





Abstract
Marine bacterial lineages associated with algal blooms, such as the Roseobacter clade, have been well characterized in ecological and genomic contexts, yet such lineages have rarely been explored in freshwater blooms. This study performed phenotypic and genomic analyses of an alphaproteobacterial lineage ‘Candidatus Phycosocius’ (denoted the CaP clade), one of the few lineages ubiquitously associated with freshwater algal blooms, and described a novel species: ‘Ca. Phycosocius spiralis.’ Phylogenomic analyses indicated that the CaP clade is a deeply branching lineage in the Caulobacterales. Pangenome analyses revealed characteristic features of the CaP clade: aerobic anoxygenic photosynthesis and essential vitamin B auxotrophy. Genome size varies widely among members of the CaP clade (2.5–3.7 Mb), likely a result of independent genome reductions at each lineage. This includes a loss of tight adherence pilus genes (tad) in ‘Ca. P. spiralis’ that may reflect its adoption of a unique spiral cell shape and corkscrew-like burrowing activity at the algal surface. Notably, quorum sensing (QS) proteins showed incongruent phylogenies, suggesting that horizontal transfers of QS genes and QS-involved interactions with specific algal partners might drive CaP clade diversification. This study elucidates the ecophysiology and evolution of proteobacteria associated with freshwater algal blooms.
Introduction
In marine environments, algal blooms often occur in early spring to early summer in the stratified euphotic zone, where nutrients are replenished annually through winter mixing. As such, marine algal blooms constitute part of marine biogeochemical cycles [1]. Each algal cell or colony in a marine bloom develops a phycosphere. This phycosphere attracts many heterotrophic bacteria that utilize dissolved organic matter, such as polysaccharides, that are rarely available in the open oligotrophic ocean [2]. Among the marine phycosphere bacteria, the Roseobacter clade is one of the most abundant bacterial groups in the pelagic environment and plays a significant role in global carbon and sulfur cycling [1]. Members of the Roseobacter clade are associated with a broad taxonomic range of algae, including dinoflagellates, diatoms, and haptophytes [1, 3]. Aside from its affinity to phytoplankton, a prominent characteristic of the Roseobacter clade is its metabolic versatility. These adaptations include anoxygenic photosynthesis, sulfur and carbon monoxide oxidations as sources of energy (chemolithotrophy), and algae-derived dimethylsulfoniopropionate (DMSP) assimilation [3]. In addition, members of the Roseobacter clade show chemotaxis toward algae or algae-derived dissolved organic matter and become sessile on the nutrient-rich algal cell surface. This is often called a ‘swim-or-stick’ lifestyle [4] and is facilitated by flagellar assembly and type IV pili expression [5]. These behaviors are controlled by a suite of phosphorelays centered on a global transcriptional regulator, CtrA, with quorum-sensing (QS) also involved in its regulation [6].
In freshwater systems, algal blooms often occur in response to increasing nitrogen and phosphorus inputs resulting from human activity, although the occurrence of blooms predates human history [7]. Climate change is also thought to accelerate freshwater blooms [7]. Freshwater bloom-forming algae include Cyanobacteria (e.g., Microcystis, Dolichospermum, and Aphanizomenon), diatoms [8], and on rare occasions, green algae (e.g., Botryococcus braunii) [9]. Cyanobacterial blooms are known to produce cyanotoxins that are health hazards for both humans and wildlife [7]. Many freshwater bloom-forming algae form carbohydrate-rich colonies that contribute to a variety of phycospheres. A growing number of studies have revealed that Alphaproteobacteria, Gammaproteobacteria, and Bacteroidetes are major constituents of freshwater phycosphere bacterial communities [10–14]. However, ecophysiological interactions between freshwater bloom-forming algae and their phycosphere bacteria have been poorly described. One study suggested the presence of metabolic complementation between Microcystis aeruginosa and Roseomonas spp. [13]. Another study suggested that B. braunii supplemented biotin to Brevundimonas sp., which in return protected B. braunii from colonization by other harmful bacteria [14].
A previous study isolated a novel alphaproteobacterium, ‘Candidatus Phycosocius bacilliformis,’ from a bloom of B. braunii, a colony-forming green alga (Chlorophyta) [15]. B. braunii is unique in that it accumulates hydrocarbons within the colony and has thus attracted attention as a feedstock for biofuel production [15]. ‘Ca. P. bacilliformis’ 16S rDNA clustered with two bacterial sequences recovered from Microcystis cultures, and similar sequences (showing >99.4% DNA similarity) were frequently detected in Botryococcus and Microcystis blooms collected from lakes in Asia and Africa [15]. A subsequent metagenomic study of Microcystis-associated bacteria in 14 US lakes revealed that relatives of ‘Ca. P. bacilliformis’ were detected in all 46 Microcystis colonies and accounted for up to 19% of bacterial abundance in the Microcystis phycosphere [16]. These results suggest that ‘Ca. P. bacilliformis’ and its relatives represent a ubiquitous lineage closely associated with freshwater phytoplankton populations. Interestingly, ‘Ca. P. bacilliformis’ possesses genes for bacteriochlorophyll a (BChl a), suggesting that this species is a photosynthetic bacterium [17]. However, knowledge of this lineage is limited due to the poor availability of genome sequences.
Previous research observed another bacterium, morphologically distinct from ‘Ca. P. bacilliformis,’ co-occurring with ‘Ca. P. bacilliformis’ in a nonaxenic culture of B. braunii [15]. ‘Ca. P. bacilliformis’ was rod-shaped, whereas the novel bacterium was spiral in shape (Fig. S1). In this study, the spiral bacterium was cultivated, characterized, and designated BOTRYCO-1. Analysis of 16S sequences revealed that BOTRYCO-1 was closely related to ‘Ca. P. bacilliformis,’ motivating further characterization of ‘Ca. Phycosocius.’ The BOTRYCO-1 genome sequence was obtained and used in a pangenome analysis of four ‘Ca. Phycosocius’ relatives. Together, these bacteria represented a unique alphaproteobacterial lineage characterized by photoheterotrophy and close association with freshwater phytoplankton blooms.
Materials and methods
Cultures
B. braunii Ba10, a nonaxenic isolate recovered from a B. braunii bloom in a reservoir in Myanmar [15], was used to cultivate a spiral bacterium, designated BOTRYCO-1. A two-membered culture of a bacterium and B. braunii Ba10 was established by inoculating algal-free bacterial suspension (obtained by filtration of Ba10 using a Minisart® syringe filter, pore size 1.2 μm, Sartorius, Göttingen, Germany) into a bacteria-free culture of Ba10 (ref. 15). The culture was maintained in liquid AF-6 medium [18] at 25 °C under continuous white light (50 μmol photon m−2s−1). The detailed cultivation protocol was as described previously [15].
Light and electron microscopic studies
Light microscopy was performed using an IX71 microscope with a DP72 camera (Olympus, Tokyo, Japan). Catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) was performed using a BAG645 probe specific to the CaP clade [15], as previously described [15]. Scanning electron microscopy (SEM) was performed using a JSM-6330F field emission SEM (JEOL, Ltd., Tokyo, Japan). Transmission electron microscopy (TEM) was performed using a TEM H-7650 (Hitachi, Tokyo, Japan). Sample preparation was as previously described for SEM [19] and TEM [15].
Whole genome analysis
BOTRYCO-1 cells were collected by Minisart® filtration of 80 mL of a two-membered liquid culture comprising BOTRYCO-1 and B. braunii Ba10. Genomic DNA was extracted using NucleoBond AXG columns with buffer set III (Macherey-Nagel, Düren, Germany). Whole-genome sequencing of BOTRYCO-1 was performed using the MiSeq platform (Illumina, San Diego, CA, USA) as previously described [17]. De novo assembly of short reads was performed using SPAdes ver. 3.14.1 (ref. 20), and assembled scaffolds were polished using Pilon ver 1.24 (ref. 21). After removing experimental contaminants (based on lower K-mer coverage and BLAST analysis), the remaining scaffolds were annotated using dFAST [22]. Genome completeness was assessed using CheckM [23]. Average nucleotide (ANI) and amino acid identities (AAI) were calculated using online ANI and AAI calculators [24].
Phylogenetic analyses
Genome sequences related to BOTRYCO-1 were retrieved from GenBank, and a phylogenomic tree was constructed using PhyloPhlAn 3.0 (ref. 25) using amino acid sequences of 400 universal marker genes (PhyloPhlAn database) [26]. The statistical confidence of each tree branch was evaluated using the bootstrap method with 100 replicates. The PufL and PufM sequences of type strains of the Caulobacterales related to BOTRYCO-1 were retrieved from GenBank and aligned using Clustal W [27]. Maximum likelihood (ML) phylogenetic analyses were performed using RaxML-NG ver. 1.0.1 (ref. 28). Modeltest-NG ver. 0.1.6 (ref. 29) was used to select the amino acid substitution model for ML phylogenetic reconstruction.
Pangenome analyses and functional assessment of coding sequences
Pangenome analyses were performed using the GET_HOMOLOGUES pipeline [30], and the result was visualized using ‘ggplot2’ in R ver. 4.0.5. Initial functional annotation of proteins was performed using eggNOG mapper v.2 (ref. 31). Using the outputs of the eggNOG mapper, KEGG pathway reconstruction [32] was used to infer metabolic pathways present for each genome. Enzyme presence was further inspected by individual BLAST searches.
Results and discussion
Phenotypic and phylogenetic characterization of a novel species of ‘Ca. Phycosocius’
TEM microscopy of B. braunii Ba10 indicated the presence of spiral bacteria inside the extracellular matrix (ECM; Fig. S2). This differed from ‘Ca. P. bacilliformis,’ which is not seen inside the ECM [15]. The spiral bacterium, BOTRYCO-1, showed no growth in a low nutrient medium in which ‘Ca. P. bacilliformis’ had exhibited transient growth (Table S1). A culture exclusively containing B. braunii and BOTRYCO-1 was established, and BOTRYCO-1 growth was abundant in cocultivation (Fig. 1A), reaching > 104 cells/μL during the early stationary growth phase of B. braunii. BOTRYCO-1 was observed actively swimming in a spiral manner in the liquid medium (Movie S1) and was aggressively motile toward B. braunii. BOTRYCO-1 burrowed through the ECM in a back-and-forth spiral motion near the surface of the B. braunii colony (Movie S2). Numerous nonmotile spiral cells were present deep inside the ECM, suggesting that, after successful colonization, the bacterium stopped moving and presumably lost its motility, reminiscent of the ‘swim-or-stick’ lifestyle of the Roseobacter clade [4]. SEM microscopy indicated that BOTRYCO-1 was 1.2–2.6 μm long (Fig. 1B), and TEM examination of the coculture confirmed that BOTRCYO-1 was present inside the ECM (Fig. 1C). Collected BOTRYCO-1 cells were burgundy-red (Fig. 1D), similar to those of ‘Ca. P. bacilliformis’ [15].
Micrographs of BOTRYCO-1.
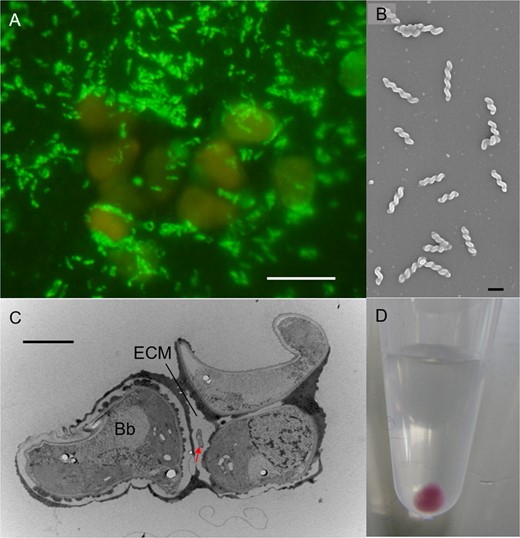
A CARD-FISH image of the two-membered culture of B. braunii (orange) and BOTRYCO-1 (yellow-green). Scale bar, 10 μm. B SEM photo of BOTRYCO-1 showing a spiral shape. Scale bar, 1 μm. C TEM photo showing a BOTRYCO-1 cell inside the extracellular matrix (ECM) of B. braunii. Arrow indicates BOTRYCO-1. Bb, B. braunii. Scale bar, 2 μm. D Collected cells of BOTRYCO-1 in a 1.5 mL microtube.
BLAST analysis indicated that the BOTRYCO-1 16S rDNA sequence had 98.9–99.3% sequence similarity to about a dozen bacterial sequences, including those of ‘Ca. P. bacilliformis,’ the recently described species Aquidulcibacter paucihalophilus TH1-2T [33], and a metagenomic assembled genome UKL13-1 recovered from an Aphanizomenon flos-aquae cyanobacterial bloom [34]. Given the morphological dissimilarities (Table S1), the close relationships observed between BOTRYCO-1 and these two species were unexpected. Phylogenetic analysis of 16S rDNA indicated that these sequences, including the BOTRYCO-1 sequence, formed a monophyletic clade, hereafter denoted the CaP (CandidatusPhycosocius) clade (Fig. S3A, B). Notably, almost all sequences of this clade were recovered from isolates or colonies of freshwater microalgae (Table S1, Fig. S3B). Our literature survey identified CaP bacteria in freshwater phycosphere microbiomes worldwide (Table S2). Furthermore, analyses of global Microcystis microbiome data [12] revealed that CaP bacteria were distributed worldwide and were one of the dominant groups of bacteria (>1% relative abundance) in 4 of 12 Microcystis blooms examined (Table S3). These results confirm the hypothesis that the CaP clade is ubiquitously associated with freshwater blooms of broad taxonomic groups of algae [15, 16]. The CaP clade and two monospecific genera, Vitreimonas [35] and Terricaulis [36], constituted a deeply branching lineage of the Caulobacterales both in phylogenomic and 16S rDNA trees, although the branching order within the CaP clade was slightly different (Fig. 2A, Fig. S3A). ‘Ca. P. bacilliformis’ 16S rDNA had 100% similarity to A. paucihalophilus; however, ANI and AAI values between the four genomes in the CaP clade were <95% (Fig. 2A), which is a proposed cutoff value used for bacterial species delineation [37, 38]. This suggests that each member of the CaP clade represents a different species. ANI values between A. paucihalophilus and ‘Ca. P. bacilliformis’ (81.4%), and A. paucihalophilus and BOTRYCO-1 (79.6%), were higher than the mean genus cutoff value (73.9 %) [39], indicating that ‘Ca. P. bacilliformis’ and BOTRYCO-1 may be affiliated with the genus Aquidulcibacter. However, the colony colors of BOTRYCO-1 and ‘Ca. P. bacilliformis’ were purplish, different from the yellowish color of A. paucihalophilus (Table S1) and from those of other known Caulobacteraceae species [36]. In addition, species affiliated with the Caulobacterales have various cell shapes, including rod, vibrioid, and fusiform, but, other than BOTRYCO-1, spiral-shaped cells have not been described [40]. On the basis of its distinct cell morphology and its genetic and phenotypic similarity to ‘Ca. P. bacilliformis,’ we tentatively propose ‘Candidatus Phycosocius spiralis’ for BOTRYCO-1. Hereafter, ‘Ca. P. spiralis’ is used for BOTRYCO-1.
Pangenome analysis of ‘Ca. Phycosocius’ and relatives.
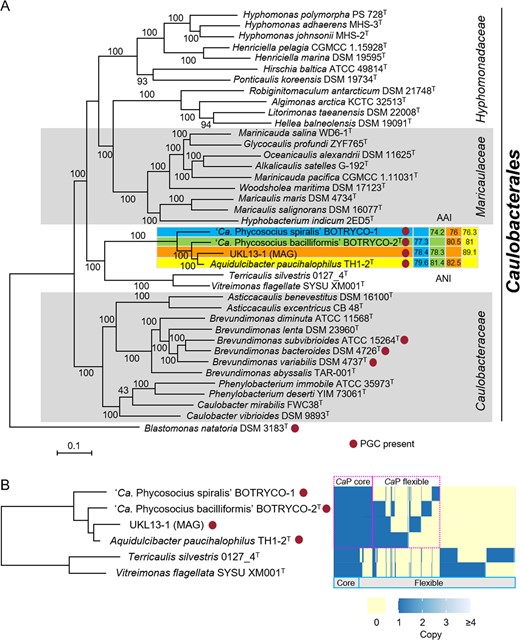
A ML phylogenomic tree rooted with Blastomonas natatoria. ML bootstrap values (>80%) are indicated at each branch. Pairwise ANI and AAI values were indicated at the cluster, including ‘Ca. P. spiralis.’ B Distribution of orthologous gene clusters in the CaP clade. A heatmap indicates the presence and absence of orthologs. Core and flexible genomes indicate orthologs present in all six genomes and less than six genomes of CaP bacteria, respectively. CaP core and flexible indicate orthologs in all and less than four CaP genomes, respectively.
Aerobic anoxygenic photosynthesis (AAnP) in the CaP clade
Whole-genome assembly of ‘Ca. P. spiralis’ produced 46 contigs with an N50 value of 149 808 bp, with the longest contig being 351 315 bp and with total contig length of 2.5 Mb. CheckM analysis indicated 100% completeness and 0% contamination, suggesting that the estimated genome size approximated the true genome size of ‘Ca. P. spiralis,’ and that the recovered scaffolds covered almost all genes. The estimated genome size differed from other members of the CaP clade, with ‘Ca. P. spiralis’ having the smallest genome and A. paucihalophilus having the largest (3.7 Mb; Table S1). To infer the genomic basis underlying the phenotypic characteristics of CaP bacteria, pangenome analyses, including the two most closely related species of the CaP clade, Terricaulis silvestris and Vitreimonas flagellata, were performed. Results indicated that each CaP bacterium possessed a substantial number of proteins that were not shared with other CaP bacteria (Fig. 2B). The differences in genome size and protein contents among the species in the CaP clade (Fig. 2B) were explained either by genome reduction or by multiple large-scale horizontal gene transfers (HGT). Accumulating evidence favors the former because the increasing association between bacteria and other organisms has often driven genome reduction in bacterial associates [41, 42]. Regardless, the genome content heterogeneity among CaP bacteria likely results from independent adaptive evolution in response to different environments such as diverse bacteria-host algae interactions.
Genome annotation of ‘Ca. P. spiralis’ revealed the presence of a complete set of genes for BChl a-based photosynthesis. These include genes for BChl a (bch) and carotenoid syntheses (crt), and light-harvesting and reaction center proteins (puf and puh). These genes were also found in the genome of ‘Ca. P. bacilliformis’ and other genomes of the CaP clade, while being completely absent in T. silvestris and V. flagellata (Table S4). The photosynthesis genes in the CaP clade were located in the photosynthetic gene cluster (PGC) and were divided between at least two subclusters separated by 20–40 kbp (Fig. 3A). Gene contents within the PGCs resembled those of other phototrophic alphaproteobacteria [43]. A characteristic feature of the PGC in the CaP clade is the lack of crtA. This encodes spheroidene monooxygenase, which catalyzes the synthesis of spheroidenone, a photosynthetic carotenoid. By contrast, the PGCs of the CaP clade contain the complete set of genes for synthesis of another carotenoid spirilloxanthin [44]. Consistent with the presence of gene sets for two photosynthetic pigments, absorption peaks indicative of BChl a and spirilloxanthin were detected in the spectral data of ‘Ca. P. spiralis’ (Fig. 3B). The purplish colony colors of ‘Ca. P. spiralis’ (Fig. 1D) and ‘Ca. P. bacilliformis’ [15] are typical for photosynthetic alphaproteobacteria [45] and are consistent with their photosynthetic potential. The yellow colony color of A. paucihalophilus (Table S1) suggests that yellow pigmented carotenoids are expressed in this species [46].
Evidence for AAnP in ‘Ca. Phycosocius’ and relatives.
![A Organization of PGC in the CaP clade. B Spectral analysis of methanol extract of photosynthesis pigments in ‘Ca. P. spiralis.’ Arrows and an arrowhead indicated putative absorbance peaks of spirilloxanthin [44] and bacteriochlorophyll a (BChl a), respectively. The protocol is described in Supplementary Methods. C ML phylogenetic tree of concatenated amino acid sequences of PufL and PufM. The tree was generated based on CPREV + G4 + F amino acid substitution model using a gap-free alignment (578 amino acids). ML bootstrap values (>80%) based on 1000 replicates are indicated at each branch.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ismecommun/3/1/10.1038_s43705-023-00228-6/3/m_43705_2023_228_fig3_html.jpeg?Expires=1748091959&Signature=ZQ81TXuSqn-BQu1QGO7Mv5VADuMvCtsFvWsBtWRLYDmFTn28xRVAHF3AJ1RnFa~ZQeyrYtwqIQq-MQAGUnn8qth~AZgQ1wqwmW5qtb~WdDvj5SW0BxYujiwWHeNTh4ymXK-A~BCq2xcz~S0X0vAzs1uD5iNxZeyfiQRvsJkG0Vhk-9z0GVfqxxVX7sJZ6UCA3IyByjGRSG8licZeBwgOJ9eqGiDU06TlDAp84pTuuvduOWihjMej28wjaCKHh7zbIEozcG0Blq-1fACvZ5QikAp3pWUVAyqDx0IBiZtBjsda-BYZelWyhLqP4yeezExPB0LxxwK-980VBUvxqaY3Gw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A Organization of PGC in the CaP clade. B Spectral analysis of methanol extract of photosynthesis pigments in ‘Ca. P. spiralis.’ Arrows and an arrowhead indicated putative absorbance peaks of spirilloxanthin [44] and bacteriochlorophyll a (BChl a), respectively. The protocol is described in Supplementary Methods. C ML phylogenetic tree of concatenated amino acid sequences of PufL and PufM. The tree was generated based on CPREV + G4 + F amino acid substitution model using a gap-free alignment (578 amino acids). ML bootstrap values (>80%) based on 1000 replicates are indicated at each branch.
Comparison of PGCs among members of the CaP clade indicated that each of the PGC genes were highly similar, and gene organization was highly conserved (Fig. 3A), suggesting common ancestry for the PGCs of the CaP clade. One exceptional difference was the lack of pufB in the UKL-13 genome (Fig. 3A). The absence of either pufA or pufB in the PGC of alphaproteobacteria was reported previously [47]. pufA and pufB encode light-harvesting protein B-875 α and β chains, respectively [48]. Given the structural similarities between pufA and pufB, the pufA product likely compensates for the function of the pufB product in ULKL-13. Notably, all the CaP genomes contained an aerobic oxidative cyclase gene, acsF, within the PGC as well as an oxygen-dependent coproporphyrinogen-III oxidase gene, hemF, both of which are hallmarks of AAnP [49, 50]. Their anaerobic counterparts, bchE and hemN, were not found in CaP genomes (Table S4). Calvin-Benson cycle enzymes (e.g., RuBisCO) were also absent in CaP genomes, whereas proteins involved in anaplerotic CO2 assimilation, phosphoenolpyruvate carboxylase, and malate dehydrogenase were present (Fig. 4 and Table S4). The latter two enzymes catalyze the formation of oxaloacetate and malate by incorporating the carboxyl group from bicarbonate into phosphoenolpyruvate and pyruvate, respectively [51]. A gene encoding a sodium-dependent bicarbonate transporter (Sbt) was found in the genomes of the CaP clade (Fig. 4 and Table S4), and this was likely involved in bicarbonate uptake for anaplerotic CO2 assimilation. Genes for lithoautotrophy were not found in the genomes of the CaP clade (Table S4). Together, these results suggest that CaP members are aerobic anoxygenic photoheterotrophic bacteria.
Metabolic modeling of CaP clade bacteria.
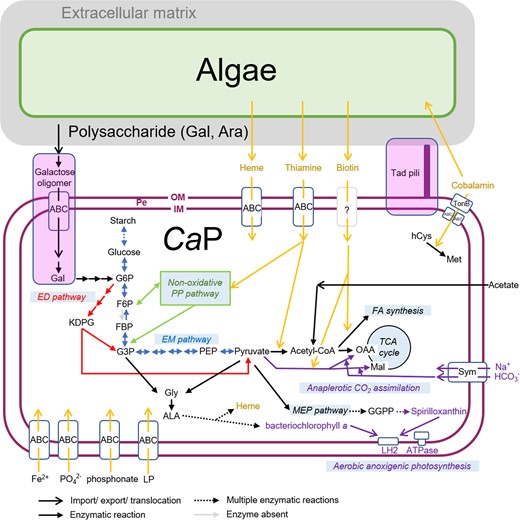
Features not shared in all CaP bacteria are indicated in the lavender box. Note that phosphofructokinase is absent in CaP bacteria (indicated by a gray arrow). PE periplasmic space, OM outer membrane, IM inner membrane, Gal galactose, Ara arabinose, G6P glucose-6-phosphate, F6P fructose 6-phosphate, FBP fructose-1,6-bisphosphate, G3P glyceraldehyde-3-phosphate, PEP phosphoenolpyruvate, OAA oxaloacetate, Mal malate, FA fatty acid, hCys homocysteine, Met methionine, Gly glycine, Ala alanine, GGPP geranylgeranyl diphosphate, Sym symporter, LP lipoprotein.
Comprehensive genome mining suggested that the PGC is rare in the Caulobacterales; only three type strains of Brevundimonas species harbor a PGC (Fig. 2A). This sporadicity can be explained either by HGT and/or by repeated loss of the gene cluster across the Caulobacterales. The phylogenomic tree (Fig. 2A) and pufLM genealogy of the Caulobacterales (Fig. 3C) were concordant. Recent phylogenetic analysis of Brevundimonas showed that Brevundimonas bacteroides was most closely related to B. variabilis, whereas B. subvibrioides was more closely related to other Brevundimonas species that lacked a PGC [52]. This favors the HGT of PGC between the CaP clade and Brevundimonas, although repeated losses of PGC in the Caulobacterales cannot be ruled out. The HGT hypothesis is further supported by frequent HGT of PGC in AAnP bacteria [53].
Metabolic features of ‘Ca. Phycosocius’ and relatives
Central carbon metabolism
All members of the CaP clade share the same nonoxidative pentose phosphate and glycolysis (Entner-Doudoroff) pathways, but the Embden-Meyerhof pathway is incomplete due to the absence of phosphofructokinase (Fig. 4). This type of glycolysis is often found in Gram-negative bacteria, in which the Embden-Meyerhof pathway is adopted for gluconeogenesis [54, 55] and is appropriate for CaP bacteria because photosynthetic ATP production can reduce the need for glycolysis [56].
Differences in culturability in synthetic media (Table S1) suggest that CaP bacteria have different nutritional requirements. Consistent with this, genes involved in galactose oligomer metabolism and transport (ganPQ, cycB, and msmX) [57] are present in ‘Ca. P. bacilliformis’ and A. paucihalophilus but are absent in ‘Ca. P. spiralis’ (Fig. 4, Table S4 and Fig. S4). The presence of these genes in ‘Ca. P. bacilliformis’ corresponds with galactose as the major component of the ECM in B. braunii [58]. ‘Ca. P. bacilliformis’ and A. paucihalophilus likely utilize galactose oligomers as one of their carbon sources, whereas ‘Ca. P. spiralis’ utilizes different carbon sources for growth.
Metabolism related to algal-bacterial interactions
Thiamine and biotin are essential B vitamins (VBs) for virtually all living organisms and are needed by enzymes involved in aldehyde and carboxyl group translocation, respectively. VB intercomplementation is one of the metabolic highlights of algal-bacterial mutualism [59]. CaP bacteria lack genes essential for thiamine and biotin synthesis. Growth experiments indicated that axenic B. braunii could grow without thiamine and biotin [14], suggesting that B. braunii can synthesize both these VBs de novo. Putative transporters involved in thiamine uptake were detected in the genomes of CaP bacteria (Table S4), but biotin transporters are yet to be identified. CaP bacteria obtain VBs from associated algae (Fig. 4), other coexisting microbes, or both. Consistent with the former hypothesis, biotin uptake from B. braunii by phycosphere bacteria has been observed previously [14], although the transporters involved in VB export in B. braunii remain to be identified.
Cobalamin functions as a cofactor for methyl-group transfer in several metabolic reactions. A hallmark of the cobalamin requirement is its role as a cofactor in methionine synthesis [60]. CaP bacteria encode a cobalamin-dependent methionine synthase (MetH) but do not encode cobalamin-independent methionine synthase (MetE). CaP bacteria also possess a set of genes for cobalamin transport, including TonB-dependent cobalamin transporter (BtuB), a periplasmic cobalamin-binding protein (BtuF), and two putative ABC transporters for cobalamin (BtuC and BtuD; Fig. 4 and Table S4). However, cobalamin synthesis genes were absent in CaP bacteria. These observations suggest that CaP bacteria utilize cobalamin produced by bacteria coexisting in the B. braunii phycosphere.
Members of the Roseobacter clade are known to synthesize bioactive secondary metabolites, such as tropodithietic acid and roseobacticide, to protect or kill associated algae [61]. Genes for tropodithietic acid and roseobacticide productions (tda) were not found in CaP bacteria (Table S4). In addition, polyketide synthase and nonribosomal peptide synthetases, which are responsible for the production of many bioactive compounds in bacteria, were not found in CaP bacteria. Nonribosomal peptide synthetases were found in several genomes of the Roseobacter clade [62, 63]. One of their products, indigoidine, was shown to act against several competing algal pathogens, including Vibrio spp. [64]. Interactions between marine algae and Roseobacter bacteria via these bioactive compounds may govern the rise and fall of marine algal blooms [61]. In this context, the lack of bioactive compounds, either beneficial or harmful to algae, in CaP bacteria is unexpected. This is indicative of a different type of interaction between CaP bacteria and competing microbes and associated algae compared to bacteria of the Roseobacter clade. Unlike the Roseobacter clade, genes for DMSP assimilation were not found in CaP bacteria (e.g., DmdA, DmdD, and DMSP lyases) [65] (Table S4). Consistent with this, substantial DMSP production was observed only rarely in freshwater algae [66]. DMSP metabolisms involving marine algal blooms and the Roseobacter clade were proposed to have an essential role in marine carbon and sulfur cycles [1]. As sulfur concentrations in freshwater are much lower than in the ocean [67], freshwater algae and associated CaP bacteria are less likely to have developed similar interactions via sulfur compounds.
QS and cell motility
Genes for QS were identified in all CaP bacterial genomes, as would be expected from the frequent observation of QS in bacterial pathogens and symbionts of eukaryotes [68]. Autoinducer synthase (LuxI) and response regulator (LuxR), a pair of proteins involved in QS [69], are present in ‘Ca. P. spiralis’ and ‘Ca. P. bacilliformis’, both of which have a green algal partner (Table S4). A. paucihalophilus and UKL-13, recovered from cyanobacteria, lack LuxI and contain only one LuxR. Such orphan LuxRs likely respond to exogenous QS molecules produced by coexisting microbes [68]. QS systems not only are involved in bacterial colonization of the algal surface [5] but also induce algal responses such as growth promotion or inhibition [70]. It is therefore possible that different modes of QS signaling might help to specify the algal partners of CaP bacteria, and this can be elucidated by examination of host specificities for the different CaP bacteria. Phylogenetic analyses revealed discordant and unresolved phylogenies of LuxI and LuxR (Fig. S5A, B), suggesting complex evolutionary histories of QS genes, including within the CaP clade. The phylogenetic complexity can most easily be explained by HGT of QS genes. This is consistent with previous research suggesting that HGT events involving QS genes are frequent in proteobacteria [68]. Therefore, HGT of QS genes might have altered CaP bacterial preferences for specific algae, which in turn may have led to the divergence of the CaP clade.
Host-bacterial colonization is governed by chemotaxis [71], and it is therefore reasonable to assume that the ‘aggressive’ motility of ‘Ca. P. spiralis’ toward B. braunii (Movie S2) occurs in response to molecules produced by the alga. This is supported by the presence of genes for chemotaxis in all CaP bacteria, namely, cheA, cheB, cheD, cheR, cheW, cheX, cheY, and cheZ in a che operon, and 2–4 chemotaxis receptor proteins (methyl-accepting chemotaxis proteins, MCP; Table S4). Different MCPs bind to different compounds, thereby directing chemotaxis [72]. Molecules implicated in bacterial chemotaxis toward algae include DMSP, amino acids, sugars, and carbohydrates [71]. At present, it is difficult to predict a chemical ligand for the CaP MCPs due to their low sequence similarities to characterized MCPs.
Genes involved in flagellum expression were found in all CaP bacteria (Table S4), although flagella were not observed in ‘Ca. P. spiralis.’ It is possible that flagella were expressed in hidden life stages, or that technical limitations hampered their observation. In the Caulobacterales, flagellar assembly is synchronized with the cell cycle, which is controlled by >20 regulatory proteins, including CtrA, PleC/D, and DivJ/K [73]. All CaP bacteria possess most of these regulatory genes (Table S4), suggesting that a similar regulatory pathway governs flagellar formation and the cell cycle.
Implications for algal-CaP bacterial interactions and their ecological impacts
Previous research showed that ‘Ca. P. bacilliformis’ promoted the growth of the host alga B. braunii during its growth, indicative of a mutualistic relationship [15]. However, preliminary growth experiments indicated that ‘Ca. P. spiralis’ had unstable effects (sometimes positive, sometimes neutral) on the growth of B. braunii (Fig. S6). It is possible that bacterial abundance relative to B. braunii abundance in the culture determines the fate of the algal-bacterial interaction [74]. The phycosphere of B. braunii in natural environments harbors many microbes in addition to ‘Ca. P. spiralis’ and likely has multiple alga-microbe interactions. For example, a nonaxenic culture of B. braunii Ba10 harbored at least four species in addition to ‘Ca. Phycosocius’ (Fig. S7). The impact of ‘Ca. P. spiralis’ on the growth of B. braunii might therefore vary in a microbiome-dependent manner. One suggestion is that the Roseobacter clade and flavobacteria synergistically remineralize phytoplankton-derived organic matter in marine environments [1], and similar interactions might occur between CaP and other bacteria.
Given the smaller genome size of ‘Ca. P. spiralis’ compared to other CaP bacteria, our initial hypothesis was that ‘Ca. P. bacilliformis’ was ancestral to ‘Ca. P. spiralis.’ However, pangenome analyses suggest independent evolution for each lineage. Increasing the nutritional and metabolic dependence of ‘Ca P. spiralis’ on associated algae may have accelerated its genome reduction, transforming ‘Ca P. spiralis’ from a mutualist into a commensal. Both theoretical and empirical data suggest that symbiotic relationships are not stable and oscillate along the continuum between mutualism and parasitism [41]. In this context, CaP bacteria fluctuate between mutualism and commensalism.
One characteristic gene set that is absent in ‘Ca. P. spiralis’ but present in other members of the CaP clade is the Tad gene cluster, which encodes tight adherence (Tad) pili (a class of type IV pili) and associated regulatory proteins (Fig. 5A) [75]. Tad pili are widespread in bacteria, including Caulobacter, a distant relative of the CaP clade [76], and are involved in surface attachment and biofilm formation [75]. Swimming and sessile cells of ‘Ca. P. spiralis’ were observed off and on host algae, respectively (Movie S2). This shift in cell motility was reminiscent of the ‘swim-or-stick’ lifestyle of algal-associated members of the Roseobacter clade [4], which use Tad pili to attach to algal partners [77]. SEM imaging captured a putative Tad pilus-like structure in ‘Ca. P. bacilliformis’ in coculture with B. braunii (Fig. 5B). This suggests that all CaP bacteria except ‘Ca. P. spiralis’ use Tad pili, allowing for a Roseobacter-like ‘swim-or-stick’ lifestyle. The loss of a Tad gene cluster in ‘Ca. P. spiralis’ likely coincides with its acquisition of spiral motility and ECM burrowing, which renders attachment to the host surface no longer necessary (Fig. 5C). ‘Ca. P. spiralis’ therefore adopts a unique strategy for its ‘swim-or-stick’ lifestyle that does not employ Tad pili for algal surface attachment. The Tad gene cluster in UKL-13 may be incomplete due to the pseudogenization of three genes, including cpeE and cpeD (Fig. 5A), and Tad pili might thus be absent in UKL-13. If so, UKL-13 might have a spiral-shaped morphology similar to that of ‘Ca P. spiralis.’
Tad pili and their possible role in algal surface attachment.
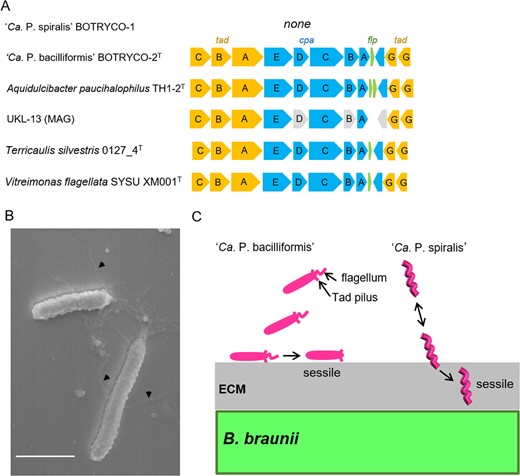
A Tad gene cluster in the CaP clade. Gray boxes indicate putative pseudogenes. B SEM image shows putative Tad pili in ‘Ca. P. bacilliformis’. Scale bar, 1 μm. C Schematic of different modes of attachment of two ‘Ca. Phycosocius’ species onto/into the algal surface.
Several features of the CaP clade, such as association with algal colonies and the metabolic potential of AAnP, point to the clade having partial ecophysiological similarity to the marine Roseobacter clade. Of these features, the shared presence of AAnP is of particular interest. A close correlation between marine AAnP bacteria and occurrence of phytoplankton blooms has been reported. The reason for this correlation is not yet fully understood [1, 46]; however, it has been suggested that freshwater bloom-associated AAnP bacteria play a role in maintaining redox balance in blooms via thiosulfate reduction and dissimilatory sulfate reduction pathways [12, 78]. CaP bacteria are unlikely to have a similar role because the genes involved in these processes (sox) are absent in CaP bacteria (Table S4). Field surveys indicate that AAnP bacterial abundance is positively correlated to temporal phytoplankton bloom dynamics [79], and CaP members likely comprise some of these AAnP bacteria. AAnP bacteria show higher growth rates than nonphotosynthetic heterotrophs when optimal light is available due to their capacity to reduce respiration in order to produce ATP [80]. AAnP bacteria are thus preferentially selected in organic matter-rich and sunlit environments like algal blooms in both marine and freshwater environments. This suggests that sunlight exposure owing to the association with blooms is one of the driving forces for the evolution of AAnP in the CaP clade. A recent report indicated that lineages in Gemmatimonadata associated with phytoplankton are photoheterotrophs [81]. Many species of the alphaproteobacterium genus Methylobacterium, which are often found on plant leaves, also possess genes for AAnP [47]. Photoheterotrophy might thus be one of the derived features in bacteria most closely associated with phototrophic organisms such as algae and plants. Conversely, close association with algae exposes associated bacteria to higher levels of sunlight-derived UV exposure. To cope with the DNA damage from UV exposure, bacteriorhodopsin-containing bacteria in marine environments, for example, possess more photolyase genes than bacteria armed with light-screening pigments [82]. With the exception of ‘Ca P. spiralis,’ which has a reduced genome, this is also the case with CaP bacteria, which have three photolyase genes. Nonphotosynthetic CaP relatives have one or no photolyase genes (Table S4).
In conclusion, this study highlights the ecological and physiological characteristics of members of the CaP clade, which have adopted photoheterotrophic and VB auxotrophic growth in close association with algal blooms in freshwater. At least two species of the CaP clade, ‘Ca. P. bacilliformis’ [15] and ‘Ca. P. spiralis,’ stick to the algal colony and subsequently become sessile. With the exception of VB auxotrophy, these ecophysiological features resemble those of the marine Roseobacter clade. In this context, the CaP clade is a freshwater counterpart of the Roseobacter clade. However, the CaP clade is genetically far less diverse (99% 16S rDNA sequence divergence) than the marine Roseobacter clade, which includes >50 genera encompassing ~89% 16S rDNA sequence diversity [1, 83]. This suggests that the CaP clade may have derived much later than the Roseobacter clade, although the possibility cannot be excluded that the diversity of the CaP clade was underestimated in this study owing to the poor availability of CaP genomes. Future studies include (1) unraveling the genomic and metabolic diversity of the CaP clade by including more strains; (2) exploring molecular interactions between CaP bacteria and their algal partners, including QS signaling; (3) investigating detailed population dynamics of CaP bacteria in relation to algal bloom dynamics; and (4) elucidating the metabolic regulation and balance between heterotrophic and phototrophic energy acquisitions of CaP bacteria. These studies will shed light on the ecological impact of the CaP clade on carbon cycling during algal blooms in freshwater environments. These studies will also allow examination of the hypothesis that evolution of photoheterotrophy in bacteria was driven by close association with phototrophic organisms, including microalgae.
Acknowledgements
We thank Nanda Kyaw Thu for the isolation of BOTRYCO-1, Mina Nomizu for the guidance of TEM sample preparation, Masahiro Koide for taking video images, and Iwane Suzuki and Makoto M. Watanabe for the use of their laboratory facilities. We also thank Katherine Cook and David Hambright for providing the raw data of their work on worldwide Microcystis blooms. This work is partly supported by Next-generation Energies for Tohoku recovery (NET) from Reconstruction Agency.
Data Availability
Genome sequence data of ‘Ca. P. spiralis’ BOTRYCO-1 are available at GenBank under the accession number BPFZ01000000.
Author contributions
YT designed the research, YT performed isolation, culture, light microscopy, pigment, whole genome, and bioinformatics analyses, HY performed NGS, MY and AK performed electron microscopy, YO performed CARD-FISH and 16S amplicon analyses, YT and YO wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
References
Poindexter JS Caulobacter. In: Trujillo ME, Dedysh S, DeVos P, Hedlund B, Kämpfer P, Rainey FA, et al. (eds.) Bergey’s Manual of Systematics of Archaea and Bacteria. 2015; https://doi.org/10.1002/9781118960608.gbm00792.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43705-023-00228-6.
Author notes
The original online version of this article was revised: Due to incorrect labels for supplementary files.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}