





Abstract
Traditional antibiotic dosing was not designed for today's escalating antibiotic resistance, lack of novel antibiotics and growing complexity in patient populations. Dosing that ensures optimal antibiotic exposures should be considered essential to increase the likelihood of effective patient treatment. Given the variability in these exposures across different patients, a ‘one-dose-fits-all’ approach is increasingly problematic. Therapeutic drug monitoring (TDM) of the β-lactams, the most widely used antibiotic class, is underutilized in certain populations. Clinical experience with β-lactam TDM remains relatively scarce. Patients most likely to benefit from such an intervention include the critically ill, the obese, the elderly and those with cystic fibrosis. Most centres actively performing β-lactam TDM target a minimum 100% of the time during the dosing interval that the free (unbound) concentration of antibiotic exceeds the MIC of the pathogen (100% fT>MIC), which is higher than a traditional target supported by in vitro data. Ideally, isolated pathogens should undergo MIC testing along with TDM on a regular basis, allowing clinicians to address the triad of bug, drug and patient (‘mug’) in equal measure.
Introduction: bug, drug and ‘mug’
Antibiotic resistance is increasing more rapidly than previously forecast.1 In earlier decades the average lag time for resistance development after an antibiotic's registration was roughly 10 years, but there is now evidence of widespread resistance to novel drugs in countries where these compounds have not yet been introduced.2 At the same time, the pipeline for antibiotics with novel mechanisms of action is nearly dry.3 This backdrop mandates the urgent optimization of the way we manage our existing antibiotics.
Meanwhile, the patient populations treated in the healthcare system are rapidly changing. The proportion of geriatric patients is growing steadily, while worldwide obesity rates have nearly doubled since 1980.4 Patients with cystic fibrosis are living longer and being exposed to more antibiotic courses.5 At the outset of the antibiotic era, patients could rarely be maintained in prolonged states of severe illness. Now they may remain critically ill for weeks or months; in that time rapid dynamic changes in physiology,6 including phenomena such as augmented renal clearance7 and also renal and hepatic dysfunction, can lead to unpredictable pharmacokinetic alterations that affect antibiotic exposure.8 Broader use of supportive extracorporeal therapies such as continuous renal replacement therapy and extracorporeal membrane oxygenation further complicate the pharmacokinetic behaviour of antibiotics.9 Meanwhile the critically ill, with their ventilatory and feeding tubes, catheters and drains, must share limited space and medical staff, creating an ideal setting for the emergence and spread of resistance. This population, whose pooled annual antibiotic consumption can be measured in tons,10 is increasingly colonized or infected with bacteria whose susceptibilities have decreased since these antibiotics were first clinically tested.
The old landscape was one of lower MICs and relatively homogeneous patient types; here the simple dichotomy of ‘bug versus drug’, informed largely by in vitro studies, sufficed. Fixed dosing regimens appeared to suffice as well. The new landscape, however, is marked by less-susceptible microorganisms, patients often suspended in previously unknown extremes of pathophysiology and an absence of novel ‘magic bullets’. This requires adjustment to a trichotomy in which the host, or ‘mug’, plays a role equal to those of bug and drug.11 Indeed, a shift towards individualized antibiotic therapy to maximize the effectiveness of our available antibiotics is inevitable12 given limited alternative options.
Yet our ignorance of the host's role in antibiotic exposure and efficacy, especially those of the β-lactams, the cornerstone of anti-infective therapy, is not negligible. Most of the pharmacokinetic data informing both clinical trials and routine practice are provided by young, healthy volunteers whose physiology differs markedly from that of sick patients. In most hospitals worldwide today, patients are given a standard, fixed-dose regimen and clinicians have little means of confirming achievement of therapeutic exposures beyond the presence or absence of clinical response days or weeks later.
Historically, therapeutic drug monitoring (TDM) for the β-lactams was not introduced into routine practice because these antibiotics do not have a narrow therapeutic window. Now, however, given increasing acquired resistance to β-lactams and its association with low antibiotic serum concentrations,13 β-lactam TDM in certain populations may bear more clinical relevance.
In this paper, we review the data relating to the rationale for β-lactam TDM, identify the populations likely to benefit from its use and describe both the pharmacokinetic/pharmacodynamic targets favoured by current proponents and the challenges that institutions may face in its implementation.
β-Lactam antibiotics and current dosing schemes
The most widely used group of antibiotics, the β-lactams have long been at the front line of patient care. Penicillin derivatives, cephalosporins, monobactams and carbapenems all contain a core β-lactam ring and target bacterial penicillin-binding proteins, inhibiting cell-wall synthesis and causing cell lysis. Pharmacokinetic/pharmacodynamic studies have consistently shown that maximum killing occurs at concentrations ∼3–4 times the MIC, with higher concentrations offering little additional bacterial killing.14 Despite this, the effectiveness of β-lactam antibiotics ultimately depends more on the duration of exposure than on the magnitude of their concentration.14 The longer a β-lactam antibiotic is present at the site of infection at a free (unbound) concentration superior to the targeted pathogen's MIC (fT>MIC), the more effectively bactericidal it is. In this context, β-lactams exhibit time-dependent activity.15–17
Current dosing schemes are primarily derived from a mixture of pre-clinical infection models and Phase 1 pharmacokinetic data. But an identical dose of a β-lactam may lead to markedly different antibiotic exposures in healthy volunteers versus patients, including critically ill, obese or elderly patients. Critically ill patients in particular are known to undergo rapid changes in physiology that fundamentally alter their pharmacokinetics.12 Patients with sepsis become hyperdynamic, with increased renal blood flow and glomerular filtration that can result in augmented renal clearance of many drugs, among them the β-lactams.18 These pharmacokinetic changes also occur with post-trauma, post-major surgery, burns and transplant patients.6,19,20 Critically ill patients may also develop capillary leak syndrome, resulting in increased interstitial fluid volumes; these in turn lead to an increased volume of distribution of most β-lactams.21 Conversely, patients may develop end-organ dysfunction and thus impaired drug distribution and clearance.22 The presence of renal replacement therapy in particular introduces further challenges to optimal dosing. The combination of these different pathophysiological effects leads to highly variable and significant changes in pharmacokinetics not only between patients, but also within the same patient. In this setting, use of loading doses to account for the increased volumes of distribution should be considered, as should altered maintenance doses that are appropriate for the possible prolongation of drug half-life.
Figure 1 demonstrates the contrast in mean imipenem serum concentrations throughout the dosing interval in healthy volunteers23 and in 54 adult patients in an ICU with at least moderate renal function at the start of therapy.24 The critically ill patients clearly have consistently lower concentrations than healthy volunteers receiving the same dose, with mean concentrations below the MICs of several ICU pathogens. Similarly discordant pharmacokinetics have been demonstrated for β-lactams in patients with febrile neutropenia.25
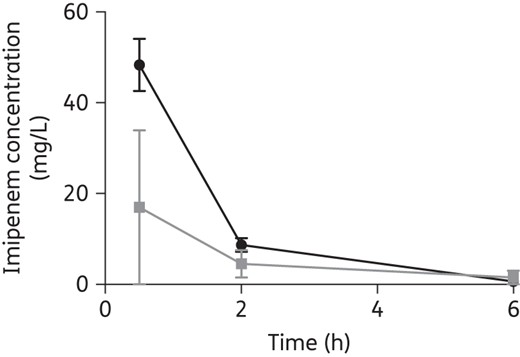
Mean (SD) serum imipenem concentrations in eight healthy volunteers (black)23 and in 54 critically ill patients (grey) throughout the dosing interval following a 0.5 g dose.
The pharmacokinetics of antibiotics in the obese remain largely unknown, though it is evident that they diverge significantly from those in the non-obese.26,27 Obese patients may have reduced total exposure of orally administered antibiotics, while most antibiotic classes demonstrate an increased volume of distribution in this population. Though obesity is associated with an increase in renal clearance, this is often countered by a higher incidence of renal dysfunction over time. For instance, the presence of comorbidities such as diabetes mellitus is associated with an elevated renal clearance in the first years of the disease, which later leads to chronic kidney disease and impaired drug clearance. This bidirectional effect and the fact that commonly used creatinine clearance calculations may be inaccurate in the obese together result in antibiotic exposure that is difficult to predict.26
Antibiotic pharmacokinetics in geriatric patients differ as well. Elderly patients have impaired homeostasis and wider inter-individual variability. While it is known that ageing is associated with reduced drug absorption from the gastrointestinal tract, protein binding and overall drug distribution,28 there are few clinical trials specifically comparing clinical outcomes as a function of drug dosage in this and younger populations. In these geriatric patients accurate descriptions of organ function in particular are required to ensure effective dosing, particularly as reducing renal clearance of drugs is likely in these patients.
Patients with cystic fibrosis are also a challenging patient group where pharmacokinetic changes for renally cleared drugs such as β-lactam antibiotics are common.29 These patients tend to display increased β-lactam clearances, whilst volume of distribution remains largely unchanged.30
When clinical testing of antibiotics in infected patients does occur, it is usually for pre-specified, mild or moderate infections in limited populations with little comorbidity or obesity. Meropenem was not tested widely in patients with pneumonia and, although approved for this indication in some countries, it is not widely approved for pneumonia even though this appears to be its most frequent use in critically ill patients worldwide. The anti-MRSA cephalosporin ceftobiprole has been studied and approved only for skin and skin structure infections (SSSIs),31 yet it is frequently used off-label in some countries for ventilator-associated pneumonia (VAP) at these recommended doses despite indications that these are unlikely to achieve pharmacodynamic targets in patients with VAP.32
In addition, the majority of currently used β-lactams underwent clinical outcome testing in Phase 3 trials several decades ago, when MICs for many organisms were lower. Longitudinal surveillance programmes document steady decreases in the susceptibility of several Gram-negative (Escherichia coli, Klebsiella spp., Pseudomonas aeruginosa etc.) and Gram-positive (Staphylococcus aureus, Streptococcus spp., Enterococcus spp. etc.) bacteria to β-lactam and other antibiotics since then.33,34
It is thus highly unlikely that optimal antibiotic exposure will be achieved with traditional doses in certain patient groups. Various options for dose optimization of β-lactam antibiotics have been explored. Dosing nomograms have proven largely unable to provide acceptable achievement of target exposures.12,35,36 Dosing based primarily on calculated creatinine clearance has also proven suboptimal,37 and even well-designed continuous or extended infusions of β-lactams may fail to consistently achieve target exposures.19,38,39 Thus TDM would appear useful, not only to avoid underdosing in patients with elevated drug clearances, but also to minimize the likelihood of drug toxicity (e.g. encephalopathy and seizures) in patients with impaired drug clearance.40
TDM of the β-lactam antibiotics: current state
The great advantage of β-lactam TDM is that it provides actual measurements of serum antibiotic exposures so that dosing can be adapted to ensure optimal exposures are achieved. Ideally, these should be interpreted in concert with the antibiotic's MIC for the causative pathogen to guide therapy, and with the nature and site of infection.
We estimate that at this time ∼30 hospitals worldwide are now performing β-lactam TDM on a routine basis, the majority for critically ill patients, with a great deal of heterogeneity in their approaches. A recent survey on the conduct of β-lactam TDM in ICU settings revealed diversity in the type of β-lactams measured, the patients selected for TDM and drug assay methods.41 Only two of the nine centres surveyed measure the unbound, and therefore active, concentrations of β-lactams (unbound measurement is likely to be more costly than total concentrations); the rest measure total concentrations with corrections for protein-bound drug based on published data from non-critically ill patients. While most β-lactam antibiotics have only low protein binding (exceptions include flucloxacillin, oxacillin, ceftriaxone, cefazolin and ertapenem),42,43 certain patient populations demonstrate significant hypoalbuminaemia. Measurement of total antibiotic concentrations in the presence of hypoalbuminaemia can lead to misinterpretation and potentially inappropriate dosing regimen adjustments or non-adjustments.44
Pharmacokinetic targets used by sites currently applying β-lactam TDM appear equally variable. The majority of centres target β-lactam concentrations with 100% fT>MIC, but some favour up to 100% fT>4×MIC, while a few target 50% fT>4×MIC or 70% fT>4×MIC for certain β-lactams.41 Indeed, whether a β-lactam's serum concentration needs to be above the MIC at all times (100% fT>MIC) requires prospective clinical validation. Although intuitively 100% fT>MIC would seem to be a safe target to ensure maximal antibiotic exposure, early in vitro pharmacokinetic/pharmacodynamic studies of some β-lactams suggest that such prolonged exposures may not be necessary.16
Time dependence and pharmacokinetic/pharmacodynamic targets
Murine infection models from the 1980s to the 2000s suggest that certain antibiotics’ fT>MIC need only be 40%–60% for certain organisms.16,17,45 Given that standard β-lactam dosing regimens target an exposure of ∼50% fT>MIC, it would seem that if a higher exposure were absolutely necessary, then the current practice should be associated with chronic underdosing and therapeutic failure. Yet there is currently only modest evidence in humans that β-lactam 100% fT>MIC concentrations translate to improved clinical outcome, possibly because the issue has yet to be investigated in a robust prospective manner, but also because of differences between studied patients in terms of how representative blood concentrations are of infection site concentrations and differences in the inoculum of different infections, as well as patient immunocompetence. The recent DALI study, a multinational point-prevalence analysis of intermediate (50% fT>MIC) and trough (100% fT>MIC) β-lactam serum concentrations in 384 patients across 68 ICUs, found an association between positive clinical outcome and an increasing 100% fT>MIC ratio (OR 1.53, P < 0.03). Though hampered by a lack of universal MIC measurements, the study found that the 16% of patients who did not achieve even 50% fT>MIC were 32% less likely to have a positive clinical outcome.19 The association between increasing antibiotic exposure above 50% fT>MIC and clinical cure may support a higher target, such as 100% fT>MIC for critically ill patients. Other populations remain understudied in this regard.
Interestingly, even altered dosing approaches intended to increase fT>MIC, such as continuous infusion of β-lactams, may not always achieve pharmacokinetic/pharmacodynamic targets. A recent trial randomized 60 patients with severe sepsis to β-lactam therapy via either intermittent dosing or continuous infusion; 82% and only 29% of those receiving continuous infusion and intermittent dosing, respectively, achieved 100% fT>MIC for targeted pathogens.46 While clinical cure was higher in the continuous infusion group (70% versus 43%; P = 0.037), ICU-free days (19.5 versus 17; P = 0.14) and survival to hospital discharge (90% versus 80%, P = 0.47) did not significantly differ between groups.
Nonetheless, like optimal antibiotic therapy, pharmacokinetic/pharmacodynamic targets must also be individualized. Sufficient tissue exposure will require different serum concentrations depending on where those tissues are and what antibiotic is being administered. For example, while cefepime appears to penetrate lung epithelial lining fluid (ELF) fully, ceftazidime has an ELF-to-serum concentration ratio of only 0.2.47,48 Pharmacokinetics change in relation to the patient's infective state itself; it has long been known that optimal CSF concentrations of most β-lactams are more likely to be achieved in the presence of meningeal inflammation.49 Furthermore, targets for bactericidal effect are different from those preventing the emergence of drug resistance, as the former often do not adequately suppress the growth of resistant subpopulations, especially in rapidly adaptive pathogens such as P. aeruginosa.50
But until validated sampling and software tools12 that would allow the identification of individual pharmacokinetic targets become widely available, serum trough concentrations of at least 100% fT>MIC can, in our view, provide a reasonably reliable confirmation of appropriate exposure in most clinical circumstances.
For practical reasons, where β-lactam trough concentrations are to be taken, the blood samples could be drawn once pharmacokinetic steady state has been reached (24 h after the start of therapy) to ensure attainment of this target.6 Alternatively and preferably, where a Bayesian adaptive feedback approach is available, sampling could occur during the first dosing interval to allow even more rapid prediction of optimized dosing.12 Open-source, web-based programs are available for this approach, which still requires clinical testing in randomized trials.12,51
β-Lactam TDM and MIC determination
According to the survey from Wong et al.,41 very few hospital centres today have instituted routine β-lactam TDM with concomitant pathogen MIC determination. The majority of centres performing TDM instead use local antibiograms or EUCAST breakpoints as surrogates for the actual MIC. Using epidemiological MIC breakpoints should ensure that all patients will achieve pharmacokinetic/pharmacodynamic targets, but clearly many patients will also have unnecessarily high doses when the infection is caused by highly susceptible pathogens. However, such high doses should be considered acceptable as these loading doses can also ensure maximal efficacy in the presence of pharmacokinetic changes and also the presence of a high inoculum of pathogens.
Indeed, timely MIC testing of the causative pathogen should be a fundamental aspect of TDM—but, as for aminoglycosides and glycopeptides, it is a frequently absent partner. EUCAST and CLSI breakpoints are essentially indicators constructed to err on the side of clinical caution, with high cut-offs for presumed susceptibility; as such they may not be applicable to local resistance patterns. Direct MIC testing is both time consuming and labour intensive. While newer automated methods such as Etest® and Vitek2® are promising, the results are apparently not frequently reported to clinicians.41 A more widespread use of β-lactam MIC testing in conjunction with TDM would allow truly individualized and optimized drug therapy.52
What are the issues preventing institutions providing β-lactam TDM?
The first issue likely to be preventing wide-scale adoption of β-lactam TDM programmes is the absence of a prospective randomized controlled trial demonstrating either a clinical or an economic benefit of such an intervention. To date, no such trial has been attempted to our knowledge. In fact, a randomized trial assessing a ‘hard clinical endpoint’ such as mortality may be exceedingly difficult to undertake because the institutions that have the β-lactam assay available may not have clinical equipoise as to the efficacy of TDM and therefore would find it ethically challenging to randomize patients to a treatment that is not the standard of care at their institution. It follows that clinical or economic evidence quantifying the benefits of β-lactam TDM may not be forthcoming in the immediate future.
The requirement for a chromatography-based method for analysing β-lactam concentrations is the second and perhaps larger issue preventing the introduction of routine β-lactam TDM services. Not only is the relative turnaround time (6–24 h) longer than those of other techniques (e.g. results of immunoassays for aminoglycosides and glycopeptides may be available within 30 min), but equipment can be expensive and requires skilled operators, providing a recurring equipment and staff cost for the institution. Given the importance of achieving effective exposures in the patient groups highlighted above, this chromatographic assay should also be available 24 h a day 7 days a week, which introduces additional infrastructure, staff and training costs. Of interest, the cost of measuring one β-lactam serum concentration varies, but is roughly USD 30/assay (20 €) in a central laboratory. Whilst some cost savings may be realized from dose decreases for infection with pathogens with lower MICs, unless changes to service utilization in terms of reduced duration of therapy or patient length of stay can be demonstrated, β-lactam TDM may always appear to represent a cost burden.
Finally, a collaborative approach between the treating team, infectious disease physicians, microbiologists, clinical pharmacists and the laboratory needs to be established with adherence to an agreed protocol for sampling and dose modification. This service can be successful and long lasting, as evidenced by the existing β-lactam TDM programmes in place at institutions around the world.
Conclusions
At present, TDM of the β-lactam antibiotics is probably not warranted with relatively mild infections and/or a low risk of less-susceptible pathogens. In populations with grossly varied and unpredictable pharmacokinetics however, or those with infections due to pathogens with high MICs, β-lactam TDM would appear highly relevant. We would conclude there is only indirect evidence supporting β-lactam TDM as an intervention leading to improved outcomes and a randomized controlled trial quantifying such a benefit is yet to be conducted.
Funding
This study was supported by internal funding.
A. H. is supported by the EU-AIDA project (grant agreement 278348). J. A. R. is funded by a Career Development Fellowship from the National Health and Medical Research Council of Australia (APP1048652).
Transparency declarations
None to declare.

{kind=link}