 36 (27.1) | 30 (22.4) |
| Item | ATV/r + 3TC (n = 133) | ATV/r + 2NUCs (n = 134) |
|---|---|---|
| HIV-1-RNA <50 copies/mL | 99 (74.4) | 99 (73.9) |
| HIV-1-RNA ≥50 copies/mL | 10 (7.5) | 7 (5.2) |
| Withdrawal of informed consent | 7 (5.3) | 9 (6.7) |
| Discontinuation because of adverse events | 7 (5.3) | 11 (8.2) |
| Loss to follow-up | 6 (4.5) | 6 (4.5) |
| Death | 1 (0.8) | 0 (0.0) |
| Other | 3 (2.3) | 2 (1.5) |
| Total | 133 (100) | 134 (100) |
| Patients with blips not leading to interruption of cART | 36 (27.1) | 30 (22.4) |
All variables are expressed as absolute number and percentage; n (%). ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two nucleos(t)ide analogue inhibitors.
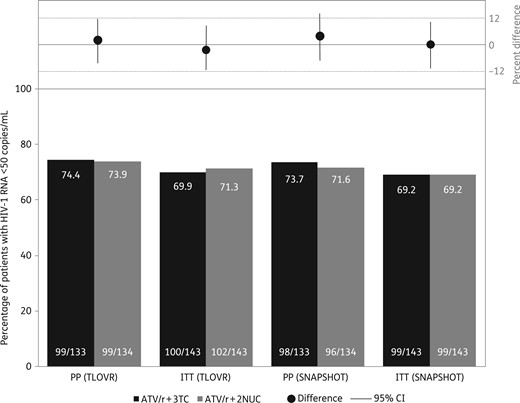
HIV-1-RNA <50 copies/mL (%) at week 96 for the PP and ITT populations.
At week 48, there had been nine cases of confirmed virological failure (five in the ATV/r + 3TC arm and four in the ATV/r + 2NUCs arm), while from week 48 to week 96 only five additional cases were found: four in the ATV/r + 3TC arm and one in the ATV/r + 2NUCs arm. Nine samples could not be amplified because of low HIV-1-RNA, four did not show resistance mutations and only one patient (ATV/r + 2NUCs arm at week 12) developed resistance mutations (M184V). No NRTI or PI resistance mutations were documented in the ATV/r + 3TC arm (Table 2). Furthermore, the frequency of blips in HIV-1-RNA not leading to treatment interruption was similar between both arms: 27.1% for ATV/r + 3TC and 22.4% for ATV/r + 2NUCs (P = 0.37). Adherence to the study medication seemed to be slightly better in the ATV/r + 3TC group (94.1%) than in the ATV/r + 2NUCs group (87.1%) (P = 0.062). Nevertheless, that difference is probably not clinically relevant given that most of the patients were taking the same number of pills in both arms and drug tolerability was similar.
Resistance mutations at failure
| Treatment arm | Time of virological failure (weeks) | First VL (copies/mL) | Second VL (copies/mL) | Resistance mutations |
|---|---|---|---|---|
| ATV/r + 2NUCs | 12 | 71 | 76 | no amplification |
| ATV/r + 2NUCs | 12 | 135 | 1058 | M184V; L63P |
| ATV/r + 2NUCs | 24 | 159 | 142 | no mutations |
| ATV/r + 2NUCs | 36 | 93 | 57 | no amplification |
| ATV/r + 2NUCs | 60 | 227 | 281 | no amplification |
| ATV/r + 3TC | 24 | 84 | 57 | no amplification |
| ATV/r + 3TC | 36 | 61 | 74 | no amplification |
| ATV/r + 3TC | 36 | 572 | 420 | no mutations |
| ATV/r + 3TC | 48 | 134 | 109 | no amplification |
| ATV/r + 3TC | 48 | 73 | 126 | no amplification |
| ATV/r + 3TC | 60 | 51 | 75 | no amplification |
| ATV/r + 3TC | 72 | 93 570 | 3700 | no mutations |
| ATV/r + 3TC | 84 | 5102 | 5497 | no mutations |
| ATV/r + 3TC | 96 | 480 | 50 | no amplification |
| Treatment arm | Time of virological failure (weeks) | First VL (copies/mL) | Second VL (copies/mL) | Resistance mutations |
|---|---|---|---|---|
| ATV/r + 2NUCs | 12 | 71 | 76 | no amplification |
| ATV/r + 2NUCs | 12 | 135 | 1058 | M184V; L63P |
| ATV/r + 2NUCs | 24 | 159 | 142 | no mutations |
| ATV/r + 2NUCs | 36 | 93 | 57 | no amplification |
| ATV/r + 2NUCs | 60 | 227 | 281 | no amplification |
| ATV/r + 3TC | 24 | 84 | 57 | no amplification |
| ATV/r + 3TC | 36 | 61 | 74 | no amplification |
| ATV/r + 3TC | 36 | 572 | 420 | no mutations |
| ATV/r + 3TC | 48 | 134 | 109 | no amplification |
| ATV/r + 3TC | 48 | 73 | 126 | no amplification |
| ATV/r + 3TC | 60 | 51 | 75 | no amplification |
| ATV/r + 3TC | 72 | 93 570 | 3700 | no mutations |
| ATV/r + 3TC | 84 | 5102 | 5497 | no mutations |
| ATV/r + 3TC | 96 | 480 | 50 | no amplification |
ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two NRTIs.
Resistance mutations at failure
| Treatment arm | Time of virological failure (weeks) | First VL (copies/mL) | Second VL (copies/mL) | Resistance mutations |
|---|---|---|---|---|
| ATV/r + 2NUCs | 12 | 71 | 76 | no amplification |
| ATV/r + 2NUCs | 12 | 135 | 1058 | M184V; L63P |
| ATV/r + 2NUCs | 24 | 159 | 142 | no mutations |
| ATV/r + 2NUCs | 36 | 93 | 57 | no amplification |
| ATV/r + 2NUCs | 60 | 227 | 281 | no amplification |
| ATV/r + 3TC | 24 | 84 | 57 | no amplification |
| ATV/r + 3TC | 36 | 61 | 74 | no amplification |
| ATV/r + 3TC | 36 | 572 | 420 | no mutations |
| ATV/r + 3TC | 48 | 134 | 109 | no amplification |
| ATV/r + 3TC | 48 | 73 | 126 | no amplification |
| ATV/r + 3TC | 60 | 51 | 75 | no amplification |
| ATV/r + 3TC | 72 | 93 570 | 3700 | no mutations |
| ATV/r + 3TC | 84 | 5102 | 5497 | no mutations |
| ATV/r + 3TC | 96 | 480 | 50 | no amplification |
| Treatment arm | Time of virological failure (weeks) | First VL (copies/mL) | Second VL (copies/mL) | Resistance mutations |
|---|---|---|---|---|
| ATV/r + 2NUCs | 12 | 71 | 76 | no amplification |
| ATV/r + 2NUCs | 12 | 135 | 1058 | M184V; L63P |
| ATV/r + 2NUCs | 24 | 159 | 142 | no mutations |
| ATV/r + 2NUCs | 36 | 93 | 57 | no amplification |
| ATV/r + 2NUCs | 60 | 227 | 281 | no amplification |
| ATV/r + 3TC | 24 | 84 | 57 | no amplification |
| ATV/r + 3TC | 36 | 61 | 74 | no amplification |
| ATV/r + 3TC | 36 | 572 | 420 | no mutations |
| ATV/r + 3TC | 48 | 134 | 109 | no amplification |
| ATV/r + 3TC | 48 | 73 | 126 | no amplification |
| ATV/r + 3TC | 60 | 51 | 75 | no amplification |
| ATV/r + 3TC | 72 | 93 570 | 3700 | no mutations |
| ATV/r + 3TC | 84 | 5102 | 5497 | no mutations |
| ATV/r + 3TC | 96 | 480 | 50 | no amplification |
ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two NRTIs.
The frequency of grade 3–4 toxicity was evenly distributed between the study groups (Table 3): 70.7% in the ATV/r + 3TC arm versus 70.2% in the ATV/r + 2NUCs arm. The rate of treatment discontinuation at week 96 was similar between the study arms (5.0% versus 7.1%; P = 0.46). Most of the 17 discontinuations were because of hyperbilirubinaemia (n = 4) or toxicity considered secondary to nucleosides (n = 6), namely renal toxicity (n = 3), osteoporosis, hypersensitivity reaction to abacavir and hypophosphataemia. There were 19 (6.8%) severe adverse events, although these were not related to the study medication. The events were lip abscess, cervical polyps, haemorrhoids, avascular hip necrosis, Hodgkin lymphoma relapse, acute pyelonephritis, Hodgkin lymphoma, colonic adenocarcinoma, alcoholic hepatitis and traumatic bone fracture in the ATV/r + 3TC arm and toxicity due to drugs of abuse, spinal stenosis, acute myocardial ischaemia, vulvar abscess, cerebral toxoplasmosis, acute tracheobronchitis with respiratory failure, cholelithiasis, lumbar spinal stenosis and acute renal failure in the ATV/r + 2NUCs arm.
Safety summary
| Toxicity-related discontinuations and AEs | ATV/r + 3TC (n = 140) | ATV/r + 2NUCs (n = 141) |
|---|---|---|
| Discontinuations due to any AEs | 7 (5.0) | 10 (7.1) |
| hyperbilirubinaemia and/or ocular icterus | 2 | 2 |
| renal toxicity | 1 | 3 |
| increased LFT results | 1 | 1 |
| nephrolithiasis | 1 | 1 |
| heartburn | 1 | 0 |
| osteoporosis | 0 | 1 |
| hypersensitivity reaction to abacavir | 0 | 1 |
| metabolic syndrome | 1 | 0 |
| hypophosphataemia | 0 | 1 |
| Grade 3–4 AEs occurring in ≥1% of patients in either group | ||
| any AEs | 99 (70.7) | 99 (70.2) |
| hyperbilirubinaemia | 91 (65.0) | 93 (65.9) |
| jaundice | 0 (0.0) | 2 (1.4) |
| increased LFT results | 3 (2.1) | 1 (0.7) |
| hyperlipidaemiaa | 3 (2.1) | 3 (2.1) |
| thrombocytopenia | 2 (1.4) | 1 (0.7) |
| Toxicity-related discontinuations and AEs | ATV/r + 3TC (n = 140) | ATV/r + 2NUCs (n = 141) |
|---|---|---|
| Discontinuations due to any AEs | 7 (5.0) | 10 (7.1) |
| hyperbilirubinaemia and/or ocular icterus | 2 | 2 |
| renal toxicity | 1 | 3 |
| increased LFT results | 1 | 1 |
| nephrolithiasis | 1 | 1 |
| heartburn | 1 | 0 |
| osteoporosis | 0 | 1 |
| hypersensitivity reaction to abacavir | 0 | 1 |
| metabolic syndrome | 1 | 0 |
| hypophosphataemia | 0 | 1 |
| Grade 3–4 AEs occurring in ≥1% of patients in either group | ||
| any AEs | 99 (70.7) | 99 (70.2) |
| hyperbilirubinaemia | 91 (65.0) | 93 (65.9) |
| jaundice | 0 (0.0) | 2 (1.4) |
| increased LFT results | 3 (2.1) | 1 (0.7) |
| hyperlipidaemiaa | 3 (2.1) | 3 (2.1) |
| thrombocytopenia | 2 (1.4) | 1 (0.7) |
All variables are expressed as absolute number and percentage; n (%). AEs, adverse events; LFT, liver function tests; ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two nucleos(t)ide analogue inhibitors.
aHypertriglyceridaemia (n = 5) and hypercholesterolaemia (n = 1; ATV/r + 2NUCs arm)
Safety summary
| Toxicity-related discontinuations and AEs | ATV/r + 3TC (n = 140) | ATV/r + 2NUCs (n = 141) |
|---|---|---|
| Discontinuations due to any AEs | 7 (5.0) | 10 (7.1) |
| hyperbilirubinaemia and/or ocular icterus | 2 | 2 |
| renal toxicity | 1 | 3 |
| increased LFT results | 1 | 1 |
| nephrolithiasis | 1 | 1 |
| heartburn | 1 | 0 |
| osteoporosis | 0 | 1 |
| hypersensitivity reaction to abacavir | 0 | 1 |
| metabolic syndrome | 1 | 0 |
| hypophosphataemia | 0 | 1 |
| Grade 3–4 AEs occurring in ≥1% of patients in either group | ||
| any AEs | 99 (70.7) | 99 (70.2) |
| hyperbilirubinaemia | 91 (65.0) | 93 (65.9) |
| jaundice | 0 (0.0) | 2 (1.4) |
| increased LFT results | 3 (2.1) | 1 (0.7) |
| hyperlipidaemiaa | 3 (2.1) | 3 (2.1) |
| thrombocytopenia | 2 (1.4) | 1 (0.7) |
| Toxicity-related discontinuations and AEs | ATV/r + 3TC (n = 140) | ATV/r + 2NUCs (n = 141) |
|---|---|---|
| Discontinuations due to any AEs | 7 (5.0) | 10 (7.1) |
| hyperbilirubinaemia and/or ocular icterus | 2 | 2 |
| renal toxicity | 1 | 3 |
| increased LFT results | 1 | 1 |
| nephrolithiasis | 1 | 1 |
| heartburn | 1 | 0 |
| osteoporosis | 0 | 1 |
| hypersensitivity reaction to abacavir | 0 | 1 |
| metabolic syndrome | 1 | 0 |
| hypophosphataemia | 0 | 1 |
| Grade 3–4 AEs occurring in ≥1% of patients in either group | ||
| any AEs | 99 (70.7) | 99 (70.2) |
| hyperbilirubinaemia | 91 (65.0) | 93 (65.9) |
| jaundice | 0 (0.0) | 2 (1.4) |
| increased LFT results | 3 (2.1) | 1 (0.7) |
| hyperlipidaemiaa | 3 (2.1) | 3 (2.1) |
| thrombocytopenia | 2 (1.4) | 1 (0.7) |
All variables are expressed as absolute number and percentage; n (%). AEs, adverse events; LFT, liver function tests; ATV/r, atazanavir/ritonavir; 3TC, lamivudine; 2NUCs, two nucleos(t)ide analogue inhibitors.
aHypertriglyceridaemia (n = 5) and hypercholesterolaemia (n = 1; ATV/r + 2NUCs arm)
We noted no significant changes from baseline to week 96 in renal function, bone mineral density or fat gain or distribution between groups. Use of tenofovir in the new regimen was not associated with a different effect on those characteristics. Similarly, concentrations of 25(OH)-vitamin D did not differ between the study groups at week 96 and were not associated with previous use of efavirenz (Table 4).
Differences in renal function, 25(OH)-vitamin D levels, bone density and fat gain/distribution at week 96 compared with baseline
| ATV/r + 3TC | ATV/r + 2NUCs | P value | |
|---|---|---|---|
| eGFR (CKD-EPI) mL/min | −0.8 (−6.2; +5.0) | −1.4 (−10.8; +3.7) | 0.10 |
| BMD (lumbar) T-score (%) | −10.3 (−40.0; +10.0) | −4.6 (−48.2; +6.8) | 0.95 |
| Trunk fat DEXA (%) | +5.3 (−8.1; +21.3) | +0.9 (−12.0; +18.7) | 0.35 |
| Limb fat DEXA (%) | +5.8 (−5.4; +14.6) | +1.6 (−8.3; +21.3) | 0.78 |
| Limb-to-trunk fat ratio (%) | −2.14 (−10.7; +7.6) | +0.64 (−6.7; +10.5) | 0.28 |
| 25(OH)-vitamin D (ng/mL) | −0.2 (−5.1; 6.4) | +3.1 (−5.0; +10.0) | 0.24 |
| ATV/r + 3TC | ATV/r + 2NUCs | P value | |
|---|---|---|---|
| eGFR (CKD-EPI) mL/min | −0.8 (−6.2; +5.0) | −1.4 (−10.8; +3.7) | 0.10 |
| BMD (lumbar) T-score (%) | −10.3 (−40.0; +10.0) | −4.6 (−48.2; +6.8) | 0.95 |
| Trunk fat DEXA (%) | +5.3 (−8.1; +21.3) | +0.9 (−12.0; +18.7) | 0.35 |
| Limb fat DEXA (%) | +5.8 (−5.4; +14.6) | +1.6 (−8.3; +21.3) | 0.78 |
| Limb-to-trunk fat ratio (%) | −2.14 (−10.7; +7.6) | +0.64 (−6.7; +10.5) | 0.28 |
| 25(OH)-vitamin D (ng/mL) | −0.2 (−5.1; 6.4) | +3.1 (−5.0; +10.0) | 0.24 |
All variables are expressed as median and IQR. BMD, bone mineral density; DEXA, dual-energy X-ray absorptiometry; eGFR, estimated glomerular filtration rate; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration.
Differences in renal function, 25(OH)-vitamin D levels, bone density and fat gain/distribution at week 96 compared with baseline
| ATV/r + 3TC | ATV/r + 2NUCs | P value | |
|---|---|---|---|
| eGFR (CKD-EPI) mL/min | −0.8 (−6.2; +5.0) | −1.4 (−10.8; +3.7) | 0.10 |
| BMD (lumbar) T-score (%) | −10.3 (−40.0; +10.0) | −4.6 (−48.2; +6.8) | 0.95 |
| Trunk fat DEXA (%) | +5.3 (−8.1; +21.3) | +0.9 (−12.0; +18.7) | 0.35 |
| Limb fat DEXA (%) | +5.8 (−5.4; +14.6) | +1.6 (−8.3; +21.3) | 0.78 |
| Limb-to-trunk fat ratio (%) | −2.14 (−10.7; +7.6) | +0.64 (−6.7; +10.5) | 0.28 |
| 25(OH)-vitamin D (ng/mL) | −0.2 (−5.1; 6.4) | +3.1 (−5.0; +10.0) | 0.24 |
| ATV/r + 3TC | ATV/r + 2NUCs | P value | |
|---|---|---|---|
| eGFR (CKD-EPI) mL/min | −0.8 (−6.2; +5.0) | −1.4 (−10.8; +3.7) | 0.10 |
| BMD (lumbar) T-score (%) | −10.3 (−40.0; +10.0) | −4.6 (−48.2; +6.8) | 0.95 |
| Trunk fat DEXA (%) | +5.3 (−8.1; +21.3) | +0.9 (−12.0; +18.7) | 0.35 |
| Limb fat DEXA (%) | +5.8 (−5.4; +14.6) | +1.6 (−8.3; +21.3) | 0.78 |
| Limb-to-trunk fat ratio (%) | −2.14 (−10.7; +7.6) | +0.64 (−6.7; +10.5) | 0.28 |
| 25(OH)-vitamin D (ng/mL) | −0.2 (−5.1; 6.4) | +3.1 (−5.0; +10.0) | 0.24 |
All variables are expressed as median and IQR. BMD, bone mineral density; DEXA, dual-energy X-ray absorptiometry; eGFR, estimated glomerular filtration rate; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration.
Regarding changes in lipid values from baseline to 96 weeks, the percentage change in triglycerides was significantly different between the study arms: 12.1% for ATV/r + 3TC versus −6.2% for ATV/r + 2NUCs (P < 0.003). These changes were related to previous therapy with PI/r in the switched treatment: 25% with no previous PI/r versus −6.2% with previous PI/r (P < 0.001). The percentage change in total cholesterol (Chol) at week 96 was also significantly different, although it was not related to previous therapy with tenofovir and/or PI/r: 5.1% for ATV/r + 3TC versus −1.9% for ATV/r + 2NUCs (P < 0.001). Similarly, the percentage change in the atherogenic index (total Chol/HDL Chol) differed significantly between the study arms −2.6% for ATV/r + 3TC versus −3.7% for ATV/r + 2NUCs (P < 0.021)—and was related to previous therapy with PI/r: 4.8% for no previous PI/r versus −4.0% for previous PI/r (P = 0.002).
Forty-seven patients in the ATV/r + 3TC arm and 45 in the ATV/r + 2NUCs arm completed the neurocognitive substudy at 96 weeks in the PP population. The overall change in neurocognitive function (GDS; five domains) was not significantly different between the study arms: −0.3 (95% CI, −0.5 to −0.1) for ATV/r + 3TC versus −0.2 (95% CI, −0.4 to −0.1) for ATV/r + 2NUCs (P = 0.47). The rates of neurocognitive impairment were also similar between the arms: 51.1% for ATV/r + 3TC versus 51.1% for ATV/r + 2NUCs (P = 0.99). This absence of differences was also observed in all cognitive tasks. Detailed results of the neurocognitive substudy have been reported elsewhere.7
Discussion
NUC-sparing antiretroviral regimens have the advantage of reducing antiretroviral-associated toxicity, easing adherence and reducing costs. In virologically suppressed HIV-1-infected patients, switching to NUC-sparing regimens maintains the efficacy and safety of triple cART. The results of the SALT trial at 96 weeks confirm the non-inferiority of dual therapy with ATV/r + 3TC at 48 weeks compared with a standard triple regimen based on ATV/r + 2NUCs in patients who needed to change treatment for reasons of toxicity, intolerance or convenience. The results of efficacy at 96 weeks were consistent across several sensitivity analyses, as they were at 48 weeks. Dual therapy showed a favourable safety profile, with no negative impact on neurocognitive performance. In addition, this regimen offers some advantages regarding cost savings that can be even higher with the use of generics. In the context of limited resources, any therapeutic intervention must be applied efficiently. Dual therapy with ATV/r plus generic lamivudine can save up to 25%–40% compared with a standard treatment based on ATV/r + 2NUCs in a fixed drug combination.8
The rate of confirmed virological failure was low in both arms at 96 weeks: 6.7% (nine failures) for the ATV/r + 3TC arm compared with 3.7% (five failures) for the ATV/r + 2NUCs arm (P = 0.26). At week 48, there were four more virological failures in the dual-therapy arm and one more in the triple-therapy arm, with no new antiretroviral resistance mutations detected. Similarly, the frequency of HIV-1-RNA blips not leading to treatment interruption was similar between the arms. Thus, the long-term global efficacy of switching to dual therapy with ATV/r + 3TC was good and did not lead to the emergence of resistance mutations.
This is the first randomized trial with 96 week results on PI/r + lamivudine as a switching option in virologically suppressed patients. To date, the only long-term results published on ATV/r + 3TC are those from the AtLaS pilot study at 144 weeks.1 AtLaS was a single-arm, non-comparative study, in which 40 patients switched from ATV/r + 2NUCs (97.5% were taking tenofovir) to ATV/r + 3TC. Treatment effectiveness was good with only two virological failures (10%) at 48 and 53 weeks, with no resistance mutations. Although these results were promising, the inherent limitations of a non-comparative pilot study called for confirmation in a well-powered randomized clinical trial. In this sense, the rate for virological failure in the SALT trial was even lower and no resistance mutations were selected in the ATV/r + 3TC arm. As for adverse events, the AtLaS study disclosed a good safety profile for ATV/r + 3TC. Most were mild to moderate and only two severe events were reported (thyroid and anal cancer), although these were not considered treatment related. Adverse events leading to discontinuation were rare (3/40 patients; 7.5%) and only two were related to treatment (recurrent renal colic in both cases). In the SALT trial, the frequency of discontinuation due to any adverse event was similar for the ATV/r + 3TC arm (7/140 patients; 5.0%) and comparable to that of the ATV/r + 2NUCs arm (10/141 patients; 7.1%). Nevertheless, nephrolithiasis was much less frequent: 5/40 cases in the AtLaS trial (two leading to interruption and treatment related) versus only 2/287 cases in the SALT trial (one leading to treatment interruption and not treatment related). These results argue against the potential risk of a higher frequency of nephrolithiasis due to an increment in the plasma levels of atazanavir when withdrawing tenofovir. The only two cases were evenly distributed between the two arms. There were no new discontinuations because of hyperbilirubinaemia in either arm beyond 48 weeks.
Unlike AtLaS, we did not observe significant changes in bone mineral density, body fat distribution or renal function between the groups from baseline to 96 weeks, possibly because of the diverse nature of the reasons for switching, which are not always necessarily related to tenofovir. In fact, after randomization, 71% of patients in the triple-therapy arm were still receiving tenofovir, indicating that patients entered the study for reasons other than tenofovir-induced toxicity. Changes in the lipid profile were more complex than in AtLaS, where they were mainly influenced by withdrawal of tenofovir. In the SALT study, changes in lipid profile were significantly related to the presence of tenofovir and/or PI/r in the antiretroviral regimen prior to study entry.
We performed a post hoc analysis to test whether the response to dual therapy with ATV/r + 3TC at 48 weeks was affected in specific groups of patients. Although our results should be interpreted in the context of a non-planned analysis, no statistically significant differences in treatment efficacy were observed in patients according to sex, coinfection with HCV, prior use of PI/r, NNRTIs or ATV/r,9 thus showing the robustness of this dual strategy.
To date, no NUC-sparing dual-therapy switching regimens other than PI/r + lamivudine2–4 have shown non-inferiority to standard triple therapy in comparative trials,10 although the only strategy with long-term data is based on ATV/r + 3TC. Results from ongoing randomized clinical trials on switching to dual therapy—both darunavir/ritonavir-based (ClinicalTrials.gov NCT02159599, NCT01792570 and NCT02486133) and integrase inhibitor-based (mainly dolutegravir) (ClinicalTrials.gov NCT02263326, NCT02429791 and NCT02422797)—are pending. Taken together, these results highlight the relevance of long-term data from the SALT study, especially considering that many of the ongoing dual-therapy studies in suppressed patients were designed for a 48 week time frame.
Boosted PI monotherapy with lopinavir/ritonavir and darunavir/ritonavir was a widely explored NUC-sparing strategy that in most cases was non-inferior to triple therapy at 48 weeks, although results for long-term endpoints were not confirmed at 96 or 144 weeks,11,12 as virological rebounds proved more frequent in the monotherapy arm. The PIVOT study,13 the longest clinical trial exploring monotherapy at 5 years, showed that 23% of patients in the monotherapy arm had virological rebounds. A recent meta-analysis on monotherapy switch studies concludes that this strategy is not universally recommended.14
In summary, the 96 week results of the SALT trial showed that switching to ATV/r + 3TC is effective, safe and non-inferior to ATV/r + 2NUCs in virologically suppressed HIV-1-infected patients who need to switch cART because of toxicity or intolerance or for simplification of their regimens. Pending results from the most recent NUC-sparing dual-therapy strategies, ATV/r + 3TC offers a good alternative to standard triple therapy and has the added advantage of being a simple and inexpensive regimen with low toxicity.
Funding
Fundación SEIMC-GeSIDA sponsored the study and, together with the main author and scientific committee, managed the trial design, monitoring, data collection, data analysis, and writing of the report. Bristol-Myers Squibb supported the trial with an unrestricted grant.
Transparency declarations
J. A. P.-M. reports grants, payment for lectures and non-financial support from Bristol-Myers Squibb, payment for lectures from ViiV and Gilead, and non-financial support from Merck Sharp and Dohme outside the submitted work. R. R. reports grants from Abbott and Janssen, and payment for lectures from Abbott, Bristol-Myers Squibb, ViiV, Gilead, Janssen, and Merck Sharp and Dohme outside the submitted work. A. R. reports grants and personal fees from Bristol-Myers Squibb, Gilead, ViiV, Merck Sharp and Dohme, Boehringer Ingelheim, Jansen, Abbott, and AbbVie outside the submitted work. J. P. reports grants and personal fees from Bristol-Myers Squibb, Janssen, Abbie, Gilead, ViiV, Merck Sharp and Dohme, Boehringer Ingelheim, and Roche Pharma outside the submitted work. I. S.-L. reports personal fees and non-financial support from Gilead and Bristol-Myers Squibb, non-financial support from ViiV and AbbVie, and personal fees from Janssen outside the submitted work. R. P. reports personal fees from AbbVie, Bristol-Myers Squibb, Gilead, Janssen, and ViiV outside the submitted work. J. S.-M. reports personal fees from AbbVie, Bristol-Myers Squibb, Gilead, Janssen, and ViiV outside the submitted work. J. T. reports personal fees from AbbVie, ViiV, Merck Sharp and Dohme, Gilead, Bristol-Myers Squibb, and Janssen outside the submitted work. A. A. reports personal fees from AbbVie, Bristol-Myers Squibb, Gilead, Merck Sharp and Dohme, ViiV, and Janssen outside the submitted work. J. N. reports personal fees from AbbVie, Bristol-Myers Squibb, GlaxoSmithKline and Merck Sharp and Dohme, and personal fees and non-financial support from Gilead, Janssen, and ViiV outside the submitted work. All other authors declare no competing interests.
Author contributions
J. A. P.-M. designed the study in consultation with R. R. and S. M. The GeSIDA-7011 Study Group enrolled patients into the study and acquired data. J. A. P.-M., R. R. and S. M. analysed the data and clinically oversaw the study. J. A. P.-M., R. R., A. R., J. P., I. S.-L., M. R., M. E., R. P., J. S.-M., J. T., A. M., A. A., J. N. and S. M. interpreted the data. H. E. managed and coordinated the study. J. A. P.-M. drafted the report and R. R. and S. M. reviewed it. All authors provided input into the report and approved the final version.
Acknowledgements
Members of the GeSIDA-7011 Study Group
Complejo Hospitalario Universitario A Coruña, A Coruña (J. D. Pedreira Andrade, M. A. Castro Iglesias, A. Mena, S. López and P. Vázquez); Fundación SEIMC-GeSIDA, Madrid (H. Esteban); Hospital Arquitecto Marcide, Ferrol (S. Sánchez and T. Caínzos); Hospital Civil de Basurto, Bilbao (J. Muñoz, O. L. Ferrero, Z. Zubero Zulibarria, S. Ibarra, J. M. Santamaría Jáuregui and J. Baraitzxaburu Artexe); Hospital Costa del Sol, Málaga (J. Olalla, A. De Arco, J. De la Torre and J. L. Prada); Hospital Clínico San Carlos, Madrid (M. J. Téllez, J. Vergas and V. Estada); Hospital Clínico San Cecilio de Granada, Granada (J. Hernández Quero, A. Peña Monje, J. Parra, M. Martínez, L. Muñoz and V. Sánchez); Hospital Clínico Universitario de Santiago, Santiago de Compostela (E. Losada Arias and A. Prieto Martínez); Hospital de Donostia, Donostia (J. A. Iribarren Loyarte, M. Ibarguren, H. Azkune Galparsoro, M. J. Bistinduy Odriozola, X. C. Ortiz de Barrón, M. A. Goenaga Sánchez, M. A. Von Wichmann De Miguel, L. Pascual Tomé and X. Camino); Hospital de la Marina Baixa de Villajoyosa, Valencia (J. Ena, F. Pascuau, C. Amador and C. Benito); Hospital de Mataró, Barcelona (P. Barrufet, L. Force and G. Bejarano); Hospital de la Santa Creu i Sant Pau, Barcelona (P. Domingo Pedrol, M. Gutierren and G. Mateo); Hospital General de Jerez de la Frontera, Cádiz (A. Terrón, D. Marín and P. Bancalero); Hospital General Universitario Gregorio Marañón, Madrid (J. Cosín, J. Berenguer, P. Miralles, M. Sánchez, J. C. López, M. Ramírez, I. Cuellar and A. Carrero); Hospital General Universitario de Alicante, Alicante (J. Portilla, V. Boix, E. Merino, S. Reus, L. Giner and M. Pampliega); Hospital General Universitario de Elche, Alicante (F. Gutierrez, M. Masía Conto, J. M. Ramos Rincón, S. Padilla Urrea and C. Robledano García); Hospital Infanta Elena and Hospital Juan Ramón Jiménez, Huelva (J. M. Fajarso Picó, M. D. Merino Muñoz, F. J. Martínez Marcos and F. J. Rodríguez Gómez); Hospital Infanta Leonor, Madrid (P. Rayan, J. Solís and N. Palomero); Hospital Universitari Germans Trias i Pujol, Barcelona (B. Clotet, A. Chamorro, E. Negredo Puigmal, P. Echevarría, A. Bonjoch, J. Moltó and J. Puig); Hospital Universitari Son Espases, Mallorca (M. A. Ribas Blanco and C. Marinescu); Hospital Universitari Vall D'Hebrón, Barcelona (M. Crespo and J. Navarro); Hospital Universitario Central de Asturias, Asturias (V. Asensi and J. A. Cartón Sanchez); Hospital Universitario Doce de Octubre, Madrid (F. Pulido, M. Matarraz, M. Lagarde, O. Bisbal and A. Portillo); Hospital Universitario La Paz, Madrid (J. R. Arribas, I. Bernardino, F. J. Zamora, M. L. Montes, J. J. González, I. Pérez and J. Castro); Hospital Universitario Príncipe de Asturias, Madrid (J. De Miguel, A. Arranz and E. Casas); Hospital Universitario Ramón y Cajal, Madrid (F. Dronda, S. Moreno, J. Fortún, M. J. Pérez-Elías, E. Navas, C. Quereda and M. A. Rodríguez-Sagrado); Hospital Universitario Reina Sofía, Córdoba (R. Jurado, M. García, A. Camacho and J. De la Torre); Hospital Universitario Virgen de las Nieves, Granada (A. Tapia, C. Hidalgo, M. A. López, R. Martínez and C. García); Hospital Universitario Virgen de la Victoria, Málaga (J. Santos, M. Márquez, J. Ruiz and E. Nuño); Hospital San Pedro, Logroño (J. A. Oteo, J. R. Blanco, M. Sanz, V. Ibarra, L. Metola and L. Pérez); and Hospital Xeral Cíes de Vigo, Pontevedra (C. Miralles, A. Ocampo, A. Rodríguez and F. Warncke).
References
Author notes
Members are listed in the Acknowledgements section.

{kind=link}
{kind=link}