





Abstract
Enterovirus 71 (EV-A71) is an important pathogen that can cause severe neurological symptoms and even death. Our aim was to identify potent anti-EV-A71 compounds and study their underlying mechanisms and in vivo activity.
We identified a potent imidazolidinone derivative (abbreviated to PR66) as an inhibitor of EV-A71 infection from the screening of compounds and subsequent structure-based modification. Time-course treatments and resistant virus selection of PR66 were employed to study the mode of mechanism of PR66. In vivo activity of PR66 was tested in the ICR strain of new-born mice challenged with EV-A71/4643/MP4.
PR66 could impede the uncoating process during viral infection via interaction with capsid protein VP1, as shown by a resistant virus selection assay. Using site-directed mutagenesis, we confirmed that a change from valine to phenylalanine in the 179th amino acid residue of the cDNA-derived resistant virus resulted in resistance to PR66. PR66 increased the virion stability of WT viruses, but not the PR66-resistant mutant, in a particle stability thermal release assay. We further showed that PR66 had excellent anti-EV-A71 activity in an in vivo mouse model of disease, with a dose-dependent increase in survival rate and in protection against virus-induced hind-limb paralysis following oral or intraperitoneal administration. This was associated with reductions of viral titres in brain and muscle tissues.
We demonstrated here for the first time that an imidazolidinone derivative (PR66) could protect against EV-A71-induced neurological symptoms in vivo by suppressing EV-A71 replication. This involved binding to and restricting viral uncoating.
Introduction
Enterovirus 71 (EV-A71) is an emerging pathogen in the Asia-Pacific region. It mainly infects children <5 years of age. Most infections are symptomless, but some infected individuals might have hand, foot and mouth diseases, herpangina, pulmonary oedema or severe CNS disorders, or may even die.
EV-A71 belongs to the genus Enterovirus of the family Picornaviridae, containing one positive single-stranded 7.4 kbp RNA genome. The genome serves as a template to translate a single polyprotein, which is further cleaved by viral proteases 2A and 3C to give rise to several mature proteins. These proteins comprise four structural viral proteins (VP1–VP4) and seven non-structural proteins (2A–C and 3A–D). The virion capsid is composed of 60 protomers, which are assembled by VP1–3, to form an icosahedral lattice.1 The crystal structures of these components are now available.2–4 These revealed that VP1–3 form the shell of the virion, as in polioviruses, to provide antigenicity and receptor binding, but VP4 remains inside the capsid and interacts with the RNA genome in stabilizing the whole virion structure. A groove or ‘canyon’ is present in each lattice, which is used for receptor binding. In addition, a hydrophobic pocket is located beneath the canyon for interaction with hydrophobic compounds.5
Although vaccines against EV-A71 have completed phase III clinical trials with good safety and efficacy,6 effective antiviral agents are still needed for non-immunized or immunocompromised individuals. According to the life cycle of EV-A71, several steps might be vulnerable for inhibiting viral replication, including attachment, uncoating, RNA replication and viral assembly.7 Pleconaril was found to substitute for natural acyl compounds in binding into the hydrophobic pocket within VP1, which leads to an alteration in virion structure and prevents the conformational change needed for uncoating.3,8 The so-called WIN compounds (named from Winthrop-based companies) and derivatives were modified and synthesized based on pleconaril and some of them were able to reduce virus-induced CPE significantly.5,9–11
Materials and methods
Cell culture, viruses and viral infection
Rhabdomyosarcoma (RD) and human foreskin fibroblast (HFF) cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FBS (JRH Biosciences, Lenexa, KS, USA). Madin–Darby canine kidney (MDCK) cells were cultured in DMEM containing 10% heat-inactivated FBS, 2 mM l-glutamine (Gibco BRL, Gaithersburg, MD, USA), 0.1 mM non-essential amino acid mixture (Gibco BRL), 100 U/mL penicillin and 0.1 mg/mL streptomycin (Sigma-Aldrich, Poole, UK). Adenocarcinomic human alveolar basal epithelial (A549) cells were grown in MEM (Gibco BRL), supplemented with 10% FBS, penicillin and streptomycin as above. All cell lines were cultured at 37°C under humidified air with 5% CO2. The EV-A71/2231/TW virus strain was generated from an infectious clone provided by Dr Mei-Shang Ho (Academia Sinica, Taipei). The infectious clone of mouse-adapted EV-A71/4643/MP4 virus was a gift from Dr Jen-Ren Wang of National Cheng Kung University.12 The clinical virus strains listed in Table 1 were from the Clinical Virology Laboratory (Chang Gung Memorial Hospital, Taiwan).13 EV-A71 propagation and titre determination by plaque-forming assays using RD cells were performed essentially as described.14 Viral infection was carried out by adsorption of EV-A71 to RD cells for 1 h from −1 to 0 h post-infection with the tested compound. The unbound virus was removed by washing twice with PBS and the cells were cultured in DMEM supplemented with 2% FBS (E2 medium) until harvested.
Inhibition spectra of PR66 against enteroviruses or influenza viruses
| Cell line or virus strain | PR66 | ||
|---|---|---|---|
| CC50 (μM)a | IC50 (μM)b | SIc | |
| Cytotoxic effectd | |||
| A549 | >100 | ||
| MDCK | 3.466 ± 2.089 | ||
| RD | >100 | ||
| Enterovirus | |||
| EV71 BrCr (genotype A) | 0.028 ± 0.009 | +++ | |
| EV71 TW/2231/98 (genotype C) | 0.019 ± 0.001 | +++ | |
| EV71 TW/4643/98 (genotype C) | 0.026 ± 0.005 | +++ | |
| EV71 TW/51126/12 (genotype B) | 0.024 ± 0.010 | +++ | |
| EV71 TW/2557/12 (genotype B) | 0.028 ± 0.010 | +++ | |
| EV71 TW/50995/12 (genotype B) | 0.058 ± 0.002 | +++ | |
| EV71 TW/51045/12 (genotype B) | 0.032 ± 0.001 | +++ | |
| EV71 TW/184/12 (genotype C) | 0.008 ± 0.003 | ++++ | |
| EV71 TW/73/12 (genotype C) | 0.006 ± 0.003 | ++++ | |
| EV-D68 | >10 | − | |
| coxsackievirus A16 | 0.188 ± 0.062 | ++ | |
| coxsackievirus B1 | >25 | − | |
| coxsackievirus B2 | >25 | − | |
| coxsackievirus B3 | >25 | − | |
| echovirus 9 | >25 | − | |
| human rhinovirus sp. | >25 | ||
| Influenza virus | |||
| A/WSN/33 (H1N1) | >5 | − | |
| B/TW/70325/05 | >5 | − | |
| Herpes simplex virus-1 | >10 | − | |
| Adenovirus | >10 | − | |
| Cell line or virus strain | PR66 | ||
|---|---|---|---|
| CC50 (μM)a | IC50 (μM)b | SIc | |
| Cytotoxic effectd | |||
| A549 | >100 | ||
| MDCK | 3.466 ± 2.089 | ||
| RD | >100 | ||
| Enterovirus | |||
| EV71 BrCr (genotype A) | 0.028 ± 0.009 | +++ | |
| EV71 TW/2231/98 (genotype C) | 0.019 ± 0.001 | +++ | |
| EV71 TW/4643/98 (genotype C) | 0.026 ± 0.005 | +++ | |
| EV71 TW/51126/12 (genotype B) | 0.024 ± 0.010 | +++ | |
| EV71 TW/2557/12 (genotype B) | 0.028 ± 0.010 | +++ | |
| EV71 TW/50995/12 (genotype B) | 0.058 ± 0.002 | +++ | |
| EV71 TW/51045/12 (genotype B) | 0.032 ± 0.001 | +++ | |
| EV71 TW/184/12 (genotype C) | 0.008 ± 0.003 | ++++ | |
| EV71 TW/73/12 (genotype C) | 0.006 ± 0.003 | ++++ | |
| EV-D68 | >10 | − | |
| coxsackievirus A16 | 0.188 ± 0.062 | ++ | |
| coxsackievirus B1 | >25 | − | |
| coxsackievirus B2 | >25 | − | |
| coxsackievirus B3 | >25 | − | |
| echovirus 9 | >25 | − | |
| human rhinovirus sp. | >25 | ||
| Influenza virus | |||
| A/WSN/33 (H1N1) | >5 | − | |
| B/TW/70325/05 | >5 | − | |
| Herpes simplex virus-1 | >10 | − | |
| Adenovirus | >10 | − | |
Values are the mean ± SD of the results from two to three independent experiments.
aCC50 was determined with an MTT assay.
bIC50 was determined with an anti-CPE (neutralization) assay.
cSelectivity index (SI) was defined as the ratio of CC50 to IC50 values: ++, SI >100; +++, SI >1000; ++++, SI >10 000; and −, not available for determination.
dCytotoxicities of enteroviruses and human rhinovirus were determined in RD cells, whereas those of influenza viruses were studied in MDCK cells and those of herpes simplex virus-1 and adenovirus were studied in A549 cells.
Inhibition spectra of PR66 against enteroviruses or influenza viruses
| Cell line or virus strain | PR66 | ||
|---|---|---|---|
| CC50 (μM)a | IC50 (μM)b | SIc | |
| Cytotoxic effectd | |||
| A549 | >100 | ||
| MDCK | 3.466 ± 2.089 | ||
| RD | >100 | ||
| Enterovirus | |||
| EV71 BrCr (genotype A) | 0.028 ± 0.009 | +++ | |
| EV71 TW/2231/98 (genotype C) | 0.019 ± 0.001 | +++ | |
| EV71 TW/4643/98 (genotype C) | 0.026 ± 0.005 | +++ | |
| EV71 TW/51126/12 (genotype B) | 0.024 ± 0.010 | +++ | |
| EV71 TW/2557/12 (genotype B) | 0.028 ± 0.010 | +++ | |
| EV71 TW/50995/12 (genotype B) | 0.058 ± 0.002 | +++ | |
| EV71 TW/51045/12 (genotype B) | 0.032 ± 0.001 | +++ | |
| EV71 TW/184/12 (genotype C) | 0.008 ± 0.003 | ++++ | |
| EV71 TW/73/12 (genotype C) | 0.006 ± 0.003 | ++++ | |
| EV-D68 | >10 | − | |
| coxsackievirus A16 | 0.188 ± 0.062 | ++ | |
| coxsackievirus B1 | >25 | − | |
| coxsackievirus B2 | >25 | − | |
| coxsackievirus B3 | >25 | − | |
| echovirus 9 | >25 | − | |
| human rhinovirus sp. | >25 | ||
| Influenza virus | |||
| A/WSN/33 (H1N1) | >5 | − | |
| B/TW/70325/05 | >5 | − | |
| Herpes simplex virus-1 | >10 | − | |
| Adenovirus | >10 | − | |
| Cell line or virus strain | PR66 | ||
|---|---|---|---|
| CC50 (μM)a | IC50 (μM)b | SIc | |
| Cytotoxic effectd | |||
| A549 | >100 | ||
| MDCK | 3.466 ± 2.089 | ||
| RD | >100 | ||
| Enterovirus | |||
| EV71 BrCr (genotype A) | 0.028 ± 0.009 | +++ | |
| EV71 TW/2231/98 (genotype C) | 0.019 ± 0.001 | +++ | |
| EV71 TW/4643/98 (genotype C) | 0.026 ± 0.005 | +++ | |
| EV71 TW/51126/12 (genotype B) | 0.024 ± 0.010 | +++ | |
| EV71 TW/2557/12 (genotype B) | 0.028 ± 0.010 | +++ | |
| EV71 TW/50995/12 (genotype B) | 0.058 ± 0.002 | +++ | |
| EV71 TW/51045/12 (genotype B) | 0.032 ± 0.001 | +++ | |
| EV71 TW/184/12 (genotype C) | 0.008 ± 0.003 | ++++ | |
| EV71 TW/73/12 (genotype C) | 0.006 ± 0.003 | ++++ | |
| EV-D68 | >10 | − | |
| coxsackievirus A16 | 0.188 ± 0.062 | ++ | |
| coxsackievirus B1 | >25 | − | |
| coxsackievirus B2 | >25 | − | |
| coxsackievirus B3 | >25 | − | |
| echovirus 9 | >25 | − | |
| human rhinovirus sp. | >25 | ||
| Influenza virus | |||
| A/WSN/33 (H1N1) | >5 | − | |
| B/TW/70325/05 | >5 | − | |
| Herpes simplex virus-1 | >10 | − | |
| Adenovirus | >10 | − | |
Values are the mean ± SD of the results from two to three independent experiments.
aCC50 was determined with an MTT assay.
bIC50 was determined with an anti-CPE (neutralization) assay.
cSelectivity index (SI) was defined as the ratio of CC50 to IC50 values: ++, SI >100; +++, SI >1000; ++++, SI >10 000; and −, not available for determination.
dCytotoxicities of enteroviruses and human rhinovirus were determined in RD cells, whereas those of influenza viruses were studied in MDCK cells and those of herpes simplex virus-1 and adenovirus were studied in A549 cells.
IC50 determination by neutralization (anti-CPE) assay
RD cells were infected with EV-A71/2231 virus (9× TCID50) in the presence of various concentrations of tested compounds in E2 as previously described.15 After incubation for 72 h, the medium was removed and the cells were stained with 0.1% Crystal Violet. IC50 was defined as the concentration of chemical that could inhibit the virus-induced CPE by 50%.
Cytotoxicity determination by MTT assay
The cytotoxicity of compounds was determined using an MTT-based assay,15 where the RD cells were incubated with serially diluted compounds at 37°C for 72 h.
Time course (time-of-addition) assay
RD cells were seeded in six-well tissue culture plates overnight. Cells were washed with PBS and then infected with EV-A71/2231 (moi of 10) for 1 h (−1 to 0 h post-infection). An aliquot of 20 nM PR66 was added at −2, −1, 0, 1, 3 and 5 h post-infection. At 8 h post-infection, the cells and culture medium were collected for determining the titre by plaque-forming assays.14
Direct inhibition of EV-A71 by PR66 determined by centrifugal filtration assay
The virus was mixed with 1 mL of DMEM in the presence of PR66 or an equal volume of DMSO and then incubated at 37°C for 1 h. The mix was then transferred to Microsep centrifugal filters with a 100 kDa cut-off (Pall Corporation, Port Washington, NY, USA), centrifuged at 1900 g at 4°C for 20 min and then washed with PBS three times to remove unbound compounds. The titre of the virus in the concentrator reservoir was then determined by plaque-forming assays.
Viral attachment assay
Inhibition of viral attachment to HFF cells was performed as described.15 Briefly, precooled HFF cells were incubated with E2 (containing 9 TCID50 of virus) along with 2-fold serially diluted PR66 (0.5–500 nM). The inhibition of viral attachment was indicated by the degree of virus-induced CPE.
Virus purification by sucrose gradient and particle stability thermal release (PaSTRy) assay
Viruses were subjected to a 15%–30% discontinuous sucrose gradient and ultracentrifuged in an SW41 Ti rotor (Beckman Instruments Inc., Fullerton, CA, USA) at 234 000 g for 2 h at 4°C. Fractions containing mature viral particles were identified by western blotting using a specific antibody against VP2 (Merck Millipore, Billerica, MA, USA). These were washed with PBS to remove sucrose and concentrated using Amicon centrifugal filters (Ultra-15 100K cut-off; Merck Millipore). The PaSTRy assay was based on a method developed by Walter and colleagues.5,16 Briefly, a master mix of purified virus with PR66 or DMSO (control) was diluted to 100 μL with PBS and treated with 40 U of RNaseOUT (Invitrogen) for 1 h at 33°C. An aliquot of 10 μL of the green fluorescent dye SYTO9 (50 μM; Molecular Probes, Eugene, OR, USA) was added and the final mix was dispensed into four wells each containing 25 μL in 96-well plates. The temperature ramp from 40°C to 92°C was achieved using a StepOnePlus RT–PCR machine (Applied Biosystems, Foster City, CA, USA) and the fluorescence reading was recorded in quadruplets at 1°C intervals. The temperature of release of viral RNA was calculated from the inflection point of the change in fluorescence intensity as a function of temperature.
Generation of recombinant viruses
We screened the resistant virus by gradually increasing the concentration of PR66 to 1 μM (16 passages) along with a vehicle control (DMSO). Three virus plaques of each PR66-resistant virus (66RV) and control virus were amplified separately for resistance validation and genome sequencing. Four sets of primers (Table S1, available as Supplementary data at JAC Online) were used to generate recombinant viruses by site-directed mutagenesis, with specific single or double mutations in VP1 (V179F, L247V and V179F/L247V). For recombinant virus production, 5 μg of plasmid was linearized with MluI (TaKaRa Bio Inc.) and then extracted with EasyPure PCR Clean-up/Gel extraction kits (Bioman Scientific Co. Ltd, Taipei, Taiwan). Viral RNA was then transcribed with the T7 RiboMAX Express Large Scale RNA production system (Promega, Madison, WI, USA) and was used to transfect RD cells to produce recombinant viruses.
Growth curve of recombinant viruses
RD cells in six-well plates were infected with recombinant viruses (moi of 5) for 1 h. Progeny viruses were collected from culture supernatants and cell debris at 4, 6, 8, 10 and 12 h post-infection for viral titre determination using plaque-forming assays.
Ethics
All animal protocols were approved by the Institutional Animal Care and Use Committee of Chang Gung University (CGU10-001). The animals were cared for in line with national guidance.
Mouse experiments
The EV-A71-infected mouse model was established by Wang et al.12 Five-day-old specific pathogen-free ICR strain mice were purchased from Lasco (Taipei, Taiwan). Starting from day −1 (6 days old), PR66 was administered orally or intraperitoneally with one dose of the indicated dosage; from day 0, two doses per day were administered for eight more days. Mice were starved for 8 h before an intraperitoneal virus challenge on day 0 (virus strain: EV-A71/4643/MP4; 2× median lethal dose: 1 × 106 pfu in 100 μL, 9–11 mice per group). PR66 was prepared at the indicated concentrations in a final volume of 100 μL containing 5% DMSO in aqueous solution. The infected mice were monitored daily for any body weight change, survival rate and any neurological symptoms. The symptoms were scored to represent the progression of viral infection as: 0, normal; 1, slow movement; 2, single hind-limb paralysis and irregular movement; 3, bilateral hind-limb paralysis; or 4, death.17 Tissues of brain and muscle (from the biceps femoris) were snap-frozen in liquid nitrogen and stored at −80°C until use. The left or right brain (∼180–200 mg) or muscles (∼30–70 mg) were ground for 5–10 min with 100 μL DMEM with a tissue grinder (pellet pestle motor; Kimble Chase, Vineland, NJ, USA) until no more solid tissue could be observed, followed by one round of freeze and thaw. After adding 400 or 1100 μL DMEM to muscle and brain homogenates, respectively, the samples were centrifuged at 12 000 g for 10 min at 4°C. The supernatants were collected and stored in small aliquots at −80°C for plaque-forming assays.
Statistical analysis
Student's two-tailed t-test was used for statistical analysis and significance was set at P < 0.05. Analysis of viral titres in tissues was based on IQR measurements: [Q1 − k(Q3 − Q1), Q3 + k(Q3 − Q1)] where Q1 and Q3 are the lower and upper quartiles, respectively, and k is defined as 2. Measurements out of the range were considered outliers. Analysis of mouse survival rates was based on the Gehan–Breslow–Wilcoxon test.
Results
Screening anti-EV-A71 agents from a chemical library
In an attempt to identify EV-A71 inhibitors, we performed screening of about 30 different synthetic compounds with diverse structures (Supplementary Data). PR53 stood out as the most potent inhibitor of EV-A71/2231, other EV-A71 strains and coxsackieviruses, with an IC50 of 0.4–3.5 μM (Supplementary Data). Using further modification of the pharmacophores of PR53 by rational design, we identified PR66 as the best inhibitor against different EV-A71 genotypes (6–58 nM), including strains isolated in 2012, and the potency was enhanced by a >70-fold increase in IC50 against EV-A71/2231 with a selectivity index >1000 (Figure 1 and Table 1). The CC50 of PR66 was >100 μM for RD and A549 cells, but it was more toxic against MDCK cells. PR66 also impaired CVA16 with an IC50 of ∼0.2 μM, which was about an order of magnitude less active than EV-A71 (Table 1). However, PR66 did not inhibit CVB1-3, echovirus 9, human rhinovirus or EV-D68, although these viruses belong to the same genus as EV-A71, indicating that PR66 is a highly specific inhibitor of EV-A71. The same findings also applied to other less closely related viruses, such as the (−)RNA influenza A and B viruses and the DNA virus human herpes simplex virus-1 and adenovirus (Table 1). While improving the anti-EV-A71 efficacy by performing a structure–activity relationship study, the anti-coxsackievirus activity was lost or barely increased in comparison with the efficacy of PR53 (Supplementary Data). In addition, PR66 also markedly inhibited the virus-induced CPE at a non-toxic concentration (Supplementary Data).
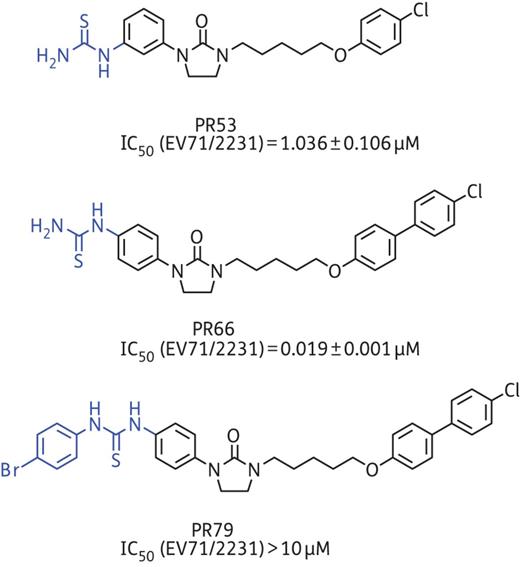
Chemical structures and corresponding IC50 of PR53, PR66 and PR79. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
PR66 might target viral particles directly as shown by time-of-addition and centrifugal filtration assays
In a time-of-addition assay, PR66 was administered to RD cells before, during or after infection (Figure 2a). The inhibitory effect of PR66 was less pronounced at 3–8 and 5–8 h post-infection, indicating that PR66 might exert its antiviral activity in the early stage of infection (Figure 2b). In addition, we performed viral attachment assays to characterize the mechanism of action of PR66 (Figure 2c). Increasing concentrations of PR66 were added to HFF cells during the viral pre-adsorption stage on ice. The unbound viruses were washed away and the medium was replenished. The protection from virus-induced cell death provided by PR66 was shown by increased cell survival in a dose-dependent manner. The IC50 obtained by the attachment assay was 38.95 ± 6.85 nM, similar to the figure of 1.265 ± 0.004 nM using an anti-CPE assay with HFF cells, indicating that PR66 might target the viral entry step, such as by inhibiting viral virions or cellular receptors. To further test whether PR66 would inhibit EV-A71 infection by directly targeting the viral particles, we employed a centrifugal filtration assay where we incubated EV-A71/2231 with increasing concentrations of PR66 on ice and then washed the preparations with PBS using centrifugal filters to remove the compounds. The results showed a considerable reduction in virus titre in a dose-dependent manner, suggesting that PR66 might inhibit the virus directly (Figure 2d).
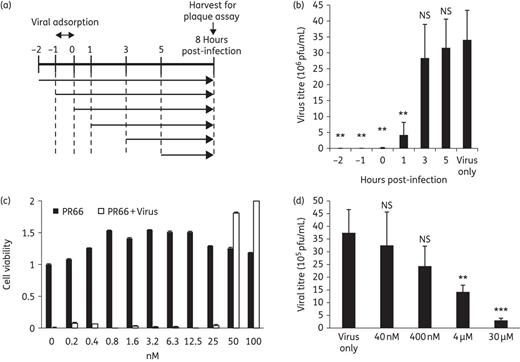
Testing how PR66 might target the early steps of viral replication. (a and b) Time-of-addition assays. (a) Schematic diagram of PR66 treatment in a life cycle of viral replication. RD cells were treated with 20 nM PR66 at the indicated timepoints post-infection and the cells and culture supernatants were collected at 8 h post-infection for titre determination by plaque-forming assays. Viral adsorption with EV-A71 with a moi of 10 was carried out at −1 to 0 h post-infection on ice in the presence or absence of PR66. Unbound virus was then removed and replaced with E2 containing PR66 if needed for that part of the experiment. (b) Bar chart of titre reduction after PR66 treatment. Data are presented as the mean ± SD from three independent experiments. NS, not significant. **P < 0.01 compared with the virus only (DMSO-treated) group. (c) Inhibition of viral attachment to HFF cells. Attachment assay was carried out by incubating increasing amounts of PR66 with virus during the viral pre-adsorption phase. Protection from virus-induced cell death conferred by PR66 was scored according to cell viability, which was normalized to the DMSO-treated control on the far left, set as 1. (d) Inactivation of EV-A71 by PR66 in a centrifugal filtration assay. Increasing concentrations of PR66 were added to EV-A71 (5 × 106 pfu/mL) in a total volume of 1 mL and incubated for 1 h at 4°C. Unbound compounds were removed by centrifugal filtration and the remaining viral titres in the retentate were determined by plaque-forming assays. Data are presented as the mean ± SD from three independent experiments. **P < 0.01 and ***P < 0.001 compared with the virus only group.
Mutation of V179F in capsid protein VP1 conferred resistance to the inhibitory effect of PR66
We employed resistant virus selection using increasing concentrations of PR66 and reciprocally examined resistance values with recombinant viruses carrying mutation sites. In the resistance validation result shown in Table 2, all three control viruses remained susceptible to PR66 (IC50 from 0.020 to 0.044 μM), whereas the 66RVs were resistant to PR66 with an IC50 >5 μM. The sequencing results revealed that two mutations (VP1_V179F and VP1_L247V) were consistently present in three 66RVs (Table 3). We generated recombinant viruses from infectious clone plasmids carrying these mutations. The anti-CPE assays showed that V179F and the double mutant were resistant to PR66, but the WT virus and L247V were still susceptible to PR66, with IC50 values of 0.025 and 0.002 μM, respectively (Table 4). The one-step growth curve showed that the V179 variant grew slightly faster than the WT virus, but there were no discernible differences among the WT and double mutant viruses (Figure 3a). Both V179F and the double mutant became resistant to the inhibition of PR66 in the centrifugal filtration assay (Figure 3b), although the V179F variant became less stable during the experiment, showing a reduced titre after centrifugation (Figure 3c). A modified PaSTRy assay was used to monitor viral uncoating, during which the closed viral protein shell undergoes reconfiguration to release viral RNA or become permeable to fluorescent dyes.5,16 PR66 appeared to increase virion stability, because the temperature needed to increase viral RNA accessibility to SYTO9 was increased from ∼57°C to ∼61°C when incubated with 500 nM PR66, but not with DMSO alone or PR79 (Figure 1), an analogue with no anti-EV-A71 activity (Figure 3d). As expected, PR66 did not increase the stability of the V179F mutant virus (Figure 3e). The V179 mutant virus was more liable to releasing viral RNA, with a lower inflection temperature of ∼45°C, compared with that of the WT virus at ∼57°C (Figure 3d and e). Collectively, these data indicate that PR66 targets the VP1 region of EV-A71 to inhibit viral uncoating.
IC50 values of resistant viruses
| Virus | IC50 (μM) |
|---|---|
| EV71/2231 | 0.019 ± 0.001 |
| Control virus R1 | 0.044 ± 0.003 |
| Control virus R2 | 0.020 ± 0.002 |
| Control virus R3 | 0.023 ± 0.001 |
| PR66 R1 | >5 |
| PR66 R2 | >5 |
| PR66 R3 | >5 |
| Virus | IC50 (μM) |
|---|---|
| EV71/2231 | 0.019 ± 0.001 |
| Control virus R1 | 0.044 ± 0.003 |
| Control virus R2 | 0.020 ± 0.002 |
| Control virus R3 | 0.023 ± 0.001 |
| PR66 R1 | >5 |
| PR66 R2 | >5 |
| PR66 R3 | >5 |
IC50 values of resistant viruses
| Virus | IC50 (μM) |
|---|---|
| EV71/2231 | 0.019 ± 0.001 |
| Control virus R1 | 0.044 ± 0.003 |
| Control virus R2 | 0.020 ± 0.002 |
| Control virus R3 | 0.023 ± 0.001 |
| PR66 R1 | >5 |
| PR66 R2 | >5 |
| PR66 R3 | >5 |
| Virus | IC50 (μM) |
|---|---|
| EV71/2231 | 0.019 ± 0.001 |
| Control virus R1 | 0.044 ± 0.003 |
| Control virus R2 | 0.020 ± 0.002 |
| Control virus R3 | 0.023 ± 0.001 |
| PR66 R1 | >5 |
| PR66 R2 | >5 |
| PR66 R3 | >5 |
Sequence analysis of resistant viruses
| P1 region | P2 region | P3 region | |
|---|---|---|---|
| Control virus R1 | VP2_K69R | 2A_N66N | 3C_G44R |
| VP1_T101T | 3C_K55R | ||
| 3D_V52I | |||
| Control virus R2 | VP2_D225G | ||
| VP1_I262M | |||
| Control virus R3 | VP2_K149M | 2A_N66N | 3A_R6R |
| 2A_S87T | 3A_E14K | ||
| 2A_Y89Y | 3C_L74Q | ||
| 2A_M99K | 3C_A97V | ||
| PR66 R1 | VP4_N17S | 2A_N66N | 3D_F191L |
| VP1_A23A | 2A_V102A | ||
| VP1_V179F | 2C_D162G | ||
| VP1_L247V | |||
| PR66 R2 | VP1_V179F | 2A_G61G | |
| VP1_L247V | 2A_N66N | ||
| 2C_I21T | |||
| 2C_H163Y | |||
| PR66 R3 | VP1_V179F | 2A_N66N | 3C_V60V |
| VP1_L247V | 2C_S61S | 3C_F113S | |
| 3D_P441Q |
| P1 region | P2 region | P3 region | |
|---|---|---|---|
| Control virus R1 | VP2_K69R | 2A_N66N | 3C_G44R |
| VP1_T101T | 3C_K55R | ||
| 3D_V52I | |||
| Control virus R2 | VP2_D225G | ||
| VP1_I262M | |||
| Control virus R3 | VP2_K149M | 2A_N66N | 3A_R6R |
| 2A_S87T | 3A_E14K | ||
| 2A_Y89Y | 3C_L74Q | ||
| 2A_M99K | 3C_A97V | ||
| PR66 R1 | VP4_N17S | 2A_N66N | 3D_F191L |
| VP1_A23A | 2A_V102A | ||
| VP1_V179F | 2C_D162G | ||
| VP1_L247V | |||
| PR66 R2 | VP1_V179F | 2A_G61G | |
| VP1_L247V | 2A_N66N | ||
| 2C_I21T | |||
| 2C_H163Y | |||
| PR66 R3 | VP1_V179F | 2A_N66N | 3C_V60V |
| VP1_L247V | 2C_S61S | 3C_F113S | |
| 3D_P441Q |
Sequences common to PR66 viruses are shown in bold.
Sequence analysis of resistant viruses
| P1 region | P2 region | P3 region | |
|---|---|---|---|
| Control virus R1 | VP2_K69R | 2A_N66N | 3C_G44R |
| VP1_T101T | 3C_K55R | ||
| 3D_V52I | |||
| Control virus R2 | VP2_D225G | ||
| VP1_I262M | |||
| Control virus R3 | VP2_K149M | 2A_N66N | 3A_R6R |
| 2A_S87T | 3A_E14K | ||
| 2A_Y89Y | 3C_L74Q | ||
| 2A_M99K | 3C_A97V | ||
| PR66 R1 | VP4_N17S | 2A_N66N | 3D_F191L |
| VP1_A23A | 2A_V102A | ||
| VP1_V179F | 2C_D162G | ||
| VP1_L247V | |||
| PR66 R2 | VP1_V179F | 2A_G61G | |
| VP1_L247V | 2A_N66N | ||
| 2C_I21T | |||
| 2C_H163Y | |||
| PR66 R3 | VP1_V179F | 2A_N66N | 3C_V60V |
| VP1_L247V | 2C_S61S | 3C_F113S | |
| 3D_P441Q |
| P1 region | P2 region | P3 region | |
|---|---|---|---|
| Control virus R1 | VP2_K69R | 2A_N66N | 3C_G44R |
| VP1_T101T | 3C_K55R | ||
| 3D_V52I | |||
| Control virus R2 | VP2_D225G | ||
| VP1_I262M | |||
| Control virus R3 | VP2_K149M | 2A_N66N | 3A_R6R |
| 2A_S87T | 3A_E14K | ||
| 2A_Y89Y | 3C_L74Q | ||
| 2A_M99K | 3C_A97V | ||
| PR66 R1 | VP4_N17S | 2A_N66N | 3D_F191L |
| VP1_A23A | 2A_V102A | ||
| VP1_V179F | 2C_D162G | ||
| VP1_L247V | |||
| PR66 R2 | VP1_V179F | 2A_G61G | |
| VP1_L247V | 2A_N66N | ||
| 2C_I21T | |||
| 2C_H163Y | |||
| PR66 R3 | VP1_V179F | 2A_N66N | 3C_V60V |
| VP1_L247V | 2C_S61S | 3C_F113S | |
| 3D_P441Q |
Sequences common to PR66 viruses are shown in bold.
IC50 values of cDNA-derived recombinant viruses
| Recombinant virus | IC50 (μM) |
|---|---|
| WT | 0.025 ± 0.000 |
| V179F mutant | >10 |
| L247V mutant | 0.002 ± 0.000 |
| Double mutant | >10 |
| Recombinant virus | IC50 (μM) |
|---|---|
| WT | 0.025 ± 0.000 |
| V179F mutant | >10 |
| L247V mutant | 0.002 ± 0.000 |
| Double mutant | >10 |
IC50 values of cDNA-derived recombinant viruses
| Recombinant virus | IC50 (μM) |
|---|---|
| WT | 0.025 ± 0.000 |
| V179F mutant | >10 |
| L247V mutant | 0.002 ± 0.000 |
| Double mutant | >10 |
| Recombinant virus | IC50 (μM) |
|---|---|
| WT | 0.025 ± 0.000 |
| V179F mutant | >10 |
| L247V mutant | 0.002 ± 0.000 |
| Double mutant | >10 |
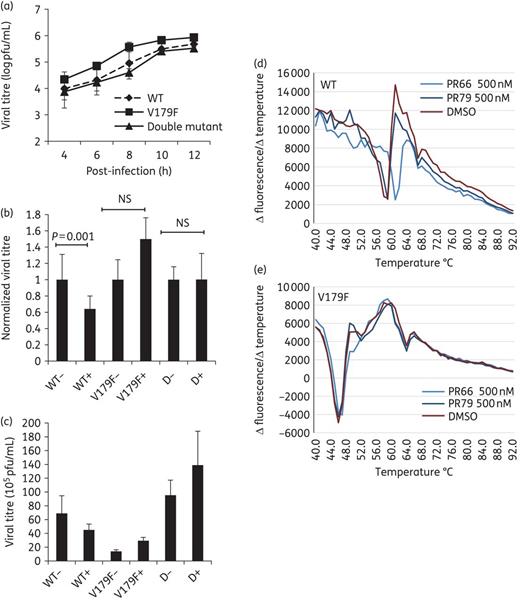
Growth kinetics of WT and mutant recombinant viruses and characterization of V179F mutant viruses. (a) RD cells were inoculated with WT, V179F and double mutant viruses (moi of 5) for 1 h. Samples were harvested at the indicated times post-infection and the viral titres were determined by plaque-forming assays. (b and c) Interaction of recombinant viruses with PR66. Recombinant viruses were incubated with 30 μM PR66 or vehicle (DMSO) control for 1 h at 4°C. Unbound PR66 was then removed using Microsep centrifugal filters and the resultant viruses in the sample reservoir were harvested for plaque-forming assays: +, with PR66; −, without PR66; D, double mutant. (b) Titres of PR66-treated groups were normalized to those of the vehicle-treated controls, set arbitrarily to 1. Data are presented as the mean ± SD (n = 3). NS, not significant. (c) Original data of (b) are shown without normalization. (d and e) PaSTRy assays using 5 μg aliquots of purified WT (d) or V179 viruses (e) were incubated with SYTO9 dye to detect the accessibility of viral RNA; 500 nM PR66 or PR79 was used in these assays. This is a representative result from three independent and reproducible experiments. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
PR66 conferred protection against EV-A71 infection in a mouse model
Because of the potent activity of PR66 in the cell-based results, we tested the proof of concept by investigating whether PR66 would also inhibit mouse-adapted EV-A71 (2231/MP4) in a mouse model. First, we examined toxicity by monitoring body weight changes in suckling mice using four different doses of PR66 in combination with two administration routes for 7 days (Supplementary Data). The mice grew normally, suggesting that PR66 had no toxicity at the specified doses up to 50 mg/kg per day given by oral or intraperitoneal routes. We did not observe any abnormal physical activity or ruffled pelage in PR66-treated mice (data not shown). We then challenged the mice with EV-A71/4643/MP4 and treated them with PR66 (Figure 4a). Two doses significantly protected mice from the lethal challenge with EV-A71/4643/MP4 in terms of both neurological signs (Figure 4b) and the survival rate, with oral or intraperitoneal administration (Figure 4c and d). Without PR66, the mice displayed EV-A71-induced neurological symptoms from day 3 and started to die from day 4 after intraperitoneal administration or from day 5 after oral administration. Intraperitoneal administration of PR66 produced better activity in alleviating neurological symptoms than oral administration (Figure 4b). A pharmacokinetic study also indicated that an intravenous injection of PR66 had twice the area under the curve (AUC) of the plasma concentration of PR66 over time compared with oral administration (Supplementary Data). In addition, both dosages of PR66 exhibited complete protection in terms of survival with either route of administration (Figure 4c and d).

Protective effect of PR66 against EV-A71/4643/MP4 infection in suckling mice. Six-day-old mice (day −1) were first administered one dose of 12.5 or 50 mg/kg per day of PR66 orally or intraperitoneally, respectively, followed by infection with 1 × 106 pfu of EV-A71/4643/MP4 in 100 μL at day 0. From day 0, the mice were treated with PR66 twice a day. Vehicle controls used 100 μL of 5% DMSO. (a) Illustration of treatment. Mean neurological disease scores (b) and mortality (c and d) were recorded daily. Mean score from each group was calculated and the numbers of mice in each group are shown in brackets. ***P < 0.0001.
To determine the half-minimal effective dose for PR66 in mice, we examined its efficacy with increasing dosages (0.1, 0.5, 2.5 or 10 mg/kg per day; Figure 5). The mean neurological disease score (Figure 5b) and the survival rate (Figure 5c) showed dose-dependent responses, where the minimum dose we tested (0.1 mg/kg per day) protected 50% of the mice effectively. In parallel, we included another post-exposure group with delayed drug administration (after 2 days of infection in the ‘cure 10’ group in Figure 5d). We also showed that PR66 not only increased the survival rate from 10% to 70% (Figure 5d), but also reduced the neurological symptoms (Figure 5b), even when administered after 2 days of infection. This suggests that PR66 has therapeutic and prophylactic efficacy. As shown in Figure 6(a and b), the copy numbers of EV-A71 were significantly ameliorated in the PR66-treated group in brain and muscle tissues.

Evaluation of the dose- and time-dependent inhibitory effect of PR66 given by the intraperitoneal route. (a) Increasing concentrations from 0.1 to 10 mg/kg per day of PR66 were administered from day −1 by the intraperitoneal route, whereas the mice in the cure 10 group were given 10 mg/kg per day of PR66 from day 2. Neurological disease scores (b) and mortality rates (c and d) were recorded daily. Mean score from each group was calculated and the numbers of mice in each group are shown in brackets. *P < 0.05 and ***P < 0.0001.

PR66 inhibited viral replication in mice. Mice were challenged with EV-A71/4643/MP4 and PR66 (2.5 mg/kg per day) or DMSO (0.5%) given by the intraperitoneal route from day −1 until they were euthanized with Zoletil. Brains on day 5 (a) or muscle tissues on day 1 (b) were collected for plaque-forming assays.
Discussion
In this study, PR66 inhibited all three genotypes (A–C) of EV-A71, demonstrating its broad applicability in the treatment of EV-A71 infection (Table 1). Although effective capsid binders have been developed to combat EV-A71, based on rational design and the crystal structures of viral particles,5,11 no animal models have been tested for their efficacy, optimal route of administration or toxicity. In this study, we demonstrated that PR66 did not display noticeable toxicity based on body weight changes or regular physical activity in mice at doses of up to 50 mg/kg per day (Supplementary Data). PR66 can be administered by both intraperitoneal and oral routes, indicating that it is tolerated and absorbed efficiently in the digestive tract, in line with the pharmacokinetic study (Supplementary Data). More importantly, administration of PR66 provided nearly complete protection against EV-A71-triggered neurological symptoms and mortality because of reduced viral replication, based on reduced viral titres in tissues (Figures 4–6). Treatment with PR66 from 2 days post-infection also showed significant protection, suggesting that PR66 can be developed as an anti-EV-A71 agent for prophylactic and curative purposes (Figure 5).
As shown in Figure 1, a novel phenylimidazolidinone derivative PR53 was identified as the initial lead for EV71 inhibitor in our screening programme. Compared with PR53, introduction of an additional phenyl group at the 4-position of the phenoxyl ring (PR66) resulted in a significant improvement in inhibitory activity against EV71. This is probably because the stronger hydrophobic interaction could be provided by a phenyl ring at this position in the binding site. Interestingly, introduction of a phenyl ring to the thiourea group (PR79) led to a complete loss of activity. This significant result demonstrated that the thiourea group seems to play a very important role in affecting inhibitory activity to this class of compounds.
From the sequencing results of the resistant viruses and follow-up neutralization assays, we showed that the V179F mutation was responsible for resistance against PR66; but why did these resistant viruses also carry the L247V mutation? We cannot exclude the possibility of spontaneous mutation arising during the screening process, although this mutation did not appear in the control group (Table 3). Alternatively, we hypothesize that this V179F mutation might have caused a defect and then L247V arose in a co-evolutionary manner to compensate for this mutation. By sequence analysis of about 9000 strains consisting of full-length (538 genomes) and partial sequences, we identified that two strains (AFQ94276 and ACO89585) carried the V179F mutation and eight strains carried the L247V mutation (data not shown). However, these strains did not carry both mutations, indicating that V179F and L247V might not co-evolve in nature. We examined whether the introduction of the V179F mutation would produce a slower growth rate and whether the addition of L247V might give a growth advantage. However, as shown in Figure 3(a), there were no significant growth rate differences among recombinant viruses of the WT strain, those carrying V179F and the double mutants. Our centrifugal filtration assay suggested that PR66 might inhibit EV-A71 by directly binding to and inactivating viral particles (Figure 2d). Accordingly, we investigated whether the V179F and double mutant were also resistant to PR66 in this assay. The relative titres of the V179F mutant and double mutant strains hardly changed in the presence of PR66, while that of the WT virus decreased significantly (P = 0.001; Figure 3b). Carefully examining the results, we found that the V179F variant, but not the double mutant, had lower titres (Figure 3c). One possible explanation was that the mutation at V179F in the VP1 virus diminished viral particle stability and that the L247V mutation might help to compensate for this. The PaSTRy data reinforced this finding because the V179F mutant virus tended to release more RNA than the WT virus at lower inflection temperatures (∼45°C versus ∼57°C) (Figure 3d and e).
The VP1 virus harbours a burrow from the surface called the hydrophobic pocket underneath the canyon, according to high-resolution crystallography studies.2–4 The putative native pocket factor was assumed to be sphingosine and compounds that replace this pocket factor to stabilize its structure are called capsid-binding inhibitors. Residue 179 is located in the interior of virions and is one of the residues comprising the hydrophobic pocket. This is similar to the V192M mutant virus, which is resistant to another capsid binder18 (see model in Figure 7). Therefore, we hypothesized that the valine mutation at amino acid 179 of VP1 when replaced with a bulky phenylalanine residue might render the pocket too small to hold PR66 and that this resulted in resistance. The L247 amino acid is distal to the hydrophobic pocket and does not interact with PR66 when subjected to a docking analysis (data not shown).

Models of the binding of PR66 with V179 and V192 in the hydrophobic pocket in VP1. (a) Molecular-docking model of PR66 in the hydrophobic pocket of VP1 was prepared using Discovery Studio (ver. 4.1; Accelrys, San Diego, CA, USA). (b) Cartoon of the docking result. Arrow indicates the interaction of PR66 with the side chains of valines at the 179 and 192 positions of VP1. V179F and V192M are mutations that orient toward the hydrophobic pocket and diminish it in size, which reduces the contact and interaction with PR66. L247 is distal to the hydrophobic pocket, but—based on the dimensions—there is no evidence suggesting that L247V is involved in the interaction between PR66 and the pocket. However, it possibly plays a role in stabilizing the overall structure. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
In conclusion, we provide the first proof of concept of an imidazolidinone derivative showing in vivo efficacy and this involved its binding to and restricting viral uncoating.
Funding
This work was supported by grants from Chang Gung Memorial Hospital (CMRPD 1E0041, 1D0322 and 1B0423) and the Ministry of Science and Technology of Taiwan (MOST-103-2325-B-182-009 and 104-2321-B-182-002).
Transparency declarations
None to declare.
Acknowledgements
We thank Dr Guang-Wu Chen for sequencing and statistical analyses. We also thank Drs Mei-Shang Ho and Jen-Ren Wang for the plasmids carrying full-length EV-A71/2231 and EV-A71/4643/MP4.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}