





Abstract
Ecogenomic investigation of a methanogenic bioreactor degrading terephthalate (TA) allowed elucidation of complex synergistic networks of uncultivated microorganisms, including those from candidate phyla with no cultivated representatives. Our previous metagenomic investigation proposed that Pelotomaculum and methanogens may interact with uncultivated organisms to degrade TA; however, many members of the community remained unaddressed because of past technological limitations. In further pursuit, this study employed state-of-the-art omics tools to generate draft genomes and transcriptomes for uncultivated organisms spanning 15 phyla and reports the first genomic insight into candidate phyla Atribacteria, Hydrogenedentes and Marinimicrobia in methanogenic environments. Metabolic reconstruction revealed that these organisms perform fermentative, syntrophic and acetogenic catabolism facilitated by energy conservation revolving around H2 metabolism. Several of these organisms could degrade TA catabolism by-products (acetate, butyrate and H2) and syntrophically support Pelotomaculum. Other taxa could scavenge anabolic products (protein and lipids) presumably derived from detrital biomass produced by the TA-degrading community. The protein scavengers expressed complementary metabolic pathways indicating syntrophic and fermentative step-wise protein degradation through amino acids, branched-chain fatty acids and propionate. Thus, the uncultivated organisms may interact to form an intricate syntrophy-supported food web with Pelotomaculum and methanogens to metabolize catabolic by-products and detritus, whereby facilitating holistic TA mineralization to CO2 and CH4.
Introduction
In methanogenic environments, fermenters, syntrophs and methanogens interact to facilitate anaerobic degradation of organic compounds to CH4 and CO2, an essential component of anaerobic biotechnology and natural global carbon cycle (McInerney et al., 2009). Fermenters and syntrophs degrade organic compounds to methanogen-utilizable substrates (for example, acetate and H2) and methanogens further convert these compounds to CH4 and CO2. Syntrophs specifically form obligate mutualistic interactions with methanogens to metabolize organic compounds whose degradation otherwise rapidly becomes thermodynamically unfavorable (ΔG>0) as by-products accumulate (Schink, 1997; Schink and Stams, 2006). Despite such low-energy conditions, engineered and natural methanogenic environments harbor a wide diversity of uncharacterized microorganisms from known phyla and candidate phyla without cultivated representatives (microbial dark matter; Rinke et al., 2013; also see Juottonen et al., 2005; Nemergut et al., 2008; Griebler and Lueders, 2009; Riviere et al., 2009; Glockner et al., 2010; Kong et al., 2010; Kim et al., 2011; Lykidis et al., 2011; Nelson et al., 2011; Rinke et al., 2013). Their presence indicates many undiscovered microbial niches and interactions. Understanding the roles and interactions of the ubiquitous uncultivated microorganisms is necessary to unravel the black-box microbial ecology of anaerobic biotechnology and the global carbon flux.
An excellent example of a methanogenic community rich in uncultivated taxa is a hypermesophilic bioreactor (46–50 °C) treating terephthalate (TA), a major waste product of plastic manufacturing (Lykidis et al., 2011). Pelotomaculum and Syntrophorhabdus are known to syntrophically metabolize TA (Wu et al., 2001; Qiu et al., 2004, 2006, 2008). In this reactor specifically, Pelotomaculum syntrophically interacts with methanogens and predominates TA degradation (Chen et al., 2004). Our recent metagenomic and proteomic studies proposed that Pelotomaculum may produce acetate, H2 and butyrate as by-products and that uncultivated taxa (Caldiserica and ‘Ca. Cloacimonetes’, formerly OP5 and WWE1) may further metabolize H2 and butyrate (Lykidis et al., 2011; Wu et al., 2013). This provided evidence that non-methanogen community members may serve as syntrophic partners (secondary degraders) to metabolize TA degradation by-products alongside aceticlastic and hydrogenotrophic methanogens. However, the sequencing technology of the time limited investigation of the other uncultivated community members’ ecological roles. Moreover, elucidation of their ecological roles is paramount as many of these taxa are consistently found across methanogenic bioprocesses and their contribution to anaerobic biotechnology remains unclear (Narihiro et al., 2014).
Although the persistent uncultivated organisms in methanogenic bioprocesses have long been suspected to contribute to anaerobic carbon flow, their fastidiousness has hindered exploration. In the TA-degrading community, some non-methanogenic taxa may also support TA degradation by metabolizing Pelotomaculum by-products (that is, acetate, butyrate or H2). Besides such catabolic by-products, other uncultivated taxa may scavenge TA metabolism anabolic products (for example, protein and lipids) bound in detrital biomass, estimated to account for up to 10% of the TA-derived carbon (Speece, 1996). Based on the known metabolic diversity of methanogenic environments (Schink, 1997; McInerney et al., 2009), uncultivated secondary degraders and scavengers must perform fermentative, syntrophic or acetogenic metabolism. To investigate the ecological roles and metabolic capabilities of the uncultivated taxa, this study used an ‘ecogenomics’ approach synthesizing single-cell genomics (Marcy et al., 2007), metagenomics (Tyson et al., 2004), metatranscriptomics (Frias-Lopez et al., 2008) and metabolic reconstruction assisted by comparative genomics of acetogen and syntroph energy conservation pathways (McInerney et al., 2007; Muller et al., 2008; Sieber et al., 2010, 2012; Nobu et al., 2014). In doing so, we provide the first look into the genomes and metabolism of many uncultivated taxa and propose how these organisms metabolically interact to achieve holistic carbon flux from TA to CH4 and CO2 in the methanogenic reactor.
Materials and methods
Metagenome, single-cell amplified genomes and metatranscriptome
Samples for metagenomic DNA, single-cell sorting and metatranscriptomic RNA were taken from biofilm carriers (that is, porous ceramic media for biofilm attachment) in a hypermesophilic (46–50 °C) bioreactor degrading TA at >80% efficiency after 1373 days of operation. The 1.2-l hybrid reactor was packed with tubular ceramic carrier (Siporax, sera GmbH, Immenhausen, Germany) and fed TA as the sole energy source (3.6 g l−1 TA per day) in an anaerobic mineral culture medium (Chen et al., 2004; Lykidis et al., 2011). For single-cell collection, biofilms were separated from the carrier, dispersed through sonication and sorted through flow cytometry. After DNA extraction and whole genome amplification of the sorted cells, cells of target clades were screened through 16S rRNA-gene-targeted PCR and amplicon sequencing (Rinke et al., 2013). For target clades, amplified genomic DNA was sequenced to generate single amplified genomes (SAGs) (Supplementary Table S2). Metagenomic DNA was extracted from dispersed biofilm using a cetyltrimethylammonium bromide-based protocol (Joint Genome Institute, Walnut Creek, CA, USA). Paired-end reads (100 bp, 3.77 × 108 reads) sequenced using a HiSeq-2000 sequencer (Illumina, San Diego, CA, USA) were trimmed and assembled as detailed in the Supplementary Note.
For constructing a metatranscriptome, total RNA was extracted from the TA-degrading reactor using mirVana RNA isolation kit (Ambion, Grand Island, NY, USA) and purified by removing DNA through the Turbo DNA-free kit (Ambion). Ribosomal RNA was also removed from the total RNA sample using a RiboMinus bacteria transcriptome isolation kit (Invitrogen, CA, USA) and the enriched messenger RNA fraction was treated with an mRNA-Seq kit for downstream RNA-Seq analysis (Illumina Inc., San Diego, CA, USA). Only fragments between 200 and 400 bp were used for final sequencing with a Genome Analyzer IIx using a single lane (Illumina Inc.). Metatranscriptomic sequences (available on MG-RAST under 4447821.3) were mapped to the metagenome and SAGs using blastn with a cutoff of 100% sequence identity.
Phylogenetic binning
Four methods were compared for phylogenetically annotating metagenomic scaffolds and, ultimately, constructing accurate TA-degrading community member draft genomes: PhylopythiaS, ClaMS, Metawatt version 1.7 and BLAST (Camacho et al., 2009; Pati et al., 2011; Patil et al., 2012; Strous et al., 2012). Metagenomic bins generated from these k-mer- and homology-based methods (detailed in Supplementary Note) were compared to attain accurate classification of contigs containing novel genes that BLAST cannot address. For comparison of the constructed bins, Multibase 2014 (Numerical Dynamics.com) was used to perform principal component analysis based on k-mer frequency. Tetramer frequencies were calculated for scaffolds longer than 1000 bp. Scaffolds longer than 10 kb were split into fragments with a minimum length of 5000 bp. Within each tetramer, the mean tetramer frequency was adjusted to zero and the other values were shifted accordingly. The frequencies were normalized to the maximum observed frequency across all tetramers. The principal component analysis results were visualized as a three-dimensional scatter plot with principal component 1, principal component 2 and scaffold read coverage as axes using TOPCAT version 4.0-1 (Taylor, 2005).
Results and discussion
Single-cell amplified genomes and metagenome
After 1373 days of operation with stable TA degradation (Supplementary Figure S1), biofilms were sampled from carriers (that is, biofilm attachment media) for downstream metagenomics and flow cytometry-based cell sorting. After screening sorted cells through 16S rRNA sequencing from whole-genome-amplified DNA, 32 cells were selected for SAG sequencing, as described previously (Rinke et al., 2013). This generated 32 SAGs to provide genomic insight into uncultivated clades, support phylogenetically annotating (‘binning’) metagenomic contigs and improve genome coverage. Shotgun sequencing of the TA-degrading community and assembly generated a metagenome of 205 310 contigs totaling to 217 Mb sequence with an N50 of 8452 bp. EMIRGE-based construction of 16S rRNA gene sequences (Miller et al., 2011) from the raw metagenomic reads revealed that the TA-degrading community is 39% Bacteria and 61% Archaea (Supplementary Table S1). Within the bacterial community (Supplementary Figure S1), Pelotomaculum-related (16.8% of bacterial community) and Syntrophorhabdus-related (6.1%) members were identified as syntrophs most likely contributing to TA degradation (Figure 2). In the archaeal community, both aceticlastic (Methanosaeta, 51.0% of archaeal community) and hydrogenotrophic (39.4% Methanolinea and 6.3% Methanospirillum) methanogens were identified to syntrophically support TA degradation through respective acetate and H2 degradation. Besides these primary TA degraders and methanogens, the community contained ‘Ca. Marinimicrobia’ (SAR406; 19.9%), Planctomycetes (15.8%), non-Syntrophorhabdaceae Deltaproteobacteria (5.2%), Synergistetes (8.5%), Thermotogae (5.4%), Spirochaetes (3.0%), ‘Ca. Cloacimonetes’ (WWE1; 3.3%), Chloroflexi (2.6%), Caldiserica (1.9%), Betaproteobacteria (0.7%), ‘Ca. Aminicenantes’ (OP8; 0.9%), ‘Ca. Hydrogenedentes’ (NKB19; 0.8%), ‘Ca. Atribacteria’ (OP9; 0.8%), candidate division AC1 (0.7%) and ‘Ca. Acetothermia’ (OP1; 0.6%) at abundance of >0.5% of the bacterial community (Supplementary Figure S1 and Supplementary Table S1). Thus, we detect the same clades as our previous study (Lykidis et al., 2011), even though the community structure slightly differs possibly because of both increase in TA loading rate and discrepancy between 16S rRNA gene-based PCR-dependent (that is, pyrosequencing) and -independent (that is, EMIRGE) analyses.
To generate high-quality draft genomes for these organisms, four binning methods (BLAST, PhylopythiaS, Metawatt and ClaMS) were compared. Although BLAST was precise, it only binned contigs that overlapped with reference genome sequences, which are incomplete SAGs for many clades. To maximize genome coverage for target clades, k-mer frequency binning methods can annotate non-overlapping contigs. Accuracy of these methods was measured by evaluating the agreement with BLAST binning results. PhylopythiaS and Metawatt agreed better with BLAST than ClaMS (Supplementary Table S3). Although PhylopythiaS generally had higher BLAST-extrapolated accuracy, its results were complemented with results from Metawatt for taxa poorly binned by PhylopythiaS. Manual curation of these binning results (Figure 1a) using essential single copy genes, k-mer frequency distribution, read coverage and BLAST generated 26 high-quality draft genomes spanning over 13 phyla (Figure 1b, Figure 2 and Supplementary Table S2).
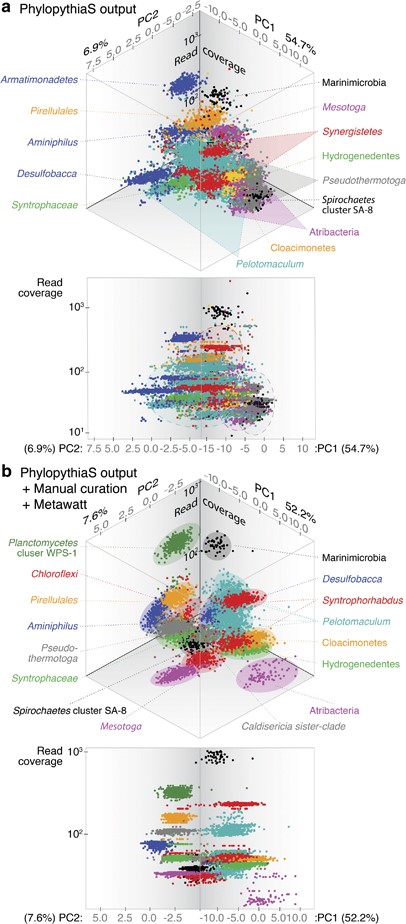
Comparison of PhylopythiaS and manually curated metagenome binning results. Principal component analysis on tetramer frequency of metagenomic contigs (>1 kb) binned through (a) PhylopythiaS and (b) combined PhylopythiaS, Metawatt and manual curation are shown with taxonomic bins differentiated by color. Angled (top) and front-face (bottom) views are shown for plots with principal components (PC1 and PC2) and contig read coverage on the horizontal and vertical axes respectively. For the raw PhylopythiaS output, several bins had high read coverage ranges (highlighted with dotted outlines) indicating poor bin quality. Relative to (a) the raw output, (b) manual curation allowed clearer separation of metagenomic bins. Some bins were manually taxonomic reclassified based on 16S rRNA gene phylogeny and BLAST.

TA reactor biofilm microbial community bin and SAG phylogeny and key anaerobic energy conservation pathways. The 16S rRNA gene-based phylogenetic tree (bootstrap 1000: >90% black node, >70% gray node with black outline and >50% gray node) contains sequences from single-cell genomes, analyzed bins (bolded, blue if methanogenic, and red or turquoise based on identified energy conservation pathways detailed below), and related isolates and clones (gray) with phyla distinguished by alternating background color. Classification of target taxa and their proposed Candidatus names are shown to the right of the tree. For each taxon, the genome completeness of SAG (gray), binned draft genome (black), and pangenome (black with *) are shown as pie charts. The following right-hand columns indicate the presence (circle) or absence (blank) of specific genes related to general (turquoise) and syntroph-associated (red) energy conservation. General energy conservation includes electron-confurcating hydrogenase (ECHyd), Rhodobacter nitrogen fixation complex (Rnf) and electron-bifurcating formate dehydrogenase, membrane-bound hydrogenase (Mbh) with or without accessory formate dehydrogenase H (FdhH), whereas syntroph-associated pathways are heterodisulfide reductase-associated putative ion-translocating ferredoxin:NADH oxidoreductase (Hdr-Ifo) and electron-transfer-flavoprotein-oxidizing hydrogenase (FixABCX). The Hdr-Ifo column indicates presence of an Hdr-Ifo gene cassette (red circle), non-adjacent Ifo and Hdr genes (light red circle with red outline) and only Hdr genes (empty red circle).

Reverse electron transport and electron confurcation mechanisms relevant to acetogen, fermenter and syntroph energy conservation. (a) The Rhodobacter nitrogen fixation complex (Rnf) reverse electron transport (RET) generates reduced ferredoxin (Fdred) for electron-confurcating hydrogenase (ECHyd) H2 generation. (b) The heterodisulfide reductase (Hdr)-associated putative ion-translocating ferredoxin:NADH oxidoreductase (Hdr-Ifo) is thought to also perform RET-driven Fdred generation to facilitate energy-conserving H2 production through either ECHyd or putative Methanothermobacter-like electron-confurcating hydrogenase associated with the Hdr-Ifo cassette. (c) The electron-transfer-flavoprotein (ETF)-oxidizing hydrogenase complex (FixABCX) takes advantage of quinol oxidation-driven H2 production. (d) Membrane-bound hydrogenases (Mbh) can conserve energy by extruding protons when performing Fdred-oxidizing H2 generation. This complex can be modulated by a formate dehydrogenase H (FdhH) to use formate (Fo) as an alternative electron donor. (e) An electron-bifurcating formate dehydrogenase (EBFdh) can facilitate energy-conserving formate metabolism; however, its reversibility remains unclear. (f) The NADH-dependent Fdred:NADP+ oxidoreductase is an electron bi(con)furcating enzyme found in organisms that utilize NADP(H) as an electron carrier (that is, acetogenesis and acetate-degrading syntrophy).
Combining SAGs and bins derived from the metagenome generated 35 draft genomes and six pangenomes (compiled bins and SAGs with >98% similarity of overlapping contigs) averaging 70% genome completeness and spanning 24 putative genera over 15 phyla, providing coverage for ∼90% of the TA-degrading bacterial community (Figure 2 and Supplementary Table S2). (Pan)genomes with at least 40% genome completeness (as described in Supplementary Note) and metatranscriptomics-based gene expression profiles available were analyzed (12 SAGs, 23 non-SAG draft genomes and 6 pangenomes; Supplementary Table S2). As an improvement to our previous study, we constructed multiple population-level genomes for methanogens and TA-degrading syntrophs: Methanolinea (3 genomes), Methanosaeta (4), Methanospirillum (2), Pelotomaculum (3), and Syntrophorhabdaceae (3) (Figure 2 and Supplementary Tables S1 and S2). This study also constructed much more complete genomes of Pseudothermotoga, Mesotoga, Caldiserica 31q06 and Cloacimonetes. Moreover, this effort generated genomes for taxa that our previous study could not address, including uncultivated phyla Atribacteria, Hydrogenedentes, Marinimicrobia and WS1; poorly understood phyla Armatimonadetes and Chloroflexi; and other uncultivated phylogenetic branches of novel Caldisericia sister clade, Syntrophus-related clade, Spirochaetes SA-8, Synergistetes TTA-B6, Planctomycetales-related clade and Phycisphaerae-related cluster WPS-1 (Chouari et al., 2005; Riviere et al., 2009; Yamada and Sekiguchi, 2009). Of these clades, pangenomes were constructed for Atribacteria, Cloacimonetes, Hydrogenedentes, Marinimicrobia, Chloroflexi and Syntrophus-related clade. Population abundance was estimated for each clade as shown in Supplementary Note and Supplementary Tables S1 and S2.
Metatranscriptomics
The 25 million read (2.0 Gb) metatranscriptome contained 78.6% rRNA and 21.4% putative mRNA reads. Blastn mapped 16.6% and 2.1% of the mRNA reads to the assembled metagenome and SAGs respectively using a 100% sequence similarity cutoff. The final draft genomes (combined SAGs and bins) accounted for 74.4% of these mapped reads. Methanogens accounted for the majority (75.6%) of the mapped data (64.4% Methanolinea, 5.7% Methanospirillum and 5.5% Methanosaeta) presumably because of the significantly larger net energy yield from methanogenesis as compared with TA degradation, especially considering that 3 m acetate and H2 are generated from 1 m TA. Among the bacterial clades, most reads were mapped to Pelotomaculum (31.7%), Marinimicrobia (22.8%), Mesotoga (11.3%), Hydrogenedentes (10.1%), Cloacimonetes (6.9%), Syntrophorhabdaceae (4.1%), Phycisphaerae-related cluster WPS-1 (3.8%), Syntrophus-related clade (3.1%) and Planctomycetales (2.1%). Atribacteria, Chloroflexi, Caldiserica, Spirochaetes, Synergistetes and Pseudothermotoga members each only accounted for 0.1–0.7% of the mapped sequences; however, this does not necessarily preclude their contribution for complete TA degradation.
Redefining syntroph and acetogen energy conservation: a comparative genomics approach
To rigorously determine the catabolic capacity of target organisms through ecogenomics, this study capitalizes on the complementarity of catabolism and energy conservation unique to methanogenic environments. Metabolism under methanogenic conditions faces two types of obstacles: thermodynamically unfavorable electron disposal and energy acquisition from limited energy margins (Thauer et al., 1977, 2008; Schink and Stams, 2013). Although H+ and CO2 are the only available exogenous electron acceptors (for example, syntrophy), coupling reoxidation of the general physiological electron carrier, NADH (E0’=−320 mV), with respective reduction of these compounds to H2 and formate (E0’=−414 and −420 mV) is thermodynamically unfavorable. To drive such endergonic reactions, anaerobic organisms are thought to employ energy from proton motive force (reverse electron transport), oxidation of a low-potential donor (electron confurcation) and reduction of a high-potential acceptor (electron bifurcation), often taking advantage of a high-energy electron carrier, ferredoxin (Fd, E0’=−453 mV). Conversely, for catabolic H2 and formate oxidation (that is, homoacetogenesis and methanogenesis), exergonic NAD+ reduction would sacrifice marginal energy yield whereas Fdox reduction is endergonic. To resolve this issue, acetogens and methanogens are thought to employ similar energy conservation strategies. As such, metabolism under methanogenic conditions necessitates complementation of substrate oxidation with electron balance and energy conservations and, thus, we genomically explore the metabolic capacity of target organisms based on this principle.
Although fermenters and methanogens have been extensively studied across a wide phylogenetic range, insight into acetogen and syntroph energy conservation still remains limited. In order to better understand energy-conserving mechanisms employed for acetogenesis and syntrophic metabolism, we survey all publically available homoacetogen and syntroph genomes (Supplementary Note). Many syntrophs appear to rely on the capacity to perform reverse electron transport-driven energy-conserving H2 generation through electron-confurcating hydrogenase (ECHyd) in combination with reduced ferredoxin (Fdred)-generating Rhodobacter nitrogen fixation (Rnf) complex (Rnf; NADH:Fd oxidoreductase) or Hdr-Ifo (heterodisulfide-reductase-associated ion-translocating Fd:NADH oxidoreductase) (Sieber et al., 2012; Nobu et al., 2014). Specifically, the syntroph-characteristic co-occurrence of Hdr-Ifo and ECHyd can serve as a good indicator for syntrophic capacity. Similarly, co-occurrence of the electron-transfer-flavoprotein-oxidizing hydrogenase (FixABCX) (Sieber et al., 2010, 2014) and carboxylate catabolism with ECHyd and Rnf or Hdr-Ifo can further implicate ability to degrade carboxylates syntrophically. As for syntrophs who do not encode Rnf and Hdr-Ifo or ECHyd, this survey reveals other reverse electron transport and electron bi(con)furcation mechanisms that syntrophs may employ. For acetogenesis, we identify that acetogens require (Schmidt et al., 2009) and encode at least one energy-conserving electron transfer between physiological electron carriers (Rnf, NAD(P)H transhydrogenase and/or NADH-dependent Fdred:NADP+ oxidoreductase) (Hugenholtz and Ljungdahl, 1989; Muller et al., 2008; Huang et al., 2012; Tremblay et al., 2012); often possess a putative clostridial sensory hydrogenase (Calusinska et al., 2010); and utilize a wide variety of hydrogenases and formate dehydrogenases. In addition, we identify five genotypes of acetogen MetF (methylenetetrahydrofolate reductase) essential for driving the homoacetogenic Wood–Ljungdahl pathway, but also postulate that undiscovered MetF homologs and genotypes must exist. Finally, in order to characterize syntrophic or acetogenic metabolism, it is paramount that we detect no electron disposal pathways for fermentative or respiration (that is, non-H+/CO2 electron acceptor). Using these findings as a foundation for genomic and metabolic properties of syntroph and acetogen, we genomically characterize syntrophic and acetogenic capacity of target taxa found in the TA-degrading reactor based on complementation between catabolic pathways and the observed energy conservation strategies.
Syntrophic TA degradation by Pelotomaculum and Syntrophorhabdus with methanogens
Organisms of the genus Pelotomaculum and family Syntrophorhabdaceae are thought to be responsible for syntrophic TA degradation in reactor systems (Lykidis et al., 2011; Wu et al., 2013). We identify diverse Pelotomaculum (bins TAPelo1–TAPelo4) predominate the community, accounting for 16.8% and 31.7% of the bacterial community and metratranscriptome. These Pelotomaculum indeed encode and express genes for syntrophic energy conservation (Hdr-Ifo and ECHyd) and a previously observed pathway for TA degradation to acetate, CO2 and H2 (Figure 2 and Supplementary Table S4) (McInerney et al., 2007; Lykidis et al., 2011). In addition, we newly identify expression of a clostridial electron-bifurcating butyryl-CoA dehydrogenase in TAPelo3 that may facilitate the previously hypothesized Sporotomaculum-like energy-conserving butyrate generation from aromatic compound degradation (Qiu et al., 2003; Buckel and Thauer, 2013; Wu et al., 2013; Nobu et al., 2014), albeit refuting the involvement of previously identified non-energy-conserving acyl-CoA dehydrogenase (Lykidis et al., 2011; Wu et al., 2013). Although butyrate-fermenting TA degradation is thermodynamically more favorable, it sacrifices precious adenosine triphosphate from substrate-level phosphorylation. Our current understanding of syntrophic TA degradation precludes determination of the energetic feasibility of butyrate fermentation, but it is plausible that butyrate serves as a supplementary electron sink during high H2 partial pressure. Although members of Syntrophorhabdaceae (bins TASrha1, TASrha2 and TASrha3) also encode and express syntrophic TA metabolism through Hdr-Ifo and ECHyd (Figure 2 and Supplementary Table S4), they comprise a smaller fraction of the bacterial community (6.1%) and metatranscriptome (4.1%). Thus, they may compete with Pelotomaculum in syntrophic TA degradation, but have a lesser contribution to total TA removal.
Supporting this TA degradation, we identify the methanogenic partners: aceticlastic Methanosaeta (30.9% of total community; 5.5% of metatranscriptome) and hydrogenotrophic Methanolinea (23.9%; 64.4%) and Methanospirillum (3.8%; 5.7%). We confirm that these organisms indeed express acetate- and H2-degrading methanogensis pathways (Supplementary Table S5). Although Methanolinea is less abundant than Methanosaeta, Methanolinea constitutes the majority of methanogen metatranscriptome and, perhaps, a larger proportion of the methanogenic activity. This discrepancy is in agreement with the higher energy yield from hydrogenotrophic methanogenesis and also suggests high importance of H2 oxidation in driving syntrophic TA degradation.
Thermotogae: unconventional syntrophic acetate degraders
Thermotogae, a phylum often found in wastewater treatment ecosystems (Narihiro et al., 2014), may be responsible for syntrophic acetate degradation, as observed for Pseudothermotoga lettingae strain TMO (Balk et al., 2002); however, the metabolic pathway remains unclear because of absence of acetyl-CoA synthase/CO dehydrogenase required for conventional syntrophic acetate catabolism through the Wood–Ljungdahl pathway (Muller et al., 2013). Interestingly, the TA-degrading community Mesotoga (bin TAMoga) encodes a potential alternative pathway mediated by the glycine cleavage system and tetrahydrofolate pathway (Supplementary Note, Supplementary Figure S2 and Supplementary Table S6), and H2-generating energy conservation complementary to syntrophic carboxylate degradation (that is, FixABCX, Rnf and ECHyd) (Figure 2). Furthermore, strain TMO and another community Thermotogae member (Pseudothermotoga, bin TAPoga) also encode the novel acetate degradation pathway with Rnf and ECHyd. Correspondingly, although the community Thermotogae member was previously inferred to oxidize butyrate-based identification of a butyryl-CoA dehydrogenase (Lykidis et al., 2011), we did not detect a complete butyrate degradation pathway. Thus, the Mesotoga and Pseudothermotoga members may syntrophically oxidize acetate through a previously uncharacterized acetate-oxidizing pathway. Although Pseudothermotoga has very low activity, the high Mesotoga activity (11.3% of the bacterial metatranscriptome or 2.8% of total) is comparable to aceticlastic Methanosaeta, suggesting that Mesotoga may specifically play a critical role in TA degradation by catabolizing acetate, one of the major by-products of TA metabolism.
Novel Syntrophaceae member: a versatile syntrophic fatty acid degrader
Our previous study (Lykidis et al., 2011) suspected that the Syntrophus-related clade (93.8% 16S rRNA similarity to Syntrophus aciditrophicus strain SB), composing 1.2% of the bacterial community and 3.1% of total metatranscriptome, may also contribute to aromatic compound degradation as characteristic of Syntrophus (Mountfort et al., 1984). Although its genome lacks the benzoate degradation pathway, we identify expression of Hdr-Ifo, ECHyd and FixABCX (Figures 2 and 3 and Supplementary Table S2), suggesting the capacity to syntrophically degrade carboxylates. In agreement, this organism expresses a butyrate degradation pathway similar to Syntrophomonas that also depends on identical energy conservation pathways (Supplementary Table S7; Sieber et al., 2010; Nobu et al., 2014). In addition, this clade expresses newly postulated syntrophic branched-chain fatty acid (that is, 2-methyl-butyrate, isovalerate and isobutyrate) degradation pathways that are consistent with previous cultivation-based studies (Conrad et al., 1974; Stieb and Schink 1986; Matthies and Schink, 1992) and share high homology with other organisms thought to degrade branched-chain fatty acids (Supplementary Note, Supplementary Figure S5 and Supplementary Table S7). Therefore, this Syntrophus-related clade likely performs syntrophic degradation of butyrate and branched-chain fatty acids (discussed later). We propose that this clade may support TA degradation through syntrophically metabolizing TA-derived butyrate. In agreement, its transcript level is much lower than Pelotomaculum and yet comparable to Syntrophorhabdaceae despite low abundance in the community.
An uncultivated Chloroflexi subphylum I member: a novel homoacetogen
Although recent studies cultivate many Chloroflexi subphylum I representatives (Yamada and Sekiguchi, 2009), much of this phylogenetic cluster remains to be characterized. For example, the TA-degrading community Chloroflexi is distantly related from the closest relatives, Anaerolinea thermophila (84.0% rRNA similarity) and Caldilinea aerophila (84.8%). Surprisingly, the pangenome of this Chloroflexi subphylum I member uniquely encodes the complete Wood–Ljungdahl pathway required for homoacetogenesis (Supplementary Table S8). Furthermore, we identify Rnf, NfnAB (NADH-dependent Fdred:NADP+ oxidoreductase) and HfsABC (putative sensory hydrogenase) that are conserved among many acetogens and are paramount for acetogenic metabolism (Supplementary Note and Supplementary Table S12). The Chloroflexi MetF, a critical gene for acetogenesis, is most likely a new type of MetF as it only shares low homology with type III acetogen MetF (<35% amino-acid similarity) and lacks MetV necessary for type III MetF function; however, discovery of a functionally novel MetF is reasonable because we have yet to discover MetF homologs for several known acetogens and characterize an acetogenic Chloroflexi. Thus, this Chloroflexi may be capable of H2-oxidizing homoacetogenesis, whereby supporting Pelotomaculum TA degradation in parallel with other H2 oxidizers (that is, Methanolinea and Methanospirillum). However, further experimental investigation is necessary as low genome coverage and gene expression levels limit definitive analysis. Although our previous study proposed that Caldiserica cluster 31q06 may perform acetogenic metabolism, reevaluation of the genes reveals that this is not likely (Supplementary Note).
‘Ca. Hydrogenedentes’ (formerly NKB19): a lipolytic glycerol degrader
Hydrogenedentes is often associated with methanogenic environments (Riviere et al., 2009; Narihiro et al., 2014), but its ecological role has remained enigmatic. While the genomic technology available for our previous study was insufficient for metagenomically capturing Hydrogenedentes, implementing modern sequencing platforms allowed construction of a Hydrogenedentes pangenome and transcriptome. These data reveal expression of extracellular lipases, type II secretion systems and the Sec system for extracellular hydrolysis of triacylglycerols to glycerol and long-chain fatty acids. This organism also expresses genes for syntrophically oxidizing glycerol to acetate, CO2, 2 NADH and Fdred and rather ECHyd than fermentative pathways for following electron disposal through H2 generation (Figure 2, Supplementary Figure S3 and Supplementary Table S9) Qatibi et al., 1991). As another NADH reoxidation mechanism, we postulate that Hydrogenedentes performs novel NADH-oxidizing H2 generation mediated by Na+-translocating NADH:quinone oxidoreductase, cytochrome bc fusion protein and periplasmic Fe-hydrogenase (Supplementary Note). In agreement, this organism expresses many Na+-transporting complexes and, thus, may rely on a transmembrane Na+ gradient metabolically and physiologically. We also identify expression of a novel Methanothermobacter electron-bifurcating hydrogenase-like gene cassette encoding MvhADG hydrogenase subunits and HdrABC (Supplementary Table S9; Kaster et al., 2011); however, its function remains unclear. Although the complete energy conservation scheme requires further investigation, we infer that Hydrogenedentes syntrophically degrades glycerol and lipids derived from detrital biomass. Despite the low Hydrogenedentes population abundance (0.8%), the strikingly high expression level (10.1% of bacterial transcriptome) suggests that Hydrogenedentes lipolysis and glycerol degradation is an important component of this TA-degrading community carbon flux. Although another clade, Planctomycetales-related clade (bin TAPire), also expresses similar lipolytic nonfermentative glycerol degradation (Supplementary Table S9), its low genome completeness prohibits accurate interpretation.
‘Ca. Marinimicrobia’ (formerly SAR406): a proteolytic amino-acid degrader
The ecological role of this typically marine-associated phylum, Marinimicrobia, in methanogenic ecosystems has remained unclear (Narihiro et al., 2014); however, modern sequencing technology has allowed us to peer into its genome that could not be addressed in our previous study. We discover expression of extracellular proteases and secretory pathways for extracellular proteolysis (Supplementary Table S10). For degrading the proteolysis-derived monomers, this organism expresses pathways for catabolizing amino acids that are strictly syntrophic (that is, Ile/Leu/Val/Pro) and also fermentable (that is, Gly/Ser/Thr/Glu/Asp/Asn) (Supplementary Table S10; Schink and Stams, 2013). More specifically, as observed in other syntrophs (Wildenauer and Winter, 1986; Fardeau et al., 1997; Baena et al., 1998; Plugge et al., 2002), Ile/Leu/Val and Pro are catabolized to branched-chain fatty acids and propionate, respectively, along with H2 and CO2 (Supplementary Figure S4 and Supplementary Table S10). To support H2-generating electron disposal, Marinimicrobia expresses Hdr-Ifo, Rnf and ECHyd (Figure 2). As for the fermentable amino acids, Gly/Ser are degraded to H2, CO2 and acetate, whereas Thr/Glu/Asp/Asn are degraded to either H2, CO2 and acetate or propionate. Interestingly, Marinimicrobia also utilizes the uncharacterized bacterial MvhADG-HdrABC found in Hydrogenedentes, suggesting that this complex may facilitate a novel energy conservation pathway found in uncultivated taxa. Based on these findings, we propose that Marinimicrobia syntrophically and fermentatively degrades amino acids through proteolysis of protein bound in the community detritus. Similar to Hydrogenedentes, Marinimicrobia also dictates a large portion of the bacterial metatranscriptome (22.8%), suggesting that Marinimicrobia proteolysis and amino-acid degradation is also a significant component of the TA-degrading community carbon flux. Although to a lesser extent, the member of Phycisphaerae-related uncultivated cluster WPS-1 (bin TAPhyc, 3.8% of metatranscriptome) may also contribute to proteolytic syntrophic (Ile/Leu/Val) and fermentative (Glu) amino-acid degradation mediated by Hdr-Ifo and ECHyd, based on the expressed pathways (Figure 2 and Supplementary Table S10). It is currently unclear why these proteolytic organisms are highly abundant and active in the community. Caldisericia sister clade (bin TACald), Aminiphilus-related clade (bin TAAmin), Synergistetes cluster TTA-B6 (SAG D21) and Spirochaetes clade SA-8 (TASpir) members also harbor proteolytic AA metabolism pathways, although with low transcriptomic coverage (see Supplementary Table S11).
‘Ca. Atribacteria’ and ‘Ca. Cloacimonetes’: syntrophic propionate degraders
This ecogenomic effort also generated a pangenome for Atribacteria, another poorly characterized candidate phylum associated with methanogenic environments (Narihiro et al., 2014) that our previous study could not address. This Atribacteria encodes propionate metabolism and specifically expresses methylmalonyl-CoA pathway genes with high homology (52–71%) to those found in Pelotomaculum thermopropionicum strain SI, a representative thermophilic propionate-degrading syntroph (Supplementary Table S11; Kosaka et al., 2008). For energy-conserving electron disposal, Atribacteria expresses an electron-bifurcating formate dehydrogenase (Wang et al., 2013) and Fdred-dependent energy-conserving membrane-bound hydrogenase (Figures 2 and 3), suggesting that both formate and H2 generation may take part in propionate degradation as observed in Syntrophobacter (Harmsen et al., 1998). Moreover, we identify that electron-bifurcating formate dehydrogenase is also encoded by Pelobacter carbinolicus (Pcar_1846-1843), a syntroph dependent on formate transfer (Dubourguier et al., 1986; Rotaru et al., 2012). Although propionate metabolism can be used in the reverse direction fermentatively, we did not detect any pathways for catabolizing fermentative substrates. Similarly, Cloacimonetes also only expresses propionate metabolism and Pelotomaculum-like methylmalonyl-CoA pathway (51–57% similarity) and energy-conserving H2 production through Rnf and ECHyd (Figures 2 and 3). Thus, Atribacteria and Cloacimonetes may perform syntrophic propionate metabolism. Compiling the amino acid, branched-chain fatty acid and propionate degradation genes expressed by the uncultivated organisms forms a comprehensive pathway metabolizing amino acids to methanogen-utilizable substrates. Based on this striking metabolic complementarity in degradation of these substrates, we speculate that chaining syntrophic interactions (secondary syntrophy) and substrate exchange may facilitate proteinaceous detritus metabolism. However, further co-culture-based studies are necessary to investigate the nature and motivation behind such compartmentalization of syntrophic and fermentative degradation in methanogenic environments.
Conclusion
Implementation of cutting-edge ecogenomics and insights into anaerobic metabolism allowed a much more robust investigation of the TA-degrading community microbial dark matter hidden behind the binary Pelotomaculum–methanogen syntrophic degradation. This study not only provided novel genomic and metabolic insights into several uncultivated phyla (Hydrogenedentes, Marinimicrobia, Atribacteria and Cloacimonetes) and clades (within Thermotogae, Syntrophaceae, Chloroflexi, Planctomycetes and Caldiserica) associated with methanogenic environments, but also shed light on their potential ecological roles and interactions. Whereas several uncultivated taxa may support TA degradation as secondary degraders metabolizing Pelotomaculum catabolism by-products (acetate, butyrate and H2), many others may contribute to scavenging detritus accounting for up to 10% of the degraded TA (Speece, 1996) through macromolecule hydrolysis, fermentation and chaining syntrophic degradation of glycerol, amino acids and branched-chain fatty acids (Figure 4). Therefore, the holistic carbon flux from TA to CH4 and CO2 may require primary TA degraders, secondary degraders, detritus scavengers and methanogens to form ‘syntrophic networks’ beyond the conventionally studied binary syntrophy. Although full validation of the proposed carbon flow model further requires effortful cultivation-based studies tracking the specific niches in the TA-degrading community, we believe that metabolic characterization of the uncultivated taxa provides a valuable step forward in improving our insight into the potential roles of these organisms in methanogenic environments and ecological synergy (that is, secondary syntrophy) facilitating degradation at the thermodynamic limit. Last, we propose Candidatus species designations (Supplementary Note) to reflect the newly discovered behavior of these elusive organisms.
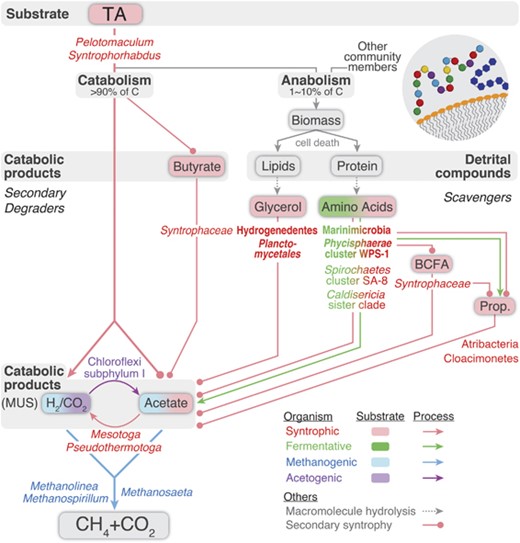
Holistic carbon flux from TA to CH4 and CO2. TA degradation generates catabolic by-products and detrital compounds. Catabolic by-products are primarily methanogen-utilizable substrates (MUS) (that is, acetate and H2) and also include butyrate. Detrital compounds consist of biological macromolecules such as lipids and protein. Syntrophy-related (red), fermentation-related (green), methanogenesis-related (blue) and acetogenesis-related (purple) taxa (name), substrates (box) and pathways (arrow) are shown. Taxa and substrates related to two processes are indicated by both colors. Syntrophic, fermentative and acetogenic secondary degraders and scavengers interact to metabolize these compounds to acetate and H2 that are finally mineralized to CH4 and CO2 by methanogens (blue). In particular, syntroph-to-syntroph substrate transfer (secondary syntrophy, circle-headed line) may play an important role in completing degradation of TA and protein. For scavenging detrital compounds, exoenzyme-producing organisms (bolded) are necessary to hydrolyze macromolecules (dotted gray arrow).
Acknowledgements
We acknowledge Peiying Hong, Xianzheng Li and Hideyuki Tamaki for sample preparation. We thank Bernhard Schink for etymological advice on Prokaryote Candidatus nomenclature. We also thank reviewers for invaluable input. The work conducted by the US Department of Energy Joint Genome Institute is supported by the US Department of Energy Office of Science under Contract No. DE-AC02-05CH11231. This work is also supported by the US Department of Energy under Award DE-SC0006771 to the University of Illinois, Urbana–Champaign, and a grant from the Energy Biosciences Institute (EBI) at the University of Illinois Urbana-Champaign (UIUC) to WTL.
Competing interests
The authors declare no conflict of interest.
References
Narihiro T, Nobu MK, Kim NK, Kamagata Y, Liu WT . (2014). The nexus of syntrophy-associated microbiota in anaerobic digestion revealed by long-term enrichment and community survey. Environ Microbiol e-pub ahead of print 4 September 2014; doi:10.1111/1462-2920.12616.
Nobu MK, Narihiro T, Hideyuki T, Qiu YL, Sekiguchi Y, Woyke T et al. (2014). The genome of Syntrophorhabdus aromaticivorans strain UI provides new insights for syntrophic aromatic compound metabolism and electron flow. Environ Microbiol e-pub ahead of print 3 March 2014; doi:10.1111/1462-2920.12444.
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information The online version of this article (doi:10.1038/ismej.2014.256) contains supplementary material, which is available to authorized users.

{kind=link}
{kind=link}
{kind=link}
{kind=link}