





Abstract
Inhibitors of uridine diphosphate-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC, which catalyses the first, irreversible step in lipid A biosynthesis) are a promising new class of antibiotics against Gram-negative bacteria. The objectives of the present study were to: (i) compare the antibiotic activities of three LpxC inhibitors (LPC-058, LPC-011 and LPC-087) and the reference inhibitor CHIR-090 against Gram-negative bacilli (including MDR and XDR isolates); and (ii) investigate the effect of combining these inhibitors with conventional antibiotics.
MICs were determined for 369 clinical isolates (234 Enterobacteriaceae and 135 non-fermentative Gram-negative bacilli). Time–kill assays with LPC-058 were performed on four MDR/XDR strains, including Escherichia coli producing CTX-M-15 ESBL and Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii producing KPC-2, VIM-1 and OXA-23 carbapenemases, respectively.
LPC-058 was the most potent antibiotic and displayed the broadest spectrum of antimicrobial activity, with MIC90 values for Enterobacteriaceae, P. aeruginosa, Burkholderia cepacia and A. baumannii of 0.12, 0.5, 1 and 1 mg/L, respectively. LPC-058 was bactericidal at 1× or 2× MIC against CTX-M-15, KPC-2 and VIM-1 carbapenemase-producing strains and bacteriostatic at ≤4× MIC against OXA-23 carbapenemase-producing A. baumannii. Combinations of LPC-058 with β-lactams, amikacin and ciprofloxacin were synergistic against these strains, albeit in a species-dependent manner. LPC-058's high efficacy was attributed to the presence of the difluoromethyl-allo-threonyl head group and a linear biphenyl-diacetylene tail group.
These in vitro data highlight the therapeutic potential of the new LpxC inhibitor LPC-058 against MDR/XDR strains and set the stage for subsequent in vivo studies.
Introduction
The emergence of MDR and XDR Gram-negative bacilli is weakening the efficacy of antimicrobial agents currently used to treat infections caused by these microorganisms.1–3 This alarming situation emphasizes the need to develop novel antibiotics directed at previously unexploited targets in order to overcome resistance to existing antibiotic classes. The development of novel antibiotics targeting uridine diphosphate-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) appears to be a promising strategy, as this essential enzyme catalyses the first, irreversible step in the biosynthesis of lipid A—the membrane anchor of lipopolysaccharide and the predominant lipid species in the outer leaflet of the Gram-negative outer membrane.4
In the late 1990s, phenyloxazoline-based LpxC inhibitors were found to be active against Escherichia coli both in vitro and in vivo, thus validating LpxC as a viable antibiotic target.5 The subsequent discovery of the compound CHIR-090 (characterized by a threonyl-hydroxamate head group coupled to a morpholine-conjugated biphenyl-acetylene tail group) led to the development of the first, active, LpxC-targeting antibiotic against Pseudomonas aeruginosa (Figure 1).6,7 Replacing the biphenyl-acetylene tail of CHIR-090 with a linear biphenyl-diacetylene group has yielded the compounds LPC-009 and LPC-011, which have enhanced antibacterial activity (Figure 1).8,9 Recently, another promising class of LpxC inhibitors (pyridine-methylsulfone-hydroxamate-based compounds, e.g. PF-5081090) was also found to show potent activity against Enterobacteriaceae and P. aeruginosa in vitro and in murine models of acute septicaemia and pulmonary infection.10 However, none of these compounds displayed activity against Acinetobacter baumannii, a pathogen often associated with nosocomial infections and outbreaks. Recently, the use of NMR spectroscopy to probe the dynamic interactions between LpxC and its bound inhibitors in solution led to the development of the difluoromethyl-allo-threonyl-hydroxamate-based compound LPC-058, which contains a para-amino substituted biphenyl-diacetylene tail group (Figure 1). LPC-058 was demonstrated to be 5- to 100-fold more active than CHIR-090 against representative strains of Enterobacteriaceae, P. aeruginosa, Vibrio cholerae and Chlamydia trachomatis.11 Most importantly, LPC-058 was the first LpxC inhibitor to display potent antibiotic activity against the A. baumannii ATCC 17978 strain; this finding indicated the feasibility of developing LpxC-targeting antibiotics against this notorious, opportunistic pathogen. Despite this exciting progress, the antibiotic activity of LPC-058 has only been determined against reference strains that lack well-characterized resistance mechanisms and thus are not representative of MDR and XDR strains. In the present study, we evaluated the potency of LPC-058 against a broad panel of Gram-negative clinical isolates and compared it with those of the reference compound CHIR-090 and two other biphenyl-diacetylene-based LpxC inhibitors that differed from LPC-058 in terms of the head group (LPC-011) or the tail group (LPC-087, which has an additional morpholine group). We hypothesized that studying clinical strains would provide information on the effects of antibiotic resistance mechanisms [such as ESBL, carbapenemase or overexpression of efflux resistance–nodulation–division (RND) pumps] on LpxC inhibitor potency. Lastly, we investigated the putative synergy between LpxC inhibitors and the conventional antibiotics commonly used to treat serious infections caused by MDR strains.12,13
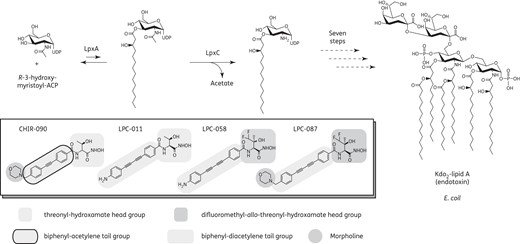
Antibiotics targeting LpxC in the lipid A biosynthetic pathway in Gram-negative bacteria. The LpxC inhibitor head groups, tail groups and morpholine moiety (if present) are highlighted in grey.
Materials and methods
Bacterial strains
Reference strains of E. coli (ATCC 25922), P. aeruginosa (ATCC 27853) and A. baumannii (ATCC 17978) were studied, along with 369 Gram-negative clinical isolates from Lille University Hospital (Lille, France). A total of 34 MDR (9%) and 44 XDR (12%) strains were identified, according to the interim standard definitions for acquired resistance.14 Two MDR strains (CTX-M-15 ESBL-producing E. coli and KPC-2 carbapenemase-producing Klebsiella pneumoniae) and two XDR strains (VIM-1 carbapenemase-producing P. aeruginosa and OXA-23 carbapenemase-producing A. baumannii) were used in time–kill assays. Lastly, Enterobacter aerogenes, P. aeruginosa and A. baumannii strains overexpressing the AcrAB-TolC, MexAB-OprM/MexCD-OprJ and AdeABC efflux pumps, respectively, were also examined, along with the corresponding parental strains.15–18
Antimicrobial compounds
Cefotaxime, ceftazidime, imipenem, ciprofloxacin and amikacin were purchased from Sigma–Aldrich (Lyon, France). The LpxC inhibitor CHIR-090 and the biphenyl-diacetylene-based LpxC inhibitors LPC-058 and LPC-011 (Figure 1) were prepared at the Duke University Small Molecule Synthesis Facility (Durham 27710, NC, USA), according to published procedures.7–9 The synthesis of LPC-087 is described in the Supplementary methods (available as Supplementary data at JAC Online). Stock solutions of conventional antibiotics and LpxC inhibitors were stored at −80°C until use.
MIC assays
Susceptibility of the bacterial strains to LpxC inhibitors at final concentrations ranging from 0.015 to 64 mg/L was determined using the CLSI standard agar dilution and broth microdilution methods in CAMHB.19 An inoculum of ∼104 cfu/spot or 5 × 105 cfu/mL was used for the agar dilution and microdilution methods, respectively. The inoculated agar and 96-well plates were subsequently incubated for 24 h at 37°C (except for Yersinia species, which were incubated for 48 h at 28°C). Each assay was performed at least twice on separate days and three quality control strains (E. coli ATCC 25922, P. aeruginosa ATCC 27853 and A. baumannii ATCC 17978) were included on each day of testing (depending on the species investigated). The MIC was defined as the lowest concentration of drug that prevented visible growth after incubation.
Time–kill assays
The bactericidal activity of LpxC inhibitors was investigated with the following strains: CTX-M-15 ESBL-producing E. coli, KPC-2 carbapenemase-producing K. pneumoniae, VIM-1 carbapenemase-producing P. aeruginosa and OXA-23 carbapenemase-producing A. baumannii. Assays were performed using LPC-058 alone at concentrations of 1× to 8× MIC or mixed at 0.25× to 1× MIC with the following conventional antibiotics: cefotaxime, ceftazidime, imipenem, ciprofloxacin and amikacin. For time–kill assays, the MIC of each antibiotic was determined by using the CLSI reference microdilution method.19 The various combinations were added to an inoculum of 5 × 105 cfu/mL from a bacterial culture growing exponentially in CAMHB. After 0, 3, 6, 9 and 24 h of incubation at 37°C with shaking, aliquots were removed and subcultured in triplicate onto bromocresol purple agar plates. Viable cells were then counted. Bactericidal activity was defined as a 3 log10cfu/mL reduction, relative to the initial inoculum. Synergy and indifference were, respectively, defined as a ≥2 log10 and <2 log10 decrease in the cfu count when the combination was compared with the most active single drug after 24 h incubation. The limit of detection (LOD) was ≤1.3 log10 cfu/mL. Each assay was repeated at least twice.
Results
Inhibitory activity of the LpxC inhibitors against clinical isolates of Enterobacteriaceae and non-fermentative Gram-negative bacilli
The antimicrobial activities of LPC-058, LPC-011 and LPC-087 were tested against 369 clinical isolates (234 Enterobacteriaceae and 135 non-fermentative Gram-negative bacilli) and compared with that of the reference compound CHIR-090 (Table 1 and Supplementary Data). All the biphenyl-diacetylene-based compounds showed a high level of activity against common Enterobacteriaceae, with MIC50 values of ≤0.25 mg/L and MIC90 values of ≤0.5 mg/L. LPC-058 was the most active compound, with MIC50 (0.06 mg/L) and MIC90 (0.12 mg/L) values that were 6- to 8-fold lower than those of the reference compound CHIR-090 (with an MIC50 of 0.5 mg/L and MIC90 of 2 mg/L). The second most active compound was LPC-011, with MIC50 (0.12 mg/L) and MIC90 (0.25 mg/L) values ∼2-fold higher than those of LPC-058 for the various Enterobacteriaceae strains (Supplementary Data); however, its MIC50 and MIC90 values were 2-fold lower than those of LPC-087 (MIC50 = 0.25 mg/L and MIC90 = 0.5 mg/L). A similar trend in antibacterial activities was observed for clinical isolates of P. aeruginosa (Table 1). LPC-058 and LPC-011 were more active (MIC50 ≤0.5 mg/L and MIC90 ≤1 mg/L) than LPC-087, which had similar MIC50 and MIC90 values as the reference compound CHIR-090 (2 and 4 mg/L, respectively). Overall, the activities of the compounds against Enterobacteriaceae and P. aeruginosa could be ranked (in decreasing order) as follows: LPC-058 > LPC-011 > LPC-087 > CHIR-090.
Activities of LPC-058, LPC-011, LPC-087 and the reference compound CHIR-90 against 234 clinical isolates of Enterobacteriaceae and 135 clinical isolates of non-fermentative Gram-negative bacilli
| Organism group (number of isolates tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae (234) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 2 | |
| range | ≤0.015–0.5 | ≤0.015–1 | ≤0.015–2 | 0.03–8 | |
| P. aeruginosa (51) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.12–1 | 0.25–8 | 0.5–8 | |
| A. baumannii (26) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | — | 0.5 to ≥64 | — | |
| B. cepacia (22) | MIC50 | 0.5 | 4 | ≥64 | ≥64 |
| MIC90 | 1 | 8 | ≥64 | ≥64 | |
| range | 0.12–2 | 1–8 | — | — | |
| S. maltophilia (20) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — | |
| A. xylosoxidans (16) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — |
| Organism group (number of isolates tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae (234) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 2 | |
| range | ≤0.015–0.5 | ≤0.015–1 | ≤0.015–2 | 0.03–8 | |
| P. aeruginosa (51) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.12–1 | 0.25–8 | 0.5–8 | |
| A. baumannii (26) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | — | 0.5 to ≥64 | — | |
| B. cepacia (22) | MIC50 | 0.5 | 4 | ≥64 | ≥64 |
| MIC90 | 1 | 8 | ≥64 | ≥64 | |
| range | 0.12–2 | 1–8 | — | — | |
| S. maltophilia (20) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — | |
| A. xylosoxidans (16) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — |
Activities of LPC-058, LPC-011, LPC-087 and the reference compound CHIR-90 against 234 clinical isolates of Enterobacteriaceae and 135 clinical isolates of non-fermentative Gram-negative bacilli
| Organism group (number of isolates tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae (234) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 2 | |
| range | ≤0.015–0.5 | ≤0.015–1 | ≤0.015–2 | 0.03–8 | |
| P. aeruginosa (51) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.12–1 | 0.25–8 | 0.5–8 | |
| A. baumannii (26) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | — | 0.5 to ≥64 | — | |
| B. cepacia (22) | MIC50 | 0.5 | 4 | ≥64 | ≥64 |
| MIC90 | 1 | 8 | ≥64 | ≥64 | |
| range | 0.12–2 | 1–8 | — | — | |
| S. maltophilia (20) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — | |
| A. xylosoxidans (16) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — |
| Organism group (number of isolates tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae (234) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 2 | |
| range | ≤0.015–0.5 | ≤0.015–1 | ≤0.015–2 | 0.03–8 | |
| P. aeruginosa (51) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.12–1 | 0.25–8 | 0.5–8 | |
| A. baumannii (26) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | — | 0.5 to ≥64 | — | |
| B. cepacia (22) | MIC50 | 0.5 | 4 | ≥64 | ≥64 |
| MIC90 | 1 | 8 | ≥64 | ≥64 | |
| range | 0.12–2 | 1–8 | — | — | |
| S. maltophilia (20) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — | |
| A. xylosoxidans (16) | MIC50 | ≥64 | ≥64 | ≥64 | ≥64 |
| MIC90 | ≥64 | ≥64 | ≥64 | ≥64 | |
| range | — | — | — | — |
Interestingly, the LpxC inhibitor activity patterns against A. baumannii and Burkholderia cepacia differed distinctly from those against Enterobacteriaceae and P. aeruginosa. For A. baumannii, only the difluoromethyl-substituted compounds (LPC-058 and LPC-087) displayed a significant level of antibiotic activity (Table 1). LPC-058 displayed a higher potency (MIC50 = 0.25 mg/L and MIC90 = 1 mg/L) against clinical isolates of A. baumannii than LPC-087 (MIC50 = 1 mg/L and MIC90 = 32 mg/L), a compound that has the same head group but a different tail (with an additional morpholine moiety attached to the biphenyl-diacetylene scaffold; Figure 1). For B. cepacia, only LPC-058 and LPC-011 were active; once again, LPC-058 was the most potent antibiotic (Table 1). Since LPC-058 and LPC-011 share the same linear para-amino-substituted biphenyl-diacetylene tail group and differ from LPC-087 and CHIR-090 in terms of the absence of the morpholine unit in the tail, their activity against B. cepacia indicates that the morpholine substitution may adversely affect the activity of LpxC inhibitors. None of the LpxC inhibitors evaluated here was effective against Stenotrophomonas maltophilia and Achromobacter xylosoxidans; this lack of activity was unexpected, given the presence of LpxC enzymes in these species.
Overall, the results of MIC assays with a large collection of Gram-negative clinical strains indicated that LPC-058 (with the difluoromethyl-allo-threonyl-hydroxamate head group and a linear biphenyl-diacetylene tail group) was the most active LpxC inhibitor and had the broadest spectrum of activity.
Inhibitory activity of LpxC inhibitors against defined efflux mutants
We next examined the effect of non-specific drug-resistant mechanisms (such as the overexpression of RND efflux pumps) on the antibiotic activity of the biphenyl-diacetylene derivatives LPC-058, LPC-011 and LPC-087. For this, MIC values were determined for selective WT strains of E. aerogenes, P. aeruginosa and A. baumannii and their isogenic mutants overexpressing efflux pumps (Table 2). The MICs of LPC-058 and LPC-011 (which have different head groups but the same tail group) were moderately higher (by a factor of four) for the AcrAB-TolC overproduction mutant of E. aerogenes than for the WT strain. The MICs of the two compounds were only slightly affected by overexpression of MexAB-OprM or MexCD-OprJ efflux pumps in P. aeruginosa and the AdeABC efflux pump in A. baumannii CIP7010-1. In contrast, the difference in MICs was greater for LPC-087 (which bears the same morpholine moiety in the tail as CHIR-090) than LPC-058 and LPC-011. The MICs of LPC-087 for the efflux pump mutant strains of E. aerogenes and P. aeruginosa were, respectively, 8- and 4-fold greater than those for the isogenic parental strain. It has already been reported that CHIR-090 is a substrate of the RND efflux pumps in P. aeruginosa.10,20 These observations suggest that the morpholine group in LPC-087 and CHIR-090 may be preferentially recognized (relative to the linear para-amino-substituted biphenyl-diacetylene tail group in LPC-058 and LPC-011) by the RND efflux pump family.
MICs of LPC-058, LPC-011, LPC-087 and CHIR-090 for E. aerogenes, P. aeruginosa and A. baumannii mutant strains overexpressing RND pump efflux systems and their respective parental strains
| Strain | Efflux pump status | MIC (mg/L) | |||
|---|---|---|---|---|---|
| LPC-058 | LPC-011 | LPC-087 | CHIR-090 | ||
| E. aerogenes | |||||
| ATCC 13048 | WT | 0.12 | 0.25 | 0.5 | 0.5 |
| CM64 | AcrAB-TolC overexpressed | 0.5 | 1 | 4 | 4 |
| P. aeruginosa | |||||
| PAO1 | WT | 0.12 | 0.25 | 2 | 2 |
| PT629 | MexAB-OprM overexpressed | 0.12 | 0.25 | 8 | 8 |
| ERYR | MexCD-OprJ overexpressed | 0.25 | 0.5 | 8 | 8 |
| A. baumannii | |||||
| CIP7010 | WT | 4 | ND | ND | ND |
| CIP7010-1 | AdeABC overexpressed | 8 | ND | ND | ND |
| Strain | Efflux pump status | MIC (mg/L) | |||
|---|---|---|---|---|---|
| LPC-058 | LPC-011 | LPC-087 | CHIR-090 | ||
| E. aerogenes | |||||
| ATCC 13048 | WT | 0.12 | 0.25 | 0.5 | 0.5 |
| CM64 | AcrAB-TolC overexpressed | 0.5 | 1 | 4 | 4 |
| P. aeruginosa | |||||
| PAO1 | WT | 0.12 | 0.25 | 2 | 2 |
| PT629 | MexAB-OprM overexpressed | 0.12 | 0.25 | 8 | 8 |
| ERYR | MexCD-OprJ overexpressed | 0.25 | 0.5 | 8 | 8 |
| A. baumannii | |||||
| CIP7010 | WT | 4 | ND | ND | ND |
| CIP7010-1 | AdeABC overexpressed | 8 | ND | ND | ND |
WT, isogenic, antibiotic-susceptible parent; ND, not determined.
MICs of LPC-058, LPC-011, LPC-087 and CHIR-090 for E. aerogenes, P. aeruginosa and A. baumannii mutant strains overexpressing RND pump efflux systems and their respective parental strains
| Strain | Efflux pump status | MIC (mg/L) | |||
|---|---|---|---|---|---|
| LPC-058 | LPC-011 | LPC-087 | CHIR-090 | ||
| E. aerogenes | |||||
| ATCC 13048 | WT | 0.12 | 0.25 | 0.5 | 0.5 |
| CM64 | AcrAB-TolC overexpressed | 0.5 | 1 | 4 | 4 |
| P. aeruginosa | |||||
| PAO1 | WT | 0.12 | 0.25 | 2 | 2 |
| PT629 | MexAB-OprM overexpressed | 0.12 | 0.25 | 8 | 8 |
| ERYR | MexCD-OprJ overexpressed | 0.25 | 0.5 | 8 | 8 |
| A. baumannii | |||||
| CIP7010 | WT | 4 | ND | ND | ND |
| CIP7010-1 | AdeABC overexpressed | 8 | ND | ND | ND |
| Strain | Efflux pump status | MIC (mg/L) | |||
|---|---|---|---|---|---|
| LPC-058 | LPC-011 | LPC-087 | CHIR-090 | ||
| E. aerogenes | |||||
| ATCC 13048 | WT | 0.12 | 0.25 | 0.5 | 0.5 |
| CM64 | AcrAB-TolC overexpressed | 0.5 | 1 | 4 | 4 |
| P. aeruginosa | |||||
| PAO1 | WT | 0.12 | 0.25 | 2 | 2 |
| PT629 | MexAB-OprM overexpressed | 0.12 | 0.25 | 8 | 8 |
| ERYR | MexCD-OprJ overexpressed | 0.25 | 0.5 | 8 | 8 |
| A. baumannii | |||||
| CIP7010 | WT | 4 | ND | ND | ND |
| CIP7010-1 | AdeABC overexpressed | 8 | ND | ND | ND |
WT, isogenic, antibiotic-susceptible parent; ND, not determined.
Inhibitory activity of individual LpxC inhibitors against MDR and/or XDR strains
To assess the broader impact of multidrug resistance (i.e. other than efflux pump overproduction), we compared the MICs of LPC-058, LPC-011, LPC-087 and CHIR-90 when tested against MDR/XDR or non-MDR/XDR clinical isolates of Enterobacteriaceae, P. aeruginosa and A. baumannii. All the LpxC inhibitors had very similar MIC values for MDR and XDR strains (Table 3). In general, the MIC50 and MIC90 values of the biphenyl-diacetylene-based compounds (LPC-058, LPC-011 and LPC-087) for non-MDR/XDR clinical isolates of Enterobacteriaceae and P. aeruginosa were similar to those for MDR/XDR clinical strains (with a 2-fold difference, typically).
Activities of LpxC inhibitors as a function of the resistance phenotype in Enterobacteriaceae, P. aeruginosa and A. baumannii
| Organism group (number tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae non-MDR/XDR (196) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 1 | |
| range | ≤0.015–0.25 | ≤0.015–1 | ≤0.015–1 | 0.03–2 | |
| Enterobacteriaceae MDR or XDR (38a) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.25 | 0.5 | 1 | 2 | |
| range | ≤0.015–0.25 | 0.06–1 | 0.25–1 | 0.25–8 | |
| P. aeruginosa non-MDR/XDR (31) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.25–2 | 0.25–4 | 0.5–8 | |
| P. aeruginosa MDR or XDR (20b) | MIC50 | 0.25 | 0.5 | 4 | 4 |
| MIC90 | 0.5 | 1 | 8 | 8 | |
| range | 0.12–0.5 | 0.25–1 | 1–8 | 2–8 | |
| A. baumannii non-MDR/XDR (6) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 0.5 | ≥64 | 2 | ≥64 | |
| range | 0.12–0.5 | ≥64 | 0.5–2 | ≥64 | |
| A. baumannii MDR or XDR (20c) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | ≥64 | 0.5 to ≥64 | ≥64 |
| Organism group (number tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae non-MDR/XDR (196) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 1 | |
| range | ≤0.015–0.25 | ≤0.015–1 | ≤0.015–1 | 0.03–2 | |
| Enterobacteriaceae MDR or XDR (38a) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.25 | 0.5 | 1 | 2 | |
| range | ≤0.015–0.25 | 0.06–1 | 0.25–1 | 0.25–8 | |
| P. aeruginosa non-MDR/XDR (31) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.25–2 | 0.25–4 | 0.5–8 | |
| P. aeruginosa MDR or XDR (20b) | MIC50 | 0.25 | 0.5 | 4 | 4 |
| MIC90 | 0.5 | 1 | 8 | 8 | |
| range | 0.12–0.5 | 0.25–1 | 1–8 | 2–8 | |
| A. baumannii non-MDR/XDR (6) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 0.5 | ≥64 | 2 | ≥64 | |
| range | 0.12–0.5 | ≥64 | 0.5–2 | ≥64 | |
| A. baumannii MDR or XDR (20c) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | ≥64 | 0.5 to ≥64 | ≥64 |
aIncludes 17 ESBL-producing and 5 carbapenemase-producing strains [OXA-48 (n = 3), NDM (n = 1) and KPC (n = 1)].
bIncludes three ESBL-producing and five carbapenemase-producing strains [VIM (n = 4) and IMP (n = 1)].
cIncludes 1 ESBL-producing and 10 carbapenemase-producing strains [OXA-23 (n = 9) and OXA-40 (n = 1)].
Activities of LpxC inhibitors as a function of the resistance phenotype in Enterobacteriaceae, P. aeruginosa and A. baumannii
| Organism group (number tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae non-MDR/XDR (196) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 1 | |
| range | ≤0.015–0.25 | ≤0.015–1 | ≤0.015–1 | 0.03–2 | |
| Enterobacteriaceae MDR or XDR (38a) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.25 | 0.5 | 1 | 2 | |
| range | ≤0.015–0.25 | 0.06–1 | 0.25–1 | 0.25–8 | |
| P. aeruginosa non-MDR/XDR (31) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.25–2 | 0.25–4 | 0.5–8 | |
| P. aeruginosa MDR or XDR (20b) | MIC50 | 0.25 | 0.5 | 4 | 4 |
| MIC90 | 0.5 | 1 | 8 | 8 | |
| range | 0.12–0.5 | 0.25–1 | 1–8 | 2–8 | |
| A. baumannii non-MDR/XDR (6) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 0.5 | ≥64 | 2 | ≥64 | |
| range | 0.12–0.5 | ≥64 | 0.5–2 | ≥64 | |
| A. baumannii MDR or XDR (20c) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | ≥64 | 0.5 to ≥64 | ≥64 |
| Organism group (number tested) | MIC (mg/L) | LPC-058 | LPC-011 | LPC-087 | CHIR-090 |
|---|---|---|---|---|---|
| Enterobacteriaceae non-MDR/XDR (196) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.12 | 0.25 | 0.5 | 1 | |
| range | ≤0.015–0.25 | ≤0.015–1 | ≤0.015–1 | 0.03–2 | |
| Enterobacteriaceae MDR or XDR (38a) | MIC50 | 0.06 | 0.12 | 0.25 | 0.5 |
| MIC90 | 0.25 | 0.5 | 1 | 2 | |
| range | ≤0.015–0.25 | 0.06–1 | 0.25–1 | 0.25–8 | |
| P. aeruginosa non-MDR/XDR (31) | MIC50 | 0.25 | 0.5 | 2 | 2 |
| MIC90 | 0.5 | 1 | 4 | 4 | |
| range | 0.06–0.5 | 0.25–2 | 0.25–4 | 0.5–8 | |
| P. aeruginosa MDR or XDR (20b) | MIC50 | 0.25 | 0.5 | 4 | 4 |
| MIC90 | 0.5 | 1 | 8 | 8 | |
| range | 0.12–0.5 | 0.25–1 | 1–8 | 2–8 | |
| A. baumannii non-MDR/XDR (6) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 0.5 | ≥64 | 2 | ≥64 | |
| range | 0.12–0.5 | ≥64 | 0.5–2 | ≥64 | |
| A. baumannii MDR or XDR (20c) | MIC50 | 0.25 | ≥64 | 1 | ≥64 |
| MIC90 | 1 | ≥64 | 32 | ≥64 | |
| range | 0.12–4 | ≥64 | 0.5 to ≥64 | ≥64 |
aIncludes 17 ESBL-producing and 5 carbapenemase-producing strains [OXA-48 (n = 3), NDM (n = 1) and KPC (n = 1)].
bIncludes three ESBL-producing and five carbapenemase-producing strains [VIM (n = 4) and IMP (n = 1)].
cIncludes 1 ESBL-producing and 10 carbapenemase-producing strains [OXA-23 (n = 9) and OXA-40 (n = 1)].
The two LpxC inhibitors with activity against A. baumannii (the difluoromethyl-substituted compounds LPC-058 and LPC-087) differed in their relative potencies against MDR/XDR isolates. The activity of the morpholine-containing LPC-087 against MDR/XDR A. baumannii isolates was much lower (the MIC90 increased from 2 mg/L to 32 mg/L), whereas LPC-058 activity was only slightly affected (the MIC90 increased from 0.5 mg/L to 1 mg/L). Overall, these observations support the concept whereby LPC-058 (with a difluoromethyl-allo-threonyl-hydroxamate head group and a linear biphenyl-diacetylene tail group) can effectively overcome multidrug resistance in all the MDR/XDR strains tested here.
Bactericidal effect of LPC-058 alone
Since LPC-058 (i) was the most potent LpxC inhibitor, (ii) displayed the broadest spectrum of antimicrobial activities, and (iii) was only slightly affected by the efflux-based antibiotic resistance mechanisms, we next sought to determine its bactericidal activity against two MDR strains (CTX-M-15 ESBL-producing E. coli and KPC-2 carbapenemase-producing K. pneumoniae) and two XDR strains (P. aeruginosa and A. baumannii producing VIM-1 and OXA-23 carbapenemases, respectively). All four MDR/XDR strains exhibited a high level of resistance to β-lactams, amikacin and ciprofloxacin (Table 4). At a concentration of 1× or 2× MIC, LPC-058 alone was strongly bactericidal (with a reduction of ≥3 log10 cfu/mL) against MDR/XDR E. coli, K. pneumoniae and P. aeruginosa (Figure 2). The bactericidal activity of LPC-058 was more prominent for ESBL-producing E. coli than for carbapenemase-producing K. pneumoniae and P. aeruginosa: a full bactericidal effect was rapidly observed (within 3 h) at 1× MIC (0.06 mg/L) for ESBL-producing E. coli, but occurred more slowly (within 6 h) at 2× MIC (0.24 and 1 mg/L, respectively) for KPC-2 carbapenemase-producing K. pneumoniae and VIM-1-producing P. aeruginosa. In contrast, LPC-058 was bacteriostatic at ≤4× MIC (MIC = 0.5 mg/L) against OXA-23 carbapenemase-producing A. baumannii and was slowly bactericidal at 8× MIC (Figure 2).
MICs (in CAMHB) for the four MDR/XDR strains used in the time–kill assays
| Strain | MIC (mg/L) | |||||
|---|---|---|---|---|---|---|
| LPC-058 | CTX | CAZ | IPM | AMK | CIP | |
| E. coli with CTX-M-15 | 0.06 | 256 | ND | 0.25 | 32 | 64 |
| K. pneumoniae with KPC-2 | 0.12 | 512 | ND | 16 | 32 | 16 |
| P. aeruginosa with VIM-1 | 0.5 | ND | 64 | 256 | 4 | 16 |
| A. baumannii with OXA-23 | 0.5 | ND | 128 | 32 | 1024 | 32 |
| Strain | MIC (mg/L) | |||||
|---|---|---|---|---|---|---|
| LPC-058 | CTX | CAZ | IPM | AMK | CIP | |
| E. coli with CTX-M-15 | 0.06 | 256 | ND | 0.25 | 32 | 64 |
| K. pneumoniae with KPC-2 | 0.12 | 512 | ND | 16 | 32 | 16 |
| P. aeruginosa with VIM-1 | 0.5 | ND | 64 | 256 | 4 | 16 |
| A. baumannii with OXA-23 | 0.5 | ND | 128 | 32 | 1024 | 32 |
CTX, cefotaxime; CAZ, ceftazidime; IPM, imipenem; AMK, amikacin; CIP, ciprofloxacin; ND, not determined.
MICs (in CAMHB) for the four MDR/XDR strains used in the time–kill assays
| Strain | MIC (mg/L) | |||||
|---|---|---|---|---|---|---|
| LPC-058 | CTX | CAZ | IPM | AMK | CIP | |
| E. coli with CTX-M-15 | 0.06 | 256 | ND | 0.25 | 32 | 64 |
| K. pneumoniae with KPC-2 | 0.12 | 512 | ND | 16 | 32 | 16 |
| P. aeruginosa with VIM-1 | 0.5 | ND | 64 | 256 | 4 | 16 |
| A. baumannii with OXA-23 | 0.5 | ND | 128 | 32 | 1024 | 32 |
| Strain | MIC (mg/L) | |||||
|---|---|---|---|---|---|---|
| LPC-058 | CTX | CAZ | IPM | AMK | CIP | |
| E. coli with CTX-M-15 | 0.06 | 256 | ND | 0.25 | 32 | 64 |
| K. pneumoniae with KPC-2 | 0.12 | 512 | ND | 16 | 32 | 16 |
| P. aeruginosa with VIM-1 | 0.5 | ND | 64 | 256 | 4 | 16 |
| A. baumannii with OXA-23 | 0.5 | ND | 128 | 32 | 1024 | 32 |
CTX, cefotaxime; CAZ, ceftazidime; IPM, imipenem; AMK, amikacin; CIP, ciprofloxacin; ND, not determined.
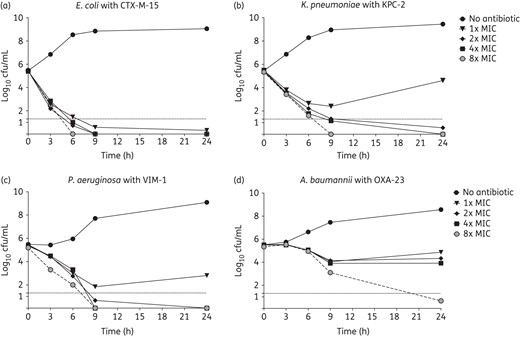
Twenty-four hour time–kill curves with LPC-058 alone at 1×, 2×, 4× and 8× MIC, as tested against CTX-M-15 ESBL-producing E. coli (a), KPC-2 carbapenemase-producing K. pneumoniae (b), VIM-1 carbapenemase-producing P. aeruginosa (c) and OXA-23 carbapenemase-producing A. baumannii (d). The broken horizontal line indicates the LOD (≤1.3 log10 cfu/mL). The bacterial count corresponds to the mean of at least two independent experiments in duplicate.
Synergistic effect of LPC-058 with conventional antibiotics
We next assessed the potential synergistic activity of LPC-058 with β-lactams, amikacin and ciprofloxacin against the above-mentioned MDR/XDR bacteria. We deliberately chose to use subinhibitory concentrations of the conventional antibiotics that could nevertheless be achieved clinically.21–25 All combinations of LPC-058 with a β-lactam were synergistic when tested against the four MDR/XDR strains (Table 5 and Supplementary Data). A higher concentration of LPC-058 (1× MIC) did not further enhance the bacterial killing of the β-lactam plus LPC-058 combinations, when compared with LPC-058 at subinhibitory concentrations (0.25× and 0.5× MIC). The synergistic killing effect of the cefotaxime plus LPC-058 combination was more rapid for ESBL-producing E. coli (at 6 or 9 h) than for KPC-2-producing K. pneumoniae (at 24 h, with an LPC-058 concentration ≥0.5× MIC). At all LPC-058 concentrations, the ceftazidime plus LPC-058 combination produced a greater killing effect with VIM-1-producing P. aeruginosa than LPC-058 alone. Impressively, the LPC-058 plus imipenem combination was effective against all carbapenemase-producing strains; this was especially true for the OXA-23-producing A. baumannii, which was killed within 3 h and did not display any regrowth (Table 5 and Supplementary Data).
Interactions between conventional antibiotics and LPC-058 at 0.25×, 0.5× and 1× MIC against E. coli producing CTX-M-15 ESBL and K. pneumoniae, P. aeruginosa and A. baumannii producing KPC-2, VIM-1 and OXA-23 carbapenemases, respectively
| Strain | Multiple of the MIC of conventional antibiotics | Combination effect, time in h | ||
|---|---|---|---|---|
| multiple of the MIC of LPC-058 | ||||
| 0.25× | 0.5× | 1× | ||
| E. coli CTX-M-15 | CTX 0.5× | synergy, 9 h | synergy, 6 h | indifference |
| AMK 0.25× | synergy, 9 h | synergy, 9 h | indifference | |
| CIP 0.5× | synergy, 6 h | synergy, 6 h | indifference | |
| K. pneumoniae KPC-2 | CTX 0.25× | indifference | synergy, 24 h | synergy, 24 h |
| IPM 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| CIP 0.5× | indifference | indifference | synergy, 24 h | |
| P. aeruginosa VIM-1 | CAZ 0.5× | synergy, 6 h | synergy, 9 h | synergy, 24 h |
| IPM 0.125× | synergy, 6 ha | synergy, 9 h | synergy, 24 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 24 h | |
| CIP 0.25× | indifference | indifference | synergy, 24 h | |
| A. baumannii OXA-23 | IPM 0.5× | ND | synergy, 3 h | synergy, 3 h |
| AMK 0.062× | ND | indifference | indifference | |
| CIP 0.5× | ND | synergy, 6 ha | synergy, 6 h | |
| Strain | Multiple of the MIC of conventional antibiotics | Combination effect, time in h | ||
|---|---|---|---|---|
| multiple of the MIC of LPC-058 | ||||
| 0.25× | 0.5× | 1× | ||
| E. coli CTX-M-15 | CTX 0.5× | synergy, 9 h | synergy, 6 h | indifference |
| AMK 0.25× | synergy, 9 h | synergy, 9 h | indifference | |
| CIP 0.5× | synergy, 6 h | synergy, 6 h | indifference | |
| K. pneumoniae KPC-2 | CTX 0.25× | indifference | synergy, 24 h | synergy, 24 h |
| IPM 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| CIP 0.5× | indifference | indifference | synergy, 24 h | |
| P. aeruginosa VIM-1 | CAZ 0.5× | synergy, 6 h | synergy, 9 h | synergy, 24 h |
| IPM 0.125× | synergy, 6 ha | synergy, 9 h | synergy, 24 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 24 h | |
| CIP 0.25× | indifference | indifference | synergy, 24 h | |
| A. baumannii OXA-23 | IPM 0.5× | ND | synergy, 3 h | synergy, 3 h |
| AMK 0.062× | ND | indifference | indifference | |
| CIP 0.5× | ND | synergy, 6 ha | synergy, 6 h | |
CTX, cefotaxime; CAZ, ceftazidime; IPM, imipenem; AMK, amikacin; CIP, ciprofloxacin; ND, not determined.
aBacterial regrowth at 24 h.
Interactions between conventional antibiotics and LPC-058 at 0.25×, 0.5× and 1× MIC against E. coli producing CTX-M-15 ESBL and K. pneumoniae, P. aeruginosa and A. baumannii producing KPC-2, VIM-1 and OXA-23 carbapenemases, respectively
| Strain | Multiple of the MIC of conventional antibiotics | Combination effect, time in h | ||
|---|---|---|---|---|
| multiple of the MIC of LPC-058 | ||||
| 0.25× | 0.5× | 1× | ||
| E. coli CTX-M-15 | CTX 0.5× | synergy, 9 h | synergy, 6 h | indifference |
| AMK 0.25× | synergy, 9 h | synergy, 9 h | indifference | |
| CIP 0.5× | synergy, 6 h | synergy, 6 h | indifference | |
| K. pneumoniae KPC-2 | CTX 0.25× | indifference | synergy, 24 h | synergy, 24 h |
| IPM 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| CIP 0.5× | indifference | indifference | synergy, 24 h | |
| P. aeruginosa VIM-1 | CAZ 0.5× | synergy, 6 h | synergy, 9 h | synergy, 24 h |
| IPM 0.125× | synergy, 6 ha | synergy, 9 h | synergy, 24 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 24 h | |
| CIP 0.25× | indifference | indifference | synergy, 24 h | |
| A. baumannii OXA-23 | IPM 0.5× | ND | synergy, 3 h | synergy, 3 h |
| AMK 0.062× | ND | indifference | indifference | |
| CIP 0.5× | ND | synergy, 6 ha | synergy, 6 h | |
| Strain | Multiple of the MIC of conventional antibiotics | Combination effect, time in h | ||
|---|---|---|---|---|
| multiple of the MIC of LPC-058 | ||||
| 0.25× | 0.5× | 1× | ||
| E. coli CTX-M-15 | CTX 0.5× | synergy, 9 h | synergy, 6 h | indifference |
| AMK 0.25× | synergy, 9 h | synergy, 9 h | indifference | |
| CIP 0.5× | synergy, 6 h | synergy, 6 h | indifference | |
| K. pneumoniae KPC-2 | CTX 0.25× | indifference | synergy, 24 h | synergy, 24 h |
| IPM 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 9 h | |
| CIP 0.5× | indifference | indifference | synergy, 24 h | |
| P. aeruginosa VIM-1 | CAZ 0.5× | synergy, 6 h | synergy, 9 h | synergy, 24 h |
| IPM 0.125× | synergy, 6 ha | synergy, 9 h | synergy, 24 h | |
| AMK 0.5× | synergy, 9 h | synergy, 9 h | synergy, 24 h | |
| CIP 0.25× | indifference | indifference | synergy, 24 h | |
| A. baumannii OXA-23 | IPM 0.5× | ND | synergy, 3 h | synergy, 3 h |
| AMK 0.062× | ND | indifference | indifference | |
| CIP 0.5× | ND | synergy, 6 ha | synergy, 6 h | |
CTX, cefotaxime; CAZ, ceftazidime; IPM, imipenem; AMK, amikacin; CIP, ciprofloxacin; ND, not determined.
aBacterial regrowth at 24 h.
Similarly, the LPC-058 plus amikacin combination displayed synergistic activity against all MDR/XDR strains (Table 5 and Supplementary Data) other than the OXA-23 carbapenemase-producing A. baumannii (which was highly resistant: MIC of amikacin = 1024 mg/L). Hence, the synergistic improvements in the activity of LPC-058 with ciprofloxacin appear to be species-dependent. For CTX-M-15-producing E. coli and OXA-23-producing A. baumannii, the synergistic effect of LPC-058 plus ciprofloxacin occurred rapidly (within 6 h), but was weaker for carbapenemase-producing P. aeruginosa and K. pneumoniae strains (with indifference at ≤0.5× MIC of LPC-058 and prevention of bacterial regrowth at 24 h with 1× MIC of LPC-058, respectively). Overall, our results highlight the ability of LPC-058 to eradicate MDR/XDR strains when combined with conventional antibiotics usually associated with a high level of resistance.
Discussion
The worldwide rise and spread of ESBLs and carbapenemases in Gram-negative bacilli has created an urgent need to develop new antimicrobial agents.1–3 Our present results show that the newly discovered difluoromethyl-allo-threonyl-hydroxamate compound LPC-058 is the most potent (in vitro) of all the biphenyl-diacetylene tail scaffold compounds tested here. It was ∼10 times more active than the reference compound CHIR-090 against a broad panel of Enterobacteriaceae and non-fermentative Gram-negative bacilli and effectively overcame resistance mechanisms in MDR/XDR strains. LPC-058 is the first inhibitor to have shown activity against both A. baumannii and B. cepacia.10,11 Furthermore, LPC-058 is an effective bactericidal antibiotic (at 1× or 2× MIC) against MDR/XDR Enterobacteriaceae and P. aeruginosa strains. Lastly, LPC-058 displays synergy with β-lactams, amikacin and ciprofloxacin.
The generally greater potency and broader spectrum of activity displayed by LPC-058, LPC-011 and LPC-087 (relative to CHIR-90) likely reflects the presence of a biphenyl-diacetylene tail (Figure 1).8,11 LpxC enzymes accommodate the acyl chain of the substrate through a hydrophobic passage, which also hosts the tail group of LpxC inhibitors. As the contour of the hydrophobic passage differs in distinct LpxC enzymes, it has been shown that the diacetylene group's elasticity and its smaller footprint within the substrate-binding hydrophobic passage of LpxC orthologues contribute to the compounds' broad-spectrum antimicrobial activity (relative to CHIR-090).8,9 However, our MIC assay data suggested that the presence of the biphenyl-diacetylene tail scaffold in LpxC inhibitors is not enough to gain activity against A. baumannii. In fact, LPC-011 has the same tail group as LPC-058 (Figure 1), but was ineffective against A. baumannii. Optimal activity of LpxC inhibitors against A. baumannii depends strictly on the presence of a difluoromethyl-allo-threonyl-hydroxamate head group (Figure 1), since only LPC-087 and LPC-058 were active against A. baumannii. Addition of the difluoromethyl moiety to the LPC-011 threonyl head strengthens the compound's electrostatic interactions with the amino acids in the LpxC active site.11 We speculate that the introduction of fluorine atoms might also increase the compound's ability to cross lipid membranes, as has previously been demonstrated for fluoroquinolone compounds.26
It is noteworthy that LPC-087 (a compound bearing the same head group as LPC-058, but with an additional morpholine moiety in the tail) was generally less active than LPC-058. It is possible that the presence of this morpholine group weakened the inhibitor–LpxC interaction or diminished the compound's ability to cross the bacterial membrane. In the present study, both LPC-087 and CHIR-090 (which also has a morpholine group) were more affected by the overexpression of efflux pumps than LPC-058 (which lacks the morpholine unit). This observation suggests that LPC-087's low activity (relative to LPC-058) may reflect the greater ability of target bacterial cells to excrete compounds containing a morpholine group.
We were very surprised to find that the broad-spectrum LpxC inhibitor LPC-058 was inactive against S. maltophilia and A. xylosoxidans, despite the presence of a high level (∼55%) of sequence homology between these LpxC orthologues and that of E. coli (Supplementary Data). Further biochemical studies are now required to establish whether or not the existing LpxC inhibitors can: (i) inhibit the target enzyme's activity; (ii) effectively penetrate the membrane barrier; or (iii) avoid efficient extrusion by the various efflux pumps present in these species.27,28
In conclusion, we found that LPC-058 (bearing a difluoromethyl-allo-threonyl-hydroxamate head group and a para-amino substituted biphenyl-diacetylene tail group) is highly effective in vitro against a broad range of Enterobacteriaceae and non-fermentative Gram-negative clinical strains. The compound retains potent activity against MDR/XDR strains. Moreover, our results showed that LPC-058 was synergistic with several classes of antibiotics against carbapenemase-producing isolates, suggesting that an LPC-058-containing combination regimen might hold promise for the clinical treatment of drug-resistant Gram-negative pathogens.
Funding
This work was funded in part by National Institutes of Health grants AI055588, AI094475 and GM115355 (awarded to P. Z.) and by the Institut National de la Santé et de la Recherche Médicale, the Institut Pasteur de Lille, the Université Lille Nord de France and the Centre National de la Recherche Scientifique.
Transparency declarations
P. Z. and E. J. T. declare a competing financial interest: a patent on difluoromethyl-allo-threonyl-hydroxamate was awarded to P. Z. and E. J. T. All other authors: none to declare.
Acknowledgements
We thank Michel Simonet (Lille, France) for critically reading the manuscript prior to submission and Thilo Köhler (Geneva, Switzerland) for the gift of strains PT629 and ERYR.
References
Author notes
Pei Zhou and Nadine Lemaitre are co-senior authors.

{kind=link}
{kind=link}