





Abstract
Emiliania huxleyi is a unicellular micro-alga that forms massive oceanic blooms and plays key roles in global biogeochemical cycles. Mounting studies demonstrate various stimulatory and inhibitory influences that bacteria have on the E. huxleyi physiology. To investigate these algal-bacterial interactions, laboratory co-cultures have been established by us and by others. Owing to these co-cultures, various mechanisms of algal-bacterial interactions have been revealed, many involving bacterial pathogenicity towards algae. However, co-cultures represent a significantly simplified system, lacking the complexity of bacterial communities. In order to investigate bacterial pathogenicity within an ecologically relevant context, it becomes imperative to enhance the microbial complexity of co-culture setups. Phaeobacter inhibens bacteria are known pathogens that cause the death of E. huxleyi algae in laboratory co-culture systems. The bacteria depend on algal exudates for growth, but when algae senesce, bacteria switch to a pathogenic state and induce algal death. Here we investigate whether P. inhibens bacteria can induce algal death in the presence of a complex bacterial community. We show that an E. huxleyi-associated bacterial community protects the alga from the pathogen, although the pathogen occurs within the community. To study how the bacterial community regulates pathogenicity, we reduced the complex bacterial community to a five-member synthetic community (syncom). The syncom is comprised of a single algal host and five isolated bacterial species, which represent major bacterial groups that are naturally associated with E. huxleyi. We discovered that a single bacterial species in the reduced community, Sulfitobacter pontiacus, protects the alga from the pathogen. We further found that algal protection from P. inhibens pathogenicity is a shared trait among several Sulfitobacter species. Algal protection by bacteria might be a common phenomenon with ecological significance, which is overlooked in reduced co-culture systems.
Introduction
Micro-algae have evolved with bacteria in their surroundings for millions of years [1]. This co-evolution gave rise to various interactions between algae and bacteria that influence the involved microorganisms and their environment [2, 3]. Research on natural bacterial communities that are associated with algae elucidated important aspects of the ecology and dynamics of algal-bacterial communities, and the influence of such communities on microbial physiology. Studies revealed the structure of bacterial communities associated with algae in the environment, the key bacterial members within these communities, and specificity between bacteria and algal hosts [4–7]. Algal derived metabolites were shown to be key in shaping the community structure and in turn, microbial physiology [7–10].
Environmental research was complemented by studies of controlled and reduced laboratory algal-bacterial co-culture systems [10–27]. Such co-cultures allow a detailed understanding of the mechanisms underlying interactions between specific algae and bacteria. The study of algal-bacterial co-cultures revealed that bacteria from the Roseobacter group [28] interact with various algal groups such as dinoflagellates, diatoms and coccolithophores, and that these interactions span from mutualism to pathogenicity [11, 14, 16, 18–27].
Studies on algal-bacterial co-cultures deepened our understanding of specific interactions between algae and bacteria, while environmental research shed light on natural algal-associated bacterial communities. Both approaches have limitations; the environmental approach is restricted in its ability to offer mechanistic insights and the co-culture approach cannot encompass the complexity of the marine environment. While some results that were achieved in the lab align with observations at sea [29], there are instances where laboratory findings contrast with environmental observations. As an example, algal-bacterial co-cultures often revealed pathogenic behavior of Roseobacter bacteria towards their algal hosts [14, 16, 19–24]. Yet many of these Roseobacters were originally isolated from natural communities associated with algae at sea, in which the pathogenic behavior that results in algal death is not observed [30–32]. Therefore, a model system with increased microbial complexity is needed in order to resolve the pathogenic nature of various Roseobacters and its relevance in the environment.
In recent years, the use of synthetic communities was highly effective in the study of complex host-microbiome interactions. Synthetic communities, or in short syncoms, are commonly used in gut and root microbiome research, due to their ability to capture some of the natural complexity yet in a simplified and controlled manner [33]. Syncoms are an intermediate model system between co-cultures and natural communities that enable the study of host-bacteria as well as bacteria-bacteria interactions in the context of a community. Syncoms have allowed the identification of keystone species and functions in the community and unveiled microbiome-mediated phenotypes [34].
Emiliania huxleyi is the most abundant coccolithophore species in modern oceans [35]. Different strains of these algae exhibit high genome variability which allows them to thrive in different environmental conditions [36]. During the spring and summer time, populations of E. huxleyi form massive annual blooms that can reach cell densities of up to 108 cells/l in temperate and subpolar regions [37]. These micro-algae contribute significantly to the oceanic carbon cycle through both photosynthesis and the formation of calcium carbonate scales [37].
In this study, we isolated bacteria associated with an E. huxleyi strain and constructed an algal-bacterial syncom to investigate bacterial behavior, with a focus on the pathogenicity of bacteria within the community. The results showed that the bacterial pathogen Phaeobacter inhibens was unable to trigger algal death when it was part of a bacterial consortium. Using the novel syncom, a single bacterial species from the abundant Sulfitobacter genus was identified as a protector of the algae from pathogenic P. inhibens bacteria. Several other species from the Sulfitobacter genus were also found to possess the ability to protect algae from the pathogen.
The bacterial ‘protector’ phenotype might be as common as the wide distribution of algal-associated Sulfitobacter species, and therefore, is of ecological significance. These findings highlight the centrality of bacteria-bacteria interactions in shaping host-microbiome dynamics and expand current knowledge on the possible roles of bacteria within a community.
Results
The alga Emiliania huxleyi harbors a bacterial community that includes members of the Roseobacter group
Often, bacterial pathogenicity is observed when the alga E. huxleyi is co-cultured with a single bacterial species that is naturally found in the algal microbiome of environmental samples [14, 18–20, 22–24]. Therefore, we sought to test the growth dynamics of a non-axenic algal strain and identify the bacteria it harbors. The algal strain E. huxleyi CCMP1516, which was isolated from the environment along with an associated bacterial community, was observed to grow in the presence of bacteria without experiencing algal death (Fig. 1A). As a comparison, the axenic isogenic strain E. huxleyi CCMP2090, which lacks bacteria, exhibited similar growth in pure culture but experienced algal death when co-cultured with pathogenic P. inhibens DSM17395 bacteria (Fig. 1A). Algal death in the co-culture resembled previous reports [20, 23, 24].
Algae grow with a bacterial community of marine bacteria.

A Growth of the non-axenic E. huxleyi CCMP1516, axenic E. huxleyi CCMP2090 and E. huxleyi CCMP2090 with the bacterium P. inhibens DSM17395 was monitored for 21 days. As can be seen, the presence of the bacterium P. inhibens DSM17395 triggered algal death, while the bacterial community that is associated with the algae (in strain E. huxleyi CCMP1516) did not impact algal growth. Each data point consists of 3 biological replicates, error bars designate ± SD. B The different bacterial taxa associated with E. huxleyi CCMP1516. Relative abundance was calculated as percentage of 16S rRNA gene sequences along three time points of algal growth. Each time point consists of 3 biological replicates (a, b and c). Bacteria were classified to the genus or family level. Two taxa with a relative abundance lower than 1% across all samples were pooled together and designated as “Others”. These taxonomies were classified as belonging to the Piscirickettsiaceae family and the Alteromonadales order.
To identify the bacteria associated with E. huxleyi CCMP1516, a 16S rRNA gene amplicon sequencing analysis was performed at three different time points representing different physiological stages of the alga: mid exponential phase (day 7), late exponential phase (day 10), and early stationary phase (day 14) (Fig. 1A). This analysis revealed that all bacteria in the community are common heterotrophic marine bacteria from the Proteobacteria phylum, belonging to five different taxonomic families including Alteromonadaceae, Marinobacteraceae, Rhodobacteraceae, Sphingomonadaceae and Piscirickettsiaceae (Fig. 1B). Two OTUs were identified as belonging to the Rhodobacteraceae family (Roseobacter group [28]), with one OTU belonging to the Sulfitobacter genus and the other not identified at the genus level. The limited diversity observed at the phylum level, as compared to environmental samples [4–7], may be attributed to the historical isolation of the algal strain E. huxleyi CCMP1516 several decades ago by the National Center for Marine Algae and Microbiota (NCMA). Consequently, its associated bacterial community likely reflects a population that has been selectively adapted to laboratory culture conditions over the years. However, it is worth noting that the bacterial composition of this algal strain does encompass the two most prevalent classes of bacteria, namely alphaproteobacteria and gammaproteobacteria, found in the deep chlorophyll maxima (DCM) and sea surface layers (SRF) of environmental samples [38]. Additionally, the presence of members of the Roseobacter group within the bacterial population is particularly noteworthy due to their known pathogenic interactions with E. huxleyi [14, 18–20, 22–24].
The pathogenicity of the P. inhibens isolate is executed in the absence of the bacterial community
The algal strain E. huxleyi CCMP1516 exhibits uninterrupted growth (Fig. 1A), suggesting that the bacteria associated with it are non-pathogenic. To determine whether indeed bacteria in the community are non-pathogenic, bacteria from E. huxleyi CCMP1516 were isolated, and each isolate was co-cultured with the isogenic axenic strain E. huxleyi CCMP2090. Six different bacterial isolates were retrieved based on colony morphology (see materials and methods), and their influence on algal growth was monitored along time (Fig. 2). One of these isolates, isolate #1, induced algal death in co-culture (Fig. 2). To identify the pathogenic isolate and the other isolates, a PacBio whole genome sequencing analysis was performed. We found that the 6 isolates belong to the taxonomic families of Alteromonadaceae, Marinobacteraceae, Rhodobacteraceae and Sphingomonadaceae (see Table 2 and materials and methods). The isolated bacteria represent four of the five taxonomic families associated with E. huxleyi CCMP1516 that we identified earlier using 16S rRNA gene sequencing (Fig. 1B). The bacterial isolates were identified at the species level (see Table 2, materials and methods, and Supplementary Table S1). The pathogenic isolate was identified as a P. inhibens strain, a well-studied E. huxleyi pathogen [20, 23, 24]. These results reveal that the isolated P. inhibens strain does have pathogenic capabilities towards algae in co-culture, however, the pathogenicity appears to be suppressed by the bacterial community in a yet unknown manner. From here on, the identified isolates are denoted by the species name followed by the mark i (for example- P. inhibens i.). Experiments involving algae were performed using axenic cultures of strain E. huxleyi CCMP2090. To enhance readability, this strain will be referred to as ‘algae’ throughout the manuscript.
Growth of algae in co-cultures with isolated bacteria.
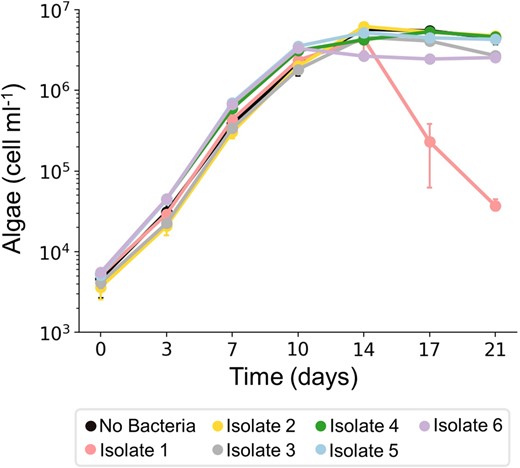
Growth of axenic E. huxleyi CCMP2090 was monitored in co-cultures, each time with a different isolate, for 21 days. As can be seen, algal death was triggered only in co-cultures with isolate #1. Each data point consists of 3 biological replicates, error bars designate ± SD. The growth of isolate #1 in the co-culture (P. inhibens i.) is presented in Supplementary Fig. S2C.
A synthetic community allows the study of community-regulation of pathogenicity
To study how a bacterial community regulates the pathogenicity of P. inhibens i. towards E. huxleyi algae, we aimed to create a synthetic community (syncom) that includes an algal host and the bacterial isolates. This new, reduced and controlled system allowed us to gain insight into the factors that govern the pathogenicity of P. inhibens i. in the context of a bacterial community. The syncom was designed to meet two criteria: (I) algae should grow with the bacterial population without induction of algal death, and (II) the pathogenic P. inhibens i. should be present and growing in the community over time. Combinations of bacteria were cultured with axenic E. huxleyi CCMP2090 until a five-member bacterial consortium was established that met these criteria. The five bacterial species in the syncom included P. inhibens i., Erythrobacter flavus i., Marinobacter sp. i., Sulfitobacter pontiacus i. and Alteromonas macleodii i. Algae indeed grew over time with the five bacterial isolates without experiencing algal death, in contrast to the abrupt algal death observed in co-cultures with only P. inhibens i. (Fig. 3A). To monitor the presence and growth of the different bacteria in the community, particularly P. inhibens i., we used a quantitative PCR (qPCR) method that we established for bacteria in the syncom (see the material and methods section for detailed information). The accurate genomes that we sequenced allowed us to identify a unique genomic locus in each isolate and generate specific primers for each species in the community (see Table 3 and material and methods). We note that our qPCR method does not differentiate live and dead bacteria. Yet, calibration experiments with pure bacterial cultures showed good agreement between counts of colony forming units (CFUs) and the qPCR method (Supplementary Fig. S1). Thus, the qPCR method provides a good approximation of total abundance of different bacteria in the syncom. To monitor bacterial growth in the syncom, we chose three time points along the algal growth curve which represent different algal physiological states: mid exponential phase (day 7), late exponential phase (day 10), and early stationary phase (day 14) (Fig. 3A). Results of bacterial qPCR monitoring showed that P. inhibens i. was present and growing in the syncom over time (Fig. 3B), reaching its highest density during algal early stationary phase (day 14). The other four isolates were also present in the community and exhibited various growth dynamics over time. Overall, the syncom recapitulated the phenotype observed in the original bacterial population of E. huxleyi CCMP1516, where the presence of pathogenic bacteria does not trigger abrupt algal demise. The constrained and regulated composition of the novel algal-bacterial syncom offers an opportunity to explore how the bacterial cohort provides protection to algae against pathogens within the community.
Algae grow in a syncom that contains the isolated bacterial pathogen.
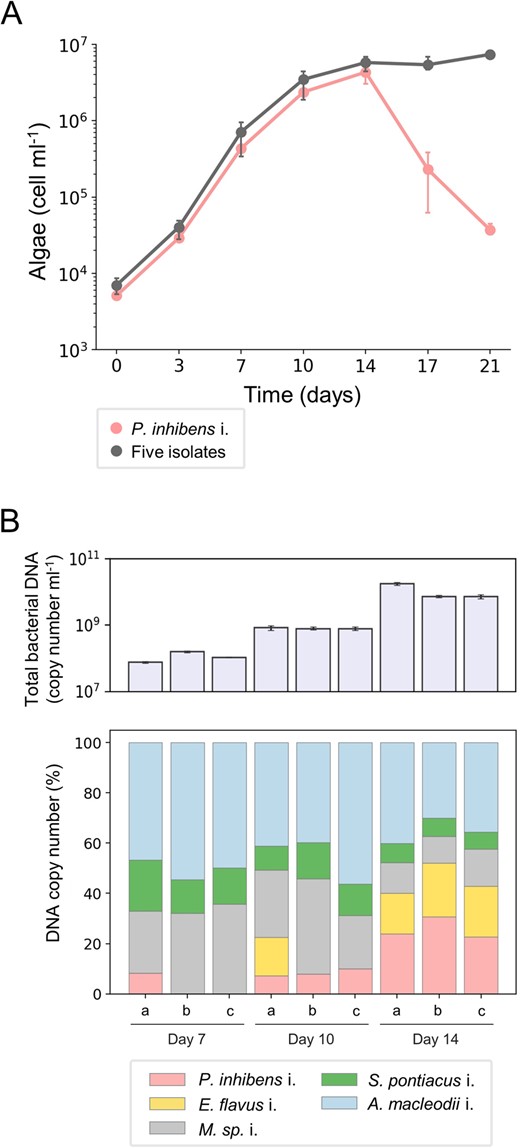
A Growth of E. huxleyi CCMP2090 with P. inhibens i. or with the five-member bacterial consortium (syncom) was monitored for 21 days. The data show that algal death is not triggered by the bacterial consortium although it contains the bacterial pathogen. Each data point consists of 3 biological replicates, error bars designate ± SD. The corresponding growth of P. inhibens i. in co-culture can be seen in Supplementary Fig. S2C. B Upper panel – total bacterial copy number was quantified using a specific DNA locus for each species in the syncom (see materials and methods) along three time points of algal growth. Each time point consists of 3 biological replicates (a, b and c), error bars designate ± SD. Lower panel - the relative abundance of the five isolated bacteria in the syncom was calculated as a percentage of DNA copy number (derived from the total DNA copy number presented in the upper panel) at three time points during algal growth. Of note, the relative abundance of the P. inhibens i. appears to increase although pathogenicity is not executed, evident by no algal death. Each time point consists of 3 biological replicates (a, b and c).
A single bacterial species in the syncom alleviates the pathogenicity of the P. inhibens isolate
The syncom is a reduced and controlled model system in which a bacterial community protects an algal host from a pathogen. To determine whether protection is mediated by the entire community or by a single bacterial species, we systematically omitted from the syncom each bacterial species one at a time, while the pathogen remained present (Fig. 4A). We found that when S. pontiacus i. was absent from the syncom, the pathogenic P. inhibens i. induced algal death. These observations suggest that a single bacterial species in the community is responsible for algal protection. To further validate these findings, we established tri-cultures in which algae were cultured with the pathogen and one additional bacterial species from the syncom (Fig. 4B). Our data showed that in tri-cultures containing algae with the pathogen and S. pontiacus i., no pathogenicity was observed and algae survived. These results demonstrate that a bacterial species belonging to the Sulfitobacter genus has the capacity to protect algae from a pathogen.
Algal growth with reduced bacterial consortium.
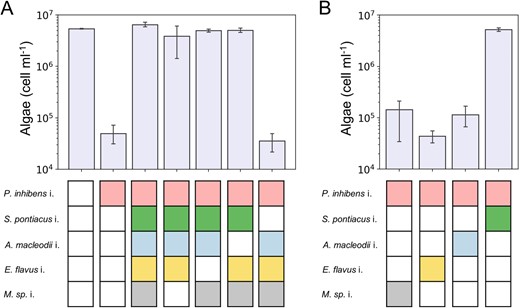
AE. huxleyi CCMP2090 were cultivated with reduced bacterial consortium in which each species was omitted systematically, giving rise to four different 4-member bacterial communities. Shown are algal cell counts on day 21 of growth. In control cultures, algae were cultivated with no bacteria, only with the pathogen P. inhibens i., and with the full five-member syncom. The data show that omission of S. pontiacus i. from the syncom results in algal death. B Algae were cultivated with the pathogen P. inhibens i. and with one additional species from the syncom. These cultures of algae with two bacterial species are called tri-cultures. Shown are algal cell counts on day 21 of the tri-cultures. These results show that S. pontiacus i. bacteria protect algae from pathogenic P. inhibens i. bacteria. The colored rectangles under each bar denote the bacterial species introduced into the algal culture. A white rectangle indicates that the species was not added. Each data point consists of 3 biological replicates, error bars designate ± SD.
Algal protection from bacterial pathogenicity is common in the Sulfitobacter genus
We sought to determine the prevalence of the protection capability among the Sulfitobacter genus by testing 10 species from this genus. Eight species were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ), and we isolated two species from E. huxleyi CCMP1516 (S. pontiacus i. and S. geojensis i.). To confirm that these various Sulfitobacter species are themselves non-pathogenic towards E. huxleyi, we co-cultured each Sulfitobacter species with algae (Fig. 5A). None of the Sulfitobacter species tested were found to be pathogenic. We then monitored tri-cultures in which algae were cultivated with the pathogenic P. inhibens i. and one Sulfitobacter species (Fig. 5B). Our results revealed that six Sulfitobacter species were able to protect algae from the pathogen, while four species were neutral and did not prevent algal death. Importantly, the cell density of protector or neutral Sulfitobacter species was not influenced by the presence of the pathogen (Supplementary Fig. S2A, B). However, the cell density of the pathogen was slightly reduced by both protector and neutral Sulfitobacter species regardless of algal fate (Supplementary Fig. S2C). These results suggest that the mechanism of protection is independent from the observed changes in cell density of the pathogen. A phylogenetic tree of the Sulfitobacter species was constructed to test whether the protection phenotype has a common evolutionary ancestor (Supplementary Fig. S3). This analysis showed that the protector phenotype is scattered throughout the tree and that protector Sulfitobacter species do not have a distinct, close phylogenetic relationship with each other. Next, we tested whether the genomes of the Sulfitobacter species could be used to identify functions related to the protection phenotype (Supplementary Fig. S4). We focused on traits that are present in all protector Sulfitobacter species but absent from all neutral Sulfitobacter species. The functional analysis did not reveal common features that are unique to protector Sulfitobacter species. While the mechanism of Sulfitobacter protection remains to be determined, this capacity is common among this genus and may therefore present an advantage for harboring Sulfitobacter species in the algal microbiome.
Algal protection in the Sulfitobacter genus.
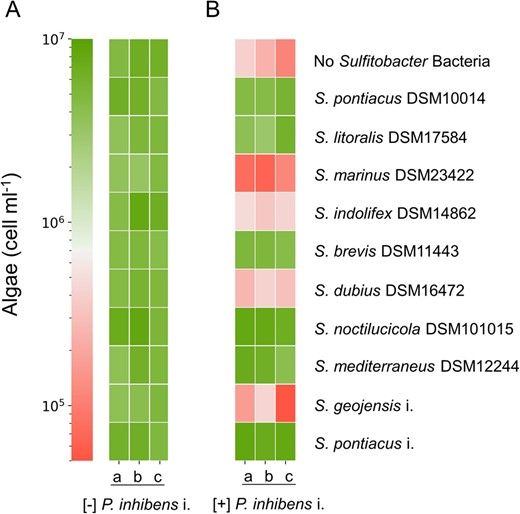
AE. huxleyi CCMP2090 cultured with various Sulfitobacter species. BE. huxleyi CCMP2090 cultured with various Sulfitobacter species and with the pathogen P. inhibens i. Shown are algal cell counts on day 21 of growth as indicated by the color scale. Each culture was tested using three biological replicates (a, b, c). The corresponding growth of selected Sulfitobacter species in co-cultures and tri-cultures can be seen in Supplementary Fig. S2A, B.
Sulfitobacter species are commonly associated with phytoplankton in the environment
To determine whether Sulfitobacter species tend to co-occur with microalgae in the environment, we analyzed data from the Tara expedition using the Ocean Barcode Atlas web server [39]. We analyzed the most abundant Roseobacters that are found co-occurring with phytoplankton. Since photosynthesizing microbes inhabit the surface waters and the DCM, we focused our analyses on samples that were retrieved from these water layers and were collected using a small pore size (0.22–3 µm) suitable for capturing bacteria. The data indicated that Sulfitobacter species were identified in 34 out of 65 surface samples and in 21 out of 37 DCM samples (Supplementary Fig. S5A), with a relative abundance ranging 0.1–14% of the Roseobacters species in these samples (Supplementary Fig. S5B). These data show that Sulfitobacter species commonly occur in waters known to inhabit phytoplankton, yet their average relative abundance is quite low. To determine whether low Sulfitobacter abundance confers protection in laboratory settings, we monitored algal cultures in which protector bacteria had an initial abundance ranging from 0.01 to 1% relative to the pathogenic bacteria abundance (Supplementary Fig. S6). Our results showed that protector Sulfitobacter bacteria could still protect algae even at extremely low initial abundance compared to the pathogen. These data indicate that the protection capability is robust even when protector bacteria are outnumbered by pathogenic bacteria.
Sulfitobacter species are often present in environmental marine samples and algal microbiomes [5, 7, 17, 32, 40–42]. Our data point to the advantageous properties of Sulfitobacter species in algal microbiomes where they potentially protect algae from bacterial pathogenicity.
Discussion
In this study, we present an exploration of algal-bacterial interactions through the use of an environmentally inspired synthetic community (syncom). This approach allowed us to investigate intricate interactions with increased bacterial complexity and to uncover previously unknown ecological roles within these interactions. While simplified algal-bacterial co-cultures are valuable for studying the mechanisms underlying these interactions, the enhanced complexity of syncoms [43] allowed the detection of the novel protector role.
Our results reveal a key bacterial species, S. pontiacus i., which protects algae from a pathogen present within the community. Our syncom is environmentally relevant, as the bacteria were isolated from the microbiome of an algal strain (Fig. 1B, Supplementary Table S1). The reduced and controlled nature of the syncom allowed us to systematically eliminate bacteria from the community and identify the protection phenotype in a single bacterial species (Fig. 4A, B). Importantly, due to the high level of genomic diversity in E. huxleyi strains [36], our results could represent a strain-specific interaction between algae and bacteria. Notably, previous research has demonstrated that specific characteristics of algal-bacterial interactions play a role in multiple algal-bacterial pairs. For instance, the production of the phytohormone indole-3-acetic acid (IAA) by algal-associated bacteria has been found to impact interactions with various phytoplankton species, including coccolithophores [20], diatoms [17], and chlorophytes [44]. Moreover, the pathogenicity of bacteria towards algae is not exclusively determined by specific algal-bacterial pairs. This has been substantiated by studies illustrating the ability of certain bacterial species to exhibit pathogenic behavior towards various algal strains [20, 22, 45]. Further studies on the protection phenotype using other E. huxleyi strains and additional phytoplankton groups will provide insights into the universality of this interaction.
Bacteria protecting their host is a known phenomenon in various natural systems [33, 46, 47]. Our study is the first to show this behavior in algal-bacterial interactions. Mechanisms of host protection by bacteria include competition between bacteria through antibiotic secretion [48], promotion of host defense systems [49], and interference with quorum sensing molecules required for virulence of pathogens [50]. In the current study, both protector and neutral bacteria slightly reduced the cell density of the pathogen (Supplementary Fig. S2C). Therefore, this inhibitory effect cannot explain the protection phenotype. The lack of commonalities in the phylogenetic and functional analyses among protector bacteria (Supplementary Figs. S3 and S4) suggests that the regulation of the protector phenotype could therefore be on a different cellular level (e.g., gene variance, transcription, protein expression). We made initial attempts to elucidate possible Sulfitobacter protection mechanisms that interfere with known pathogenicity routes of P. inhibens such as secretion of the IAA [20] and nitric oxide [45]. While our initial investigations have not yet revealed the precise mechanism of Sulfitobacter protection, ongoing research in our laboratory aims to decipher this complex interplay.
Considering the ubiquity of Sulfitobacter species in the marine environment (Supplementary Fig. S5), the observed protection behavior, which was found to be common among the species we tested (Fig. 5), may be of ecological significance. The ability of these bacteria to provide protection even at low initial abundances (Supplementary Fig. S6) raises questions about their role in maintaining algal health and influencing population dynamics. Our study, together with previous findings [22], reveals the multifaceted interactions of Sulfitobacter species with the coccolithophore microalga E. huxleyi—including pathogenic [22], neutral, and protector roles. This complexity prompts inquires into the factors governing the nature of Sulfitobacter-algal interactions and whether the environmental context can modulate these interactions. With advances in in situ genetic manipulation techniques [51], hypotheses about the protection mechanism and its environmental significance could be explored.
As our oceans experience global transformations, populations of marine microorganisms shift as well [52]. Perturbations in environmental factors such as salinity [53], pH [54] and temperature [55] could potentially disrupt the balance between pathogens and protectors in natural microbial communities. Controlled laboratory settings provide a platform to investigate the protection phenotype under conditions mimicking anticipated oceanic changes, advancing our understanding of algal-bacterial ecophysiology in changing environments.
Materials and methods
Strains and general growth conditions
The algal strains of E. huxleyi CCMP1516 (non-axenic) and CCMP2090 (axenic re-isolate of CCMP1516) were purchased from the National Center for Marine Algae and Microbiota (Bigelow Laboratory for Ocean Sciences, Maine, USA). Algae were grown in L1 medium without Na2SiO3 (L1-Si), based on filtered and autoclaved seawater (FSW) according to Guillard and Hargraves [56]. Algae were grown in standing cultures in a growth room at 18 °C under a light/dark cycle of 16/8 h. Illumination intensity during the light period was 130 μmol photons m−2 s−1. Absence of bacteria in axenic algal cultures was monitored weekly both by plating on ½ YTSS plates and under the microscope.
The bacterial strains of Phaeobacter inhibens DSM 17395, Sulfitobacter noctilucicola DSM 101015, Sulfitobacter indolifex DSM 14862, Sulfitobacter dubius DSM 16472, Sulfitobacter litoralis DSM 17584, Sulfitobacter pontiacus DSM 10014, Sulfitobacter marinus DSM 23422, Sulfitobacter mediterraneus DSM 12244 and Sulfitobacter brevis DSM 11443 were purchased from the German collection of microorganisms and cell cultures (DSMZ, Braunschweig, Germany). All bacteria were grown in Marine Broth 2216 medium (MB) (Difco, USA) in liquid cultures and agar plates. Phaeobacter inhibens DSM 17395 was grown in ½ YTSS medium (2 g yeast extract, 1.25 g tryptone and 20 g sea salt per liter. Sigma-Aldrich, USA) in liquid cultures and agar plates.
Algal counts
Algal growth in cultures was monitored by a CellStream CS-100496 flow cytometer (Merck, Darmstadt, Germany), using 561 nm excitation and 702 nm emission. For each sample 50,000 events were recorded. Algal cells were gated according to event size and fluorescence intensity.
Algal culture conditions
E. huxleyi was cultured as follows: algal cell concentrations from one week-old E. huxleyi culture were determined as described above. An inoculum of 104 algal cells was introduced into 30 ml of L1-Si medium and incubated as described above. In all experiments the bacterial strains were inoculated on the fourth day of algal cultivation and this point was designated as day 0, including control experiments where the alga was cultivated axenically.
Inoculation of bacteria to axenic E. huxleyi CCMP2090 was performed as follows: Bacteria were plated from a glycerol stock on either ½ YTSS or MB plates according to Table 1 and incubated at 30 °C for 48 h. Biomass from the plate was then transferred to liquid ½ YTSS or MB and incubated shaking at 30 °C for 48 h. One ml from the liquid bacterial culture were centrifuged at 10,000 rpm for 1 min and washed with FSW three times. The resulting washed pellet was resuspended in 1 ml FSW and diluted to an OD600 of 0.01. The diluted sample was then further diluted by a factor indicated in Table 1. From the final dilution, 20 µl were introduced into 30 ml of algal culture grown for four days as described above. Inoculation of multiple bacteria to an algal culture (syncom or tri-culture) followed the same protocol as described above with the addition of a bacterial mixing step: from the final dilution, 20 µl from each bacteria were transferred to an eppendorf tube and mixed. The inoculation volume to the algal culture corresponded to the number of desired bacteria (i.e. if two bacteria are inoculated, 40 µl was transferred to the algal culture). The bacterial starting cell concentration was verified by plating and counting colony forming units (CFUs), resulting in 5-30 CFUs per 20 µl inoculum.
List of bacteria used in this study and culturing conditions.
| Strain | Origin | Reference genome | Growth medium | Dilution factor | |
|---|---|---|---|---|---|
| 1 | Phaeobacter inhibens DSM 17395 | DSMZ, Braunschweig, Germany | GCF_000154765.2 | ½ YTSS | x102 |
| 2 | Sulfitobacter noctilucicola DSM 101015 | DSMZ, Braunschweig, Germany | GCF_014197555.1 | Marine broth | x102 |
| 3 | Sulfitobacter indolifex DSM 14862 | DSMZ, Braunschweig, Germany | GCF_022788655.1 | Marine broth | x103 |
| 4 | Sulfitobacter dubius DSM 16472 | DSMZ, Braunschweig, Germany | GCF_900113435.1 | Marine broth | x103 |
| 5 | Sulfitobacter litoralis DSM 17584 | DSMZ, Braunschweig, Germany | GCF_900103185.1 | Marine broth | x103 |
| 6 | Sulfitobacter pontiacus DSM 10014 | DSMZ, Braunschweig, Germany | GCF_900106935.1 | ½ YTSS | x103 |
| 7 | Sulfitobacter marinus DSM 23422 | DSMZ, Braunschweig, Germany | GCF_900116285.1 | Marine broth | x103 |
| 8 | Sulfitobacter mediterraneus DSM 12244 | DSMZ, Braunschweig, Germany | GCF_003054325.1 | Marine broth | x103 |
| 9 | Sulfitobacter brevis DSM 11443 | DSMZ, Braunschweig, Germany | GCF_900112755.1 | Marine broth | x103 |
| 10 | Phaeobacter inhibens i (iR1) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520018 | ½ YTSS | x102 |
| 11 | Erythrobacter flavus i (iR2) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520027 | ½ YTSS | x103 |
| 12 | Marinobacter sp. i (iR3) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520020 | ½ YTSS | x102 |
| 13 | Sulfitobacter pontiacus i (iR4) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520022 | ½ YTSS | x103 |
| 14 | Alteromonas macleodii i (iR5) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7519869 | ½ YTSS | x104 |
| 15 | Sulfitobacter geojensis i (iR6) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520006 | Marine broth | x103 |
| Strain | Origin | Reference genome | Growth medium | Dilution factor | |
|---|---|---|---|---|---|
| 1 | Phaeobacter inhibens DSM 17395 | DSMZ, Braunschweig, Germany | GCF_000154765.2 | ½ YTSS | x102 |
| 2 | Sulfitobacter noctilucicola DSM 101015 | DSMZ, Braunschweig, Germany | GCF_014197555.1 | Marine broth | x102 |
| 3 | Sulfitobacter indolifex DSM 14862 | DSMZ, Braunschweig, Germany | GCF_022788655.1 | Marine broth | x103 |
| 4 | Sulfitobacter dubius DSM 16472 | DSMZ, Braunschweig, Germany | GCF_900113435.1 | Marine broth | x103 |
| 5 | Sulfitobacter litoralis DSM 17584 | DSMZ, Braunschweig, Germany | GCF_900103185.1 | Marine broth | x103 |
| 6 | Sulfitobacter pontiacus DSM 10014 | DSMZ, Braunschweig, Germany | GCF_900106935.1 | ½ YTSS | x103 |
| 7 | Sulfitobacter marinus DSM 23422 | DSMZ, Braunschweig, Germany | GCF_900116285.1 | Marine broth | x103 |
| 8 | Sulfitobacter mediterraneus DSM 12244 | DSMZ, Braunschweig, Germany | GCF_003054325.1 | Marine broth | x103 |
| 9 | Sulfitobacter brevis DSM 11443 | DSMZ, Braunschweig, Germany | GCF_900112755.1 | Marine broth | x103 |
| 10 | Phaeobacter inhibens i (iR1) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520018 | ½ YTSS | x102 |
| 11 | Erythrobacter flavus i (iR2) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520027 | ½ YTSS | x103 |
| 12 | Marinobacter sp. i (iR3) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520020 | ½ YTSS | x102 |
| 13 | Sulfitobacter pontiacus i (iR4) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520022 | ½ YTSS | x103 |
| 14 | Alteromonas macleodii i (iR5) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7519869 | ½ YTSS | x104 |
| 15 | Sulfitobacter geojensis i (iR6) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520006 | Marine broth | x103 |
List of bacteria used in this study and culturing conditions.
| Strain | Origin | Reference genome | Growth medium | Dilution factor | |
|---|---|---|---|---|---|
| 1 | Phaeobacter inhibens DSM 17395 | DSMZ, Braunschweig, Germany | GCF_000154765.2 | ½ YTSS | x102 |
| 2 | Sulfitobacter noctilucicola DSM 101015 | DSMZ, Braunschweig, Germany | GCF_014197555.1 | Marine broth | x102 |
| 3 | Sulfitobacter indolifex DSM 14862 | DSMZ, Braunschweig, Germany | GCF_022788655.1 | Marine broth | x103 |
| 4 | Sulfitobacter dubius DSM 16472 | DSMZ, Braunschweig, Germany | GCF_900113435.1 | Marine broth | x103 |
| 5 | Sulfitobacter litoralis DSM 17584 | DSMZ, Braunschweig, Germany | GCF_900103185.1 | Marine broth | x103 |
| 6 | Sulfitobacter pontiacus DSM 10014 | DSMZ, Braunschweig, Germany | GCF_900106935.1 | ½ YTSS | x103 |
| 7 | Sulfitobacter marinus DSM 23422 | DSMZ, Braunschweig, Germany | GCF_900116285.1 | Marine broth | x103 |
| 8 | Sulfitobacter mediterraneus DSM 12244 | DSMZ, Braunschweig, Germany | GCF_003054325.1 | Marine broth | x103 |
| 9 | Sulfitobacter brevis DSM 11443 | DSMZ, Braunschweig, Germany | GCF_900112755.1 | Marine broth | x103 |
| 10 | Phaeobacter inhibens i (iR1) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520018 | ½ YTSS | x102 |
| 11 | Erythrobacter flavus i (iR2) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520027 | ½ YTSS | x103 |
| 12 | Marinobacter sp. i (iR3) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520020 | ½ YTSS | x102 |
| 13 | Sulfitobacter pontiacus i (iR4) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520022 | ½ YTSS | x103 |
| 14 | Alteromonas macleodii i (iR5) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7519869 | ½ YTSS | x104 |
| 15 | Sulfitobacter geojensis i (iR6) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520006 | Marine broth | x103 |
| Strain | Origin | Reference genome | Growth medium | Dilution factor | |
|---|---|---|---|---|---|
| 1 | Phaeobacter inhibens DSM 17395 | DSMZ, Braunschweig, Germany | GCF_000154765.2 | ½ YTSS | x102 |
| 2 | Sulfitobacter noctilucicola DSM 101015 | DSMZ, Braunschweig, Germany | GCF_014197555.1 | Marine broth | x102 |
| 3 | Sulfitobacter indolifex DSM 14862 | DSMZ, Braunschweig, Germany | GCF_022788655.1 | Marine broth | x103 |
| 4 | Sulfitobacter dubius DSM 16472 | DSMZ, Braunschweig, Germany | GCF_900113435.1 | Marine broth | x103 |
| 5 | Sulfitobacter litoralis DSM 17584 | DSMZ, Braunschweig, Germany | GCF_900103185.1 | Marine broth | x103 |
| 6 | Sulfitobacter pontiacus DSM 10014 | DSMZ, Braunschweig, Germany | GCF_900106935.1 | ½ YTSS | x103 |
| 7 | Sulfitobacter marinus DSM 23422 | DSMZ, Braunschweig, Germany | GCF_900116285.1 | Marine broth | x103 |
| 8 | Sulfitobacter mediterraneus DSM 12244 | DSMZ, Braunschweig, Germany | GCF_003054325.1 | Marine broth | x103 |
| 9 | Sulfitobacter brevis DSM 11443 | DSMZ, Braunschweig, Germany | GCF_900112755.1 | Marine broth | x103 |
| 10 | Phaeobacter inhibens i (iR1) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520018 | ½ YTSS | x102 |
| 11 | Erythrobacter flavus i (iR2) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520027 | ½ YTSS | x103 |
| 12 | Marinobacter sp. i (iR3) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520020 | ½ YTSS | x102 |
| 13 | Sulfitobacter pontiacus i (iR4) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520022 | ½ YTSS | x103 |
| 14 | Alteromonas macleodii i (iR5) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7519869 | ½ YTSS | x104 |
| 15 | Sulfitobacter geojensis i (iR6) | Isolated from Emiliania huxleyi CCMP1516 | https://doi.org/10.5281/zenodo.7520006 | Marine broth | x103 |
16S rRNA amplicon sequencing and data analysis
E. huxleyi CCMP1516 was grown for 14 days as described above. On days 7, 10 and 14 three samples were harvested for genomic DNA extraction. Twenty five ml from the algal culture were centrifuged at 10,000 rpm for 20 min. The algal-bacterial pellet was then subjected to DNA extraction using the Wizard Genomic DNA Purification kit (Promega, Madison, USA) following the manufacturer protocol for bacterial DNA extraction.
Genomic DNA was PCR-amplified with primers CS1_515F and CS2_926R (ACACTGACGACATGGTTCTACAGTGTGYCAGCMGCCGCGGTAA and TACGGTAGCAGAGACTTGGTCTCCGYCAATTYMTTTRAGTTT, respectively. Underlined regions represent linker sequences. Modified from Walters et al. 2015 [57]) targeting the V4-V5 variable regions of the microbial small subunit ribosomal RNA genes. Amplicons were generated using a two-stage PCR amplification protocol as described previously [58]. The primers contained 5’ common sequence tags (known as common sequence 1 and 2, CS1 and CS2). First stage PCR amplifications were performed in 10 µl reactions in 96-well plates, using repliQa HiFi ToughMix (Quantabio, USA). PCR conditions were 98 °C for 2 min, followed by 28 cycles of 98 °C for 10 s, 50 °C for 1 s and 68 °C for 1 s. Subsequently, a second PCR amplification was performed in 10 µl reactions in 96-well plates using repliQa HiFi ToughMix. Each well received a separate primer pair with a unique 10-base barcode, obtained from the Access Array Barcode Library for Illumina (Fluidigm, South San Francisco, CA; Item# 100-4876). One microliter of PCR product from the first stage amplification was used as template for the 2nd stage, without cleanup. Cycling conditions were 98 °C for 2 min, followed by 8 cycles of 98 °C for 10 s, 60 °C for 1 s and 68 °C for 1 s. Libraries were then pooled and sequenced with a 15% phiX spike-in on an Illumina MiSeq sequencer employing V3 chemistry (2×300 base paired-end reads). Library preparation and sequencing were performed at the Genomics and Microbiome Core Facility (GMCF) at Rush University.
Microbiome bioinformatics were performed with QIIME2 2021.11 [59]. Quality of raw sequence data was checked using FastQC and the sequences were merged using PEAR. Adapter sequences were removed using the cutadapt algorithm and reads less than 300 bp were removed. Merged and length-filtered sequences were quality filtered using the q2-demux plugin followed by denoising with DADA2 (via q2-dada2) [60]. Taxonomy was assigned to ASVs using the q2-feature-classifier [61] taxonomy classifier against the SILVA 138 SSU non-redundant-99% reference database [62]. Reagent microbial contaminants were identified and removed using decontam package based on the prevalence of the ASVs in the reagent negative blank controls using default parameters. Samples were rarefied to a depth of 10,000 sequences/sample for downstream analysis.
Bacterial isolation
Bacteria were isolated from E. huxleyi CCMP1516 as follows: algae were grown as described above. On day 7, 10 and 14 of algal growth a 1 ml sample from the algal culture was serially diluted in FSW. Twenty microliters from each dilution were plated on ½ YTSS and MB agar plates using glass beads. Six single colonies with distinct morphologies were picked and re-streaked on agar plates. Single colony picking and re-streaking was performed sequentially three times. Bacterial biomass was then transferred to 20% glycerol stocks and stored at −80 °C.
PacBio library preparation, sequencing, read processing and taxonomic classification
For PacBio long-read sequencing, multiplexed microbial libraries were prepared, using SMRTBell Express Prep Kit 2.0 (Pacific Biosciences, Menlo Park, CA). DNA was extracted from the six bacterial isolates using Wizard Genomic DNA Purification kit (Promega, Madison, USA) following the manufacturer protocol for bacterial DNA extraction, resulting in 6 genomic DNA samples. A single PacBio sequencing library was constructed in order to sequence the entire genomes of the bacterial isolates. The library was used for SMRTbell template preparation as described in the protocol (Pacific Biosciences, Menlo Park, CA). The resulting SMRTbell template was annealed to the sequencing polymerase, using a Sequel binding kit 3.0. Sequencing was conducted with a 12pM on-plate concentration using a Sequel I System in CLR mode and a single SMRT Cell (1 M; PacBio). PacBio raw reads (polymerase reads) were demultiplexed and assembled using the SMRT Link software (v8.0).
The phylogenetic classification of all bacterial isolates in the current study was determined using two approaches: 16S rRNA gene sequences were analyzed using the online portal of SILVA SINA database [63], assigning classification at the genus level (Table 2). Entire genomes were analyzed to the species level using the GTDB-Tk v1.7.0 [64] software toolkit for assigning objective taxonomic classifications to bacterial and archaeal genomes (Table 2). The two different approaches yielded identical results, confirming the identity of the isolated bacteria at the species level.
Taxonomic classification of bacterial isolates using two approaches.
| SILVA SINA (16S rRNA) [58] | GTDB-Tk (whole genome) [59] | Isolate identifier in manuscript | |||
|---|---|---|---|---|---|
| Classification | Identitya (%) | Classification | Identitya (%) | ||
| 1 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter; | 99.85 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter;inhibens DSM 16374 | 97.93 | Phaeobacter inhibens i. |
| 2 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter; | 100 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter;flavus DSM 16421 | 97.93 | Erytherobacter flavus i. |
| 3 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; | 99.45 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; sp. EhC06 | 99.99 | Marinobacter sp. i. |
| 4 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.92 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;pontiacus DSM 10014 | 97.28 | Sulfitobacter pontiacus i. |
| 5 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas; | 99.79 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas;macleodii ATCC 27126 | 97.86 | Alteromonas macleodii i. |
| 6 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.49 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;geojensis MM-124 | 96.32 | Sulfitobacter geojensis i. |
| SILVA SINA (16S rRNA) [58] | GTDB-Tk (whole genome) [59] | Isolate identifier in manuscript | |||
|---|---|---|---|---|---|
| Classification | Identitya (%) | Classification | Identitya (%) | ||
| 1 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter; | 99.85 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter;inhibens DSM 16374 | 97.93 | Phaeobacter inhibens i. |
| 2 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter; | 100 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter;flavus DSM 16421 | 97.93 | Erytherobacter flavus i. |
| 3 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; | 99.45 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; sp. EhC06 | 99.99 | Marinobacter sp. i. |
| 4 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.92 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;pontiacus DSM 10014 | 97.28 | Sulfitobacter pontiacus i. |
| 5 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas; | 99.79 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas;macleodii ATCC 27126 | 97.86 | Alteromonas macleodii i. |
| 6 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.49 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;geojensis MM-124 | 96.32 | Sulfitobacter geojensis i. |
The sequence identity is computed as the number of shared bases (common base-column pairs) divided by the length of the query sequence.
Taxonomic classification of bacterial isolates using two approaches.
| SILVA SINA (16S rRNA) [58] | GTDB-Tk (whole genome) [59] | Isolate identifier in manuscript | |||
|---|---|---|---|---|---|
| Classification | Identitya (%) | Classification | Identitya (%) | ||
| 1 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter; | 99.85 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter;inhibens DSM 16374 | 97.93 | Phaeobacter inhibens i. |
| 2 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter; | 100 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter;flavus DSM 16421 | 97.93 | Erytherobacter flavus i. |
| 3 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; | 99.45 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; sp. EhC06 | 99.99 | Marinobacter sp. i. |
| 4 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.92 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;pontiacus DSM 10014 | 97.28 | Sulfitobacter pontiacus i. |
| 5 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas; | 99.79 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas;macleodii ATCC 27126 | 97.86 | Alteromonas macleodii i. |
| 6 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.49 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;geojensis MM-124 | 96.32 | Sulfitobacter geojensis i. |
| SILVA SINA (16S rRNA) [58] | GTDB-Tk (whole genome) [59] | Isolate identifier in manuscript | |||
|---|---|---|---|---|---|
| Classification | Identitya (%) | Classification | Identitya (%) | ||
| 1 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter; | 99.85 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Phaeobacter;inhibens DSM 16374 | 97.93 | Phaeobacter inhibens i. |
| 2 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter; | 100 | Bacteria;Proteobacteria;Alphaproteobacteria;Sphingomonadales;Erythrobacteraceae;Erythrobacter;flavus DSM 16421 | 97.93 | Erytherobacter flavus i. |
| 3 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; | 99.45 | Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Marinobacteraceae;Marinobacter; sp. EhC06 | 99.99 | Marinobacter sp. i. |
| 4 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.92 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;pontiacus DSM 10014 | 97.28 | Sulfitobacter pontiacus i. |
| 5 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas; | 99.79 | Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Alteromonadaceae;Alteromonas;macleodii ATCC 27126 | 97.86 | Alteromonas macleodii i. |
| 6 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter; | 99.49 | Bacteria;Proteobacteria;Alphaproteobacteria;Rhodobacterales;Rhodobacteraceae;Sulfitobacter;geojensis MM-124 | 96.32 | Sulfitobacter geojensis i. |
The sequence identity is computed as the number of shared bases (common base-column pairs) divided by the length of the query sequence.
Monitoring bacterial growth in co-cultures
Bacterial growth in co-cultures was evaluated by sampling co-cultures at different time points, as indicated above. Samples were serially diluted and plated on ½ YTSS or MB plates, depending on the bacteria. CFUs were counted and the concentration in the sampled culture was calculated.
Monitoring bacterial growth in the syncom using quantitative PCR (qPCR)
Monitoring of bacteria in the syncom with algae was conducted using a qPCR method based on DNA copy number. A species-specific set of primers was designed for each bacterial species using the structural variant calling tool of PacBio SMRT link software (Table 3).
Species-specific primer sequences for bacterial identification and bacterial genome length.
| Bacterial isolate | Forward unique primer | Reverse unique primer | Genome length (bp) |
|---|---|---|---|
| Phaeobacter inhibens i. | GCGGCATGATTACGCAGATG | CGAGGATTTGCAGATTGGGC | 4,083,657 |
| Erythrobacter flavus i. | GCAGGACAGGCCGTATACAT | CCGATCTTCGCCTTCTCCAA | 3,013,997 |
| Marinobacter sp. i. | ACTGCTTGTCCATACCCTGC | AGCGGGTCCTGTTCTACACA | 4,660,702 |
| Sulfitobacter pontiacus i. | GCCATCCGCGATCAAAACAA | TACCGTTCAGCTGCCAGAAG | 3,509,446 |
| Alteromonas macleodii i. | CGCCCACTAAACGAAAATGGA | TACAAAACCCGCTGTGTGC | 4,712,091 |
| Sulfitobacter geojensis i. | CCCAGCGCATCAAGTCTGAA | CCTAGCTGCGGCTGTATTTG | 4,278,117 |
| Bacterial isolate | Forward unique primer | Reverse unique primer | Genome length (bp) |
|---|---|---|---|
| Phaeobacter inhibens i. | GCGGCATGATTACGCAGATG | CGAGGATTTGCAGATTGGGC | 4,083,657 |
| Erythrobacter flavus i. | GCAGGACAGGCCGTATACAT | CCGATCTTCGCCTTCTCCAA | 3,013,997 |
| Marinobacter sp. i. | ACTGCTTGTCCATACCCTGC | AGCGGGTCCTGTTCTACACA | 4,660,702 |
| Sulfitobacter pontiacus i. | GCCATCCGCGATCAAAACAA | TACCGTTCAGCTGCCAGAAG | 3,509,446 |
| Alteromonas macleodii i. | CGCCCACTAAACGAAAATGGA | TACAAAACCCGCTGTGTGC | 4,712,091 |
| Sulfitobacter geojensis i. | CCCAGCGCATCAAGTCTGAA | CCTAGCTGCGGCTGTATTTG | 4,278,117 |
Species-specific primer sequences for bacterial identification and bacterial genome length.
| Bacterial isolate | Forward unique primer | Reverse unique primer | Genome length (bp) |
|---|---|---|---|
| Phaeobacter inhibens i. | GCGGCATGATTACGCAGATG | CGAGGATTTGCAGATTGGGC | 4,083,657 |
| Erythrobacter flavus i. | GCAGGACAGGCCGTATACAT | CCGATCTTCGCCTTCTCCAA | 3,013,997 |
| Marinobacter sp. i. | ACTGCTTGTCCATACCCTGC | AGCGGGTCCTGTTCTACACA | 4,660,702 |
| Sulfitobacter pontiacus i. | GCCATCCGCGATCAAAACAA | TACCGTTCAGCTGCCAGAAG | 3,509,446 |
| Alteromonas macleodii i. | CGCCCACTAAACGAAAATGGA | TACAAAACCCGCTGTGTGC | 4,712,091 |
| Sulfitobacter geojensis i. | CCCAGCGCATCAAGTCTGAA | CCTAGCTGCGGCTGTATTTG | 4,278,117 |
| Bacterial isolate | Forward unique primer | Reverse unique primer | Genome length (bp) |
|---|---|---|---|
| Phaeobacter inhibens i. | GCGGCATGATTACGCAGATG | CGAGGATTTGCAGATTGGGC | 4,083,657 |
| Erythrobacter flavus i. | GCAGGACAGGCCGTATACAT | CCGATCTTCGCCTTCTCCAA | 3,013,997 |
| Marinobacter sp. i. | ACTGCTTGTCCATACCCTGC | AGCGGGTCCTGTTCTACACA | 4,660,702 |
| Sulfitobacter pontiacus i. | GCCATCCGCGATCAAAACAA | TACCGTTCAGCTGCCAGAAG | 3,509,446 |
| Alteromonas macleodii i. | CGCCCACTAAACGAAAATGGA | TACAAAACCCGCTGTGTGC | 4,712,091 |
| Sulfitobacter geojensis i. | CCCAGCGCATCAAGTCTGAA | CCTAGCTGCGGCTGTATTTG | 4,278,117 |
PCR analyses validated that the primers do not amplify genomic DNA of E. huxleyi CCMP2090 and indeed amplify only genomic DNA of the bacterial species of interest. qPCR was conducted in 384 well plates, using SensiFAST SYBR Lo-ROX Kit (Meridian Bioscience) in a QuantStudio 5 (384-well plate) qPCR cycler (Applied Biosystems, Foster City, CA, USA). Accuracy of primers for determining DNA copy numbers was confirmed in two manners: (1) genomic DNA from the different bacteria was mixed in known DNA concentrations and ratios and served as template for the qPCR protocol. (2) Each bacterial species was grown in pure culture to an OD600 of 0.2, the culture was plated on an agar plate for CFU counting and in parallel DNA was extracted from the cultures and served as template for the qPCR protocol (Supplementary Fig. S1). The qPCR program ran according to enzyme requirements for 32 cycles. Results were analyzed using a relative standard curve using the QuantStudio 5 software. Primer efficiencies were determined by qPCR amplification of known DNA concentrations. Only primer pairs with a minimum of 80% efficiency were selected.
The syncom of E. huxleyi CCMP2090 with the six bacterial isolates was cultured for 14 days as described above. On days 7, 10 and 14, triplicate samples were harvested for genomic DNA extraction as described above. The DNA from all samples was diluted to 1 ng/µl and served as template for the qPCR reaction as described above. To convert the results from ng DNA to DNA copy number the following formula was used (see also Table 3):
6.022 × 1023 – Avogadro’s number
109 – Conversion from g to ng
650 – Average weight of a bp (g/mole)
Monitoring bacterial growth in tri-cultures
Growth monitoring of P. inhibens i. and the Sulfitobacter species cultured with E. huxleyi CCMP2090 in tri-cultures was conducted using selective plates, as follows: both bacteria grow on MB plates, but only P. inhibens i. grows on plates that contain sucrose as the sole carbon source (sucrose 5.5 mM, NH4Cl 5 mM and NaSO4 33 mM dissolved in 1 L L1-Si medium). Tri-cultures were sampled at different time points, as indicated, and the samples were serially diluted and plated on an MB plate and on a defined sucrose plate. The MB plate was used to count CFUs of both species and the sucrose plate was used to count CFUs of P. inhibens i. The Sulfitobacter CFUs were calculated by subducting the CFUs on the sucrose-containing plate from the CFUs on the MB plates.
Phylogenetic analysis of Sulfitobacter species
SSU rRNA gene sequences, representing reference Sulfitobacter species, were obtained from SILVA [62] and the novel isolates. The above sequences were used as queries in a blastn [65] search against the SILVA 138.1 SSU non-redundant-99% reference database. The seven best matches, that were not the query itself, were retained for each query, and were organized in a single non-redundant fasta file, along with the query sequences. The sequences were aligned using the L-ins-i algorithm, as implemented in MAFFT [66], and the alignments were trimmed with TrimAl [67], to exclude alignment positions containing more than 40% gaps. Regions with missing data at the beginning or the end of the alignment were trimmed manually. The most likely tree was found through a heuristic search starting with a maximum parsimony tree and the GTR + GAMMA model of sequence evolution. Branch support was computed through a rapid bootstrap approach using 100 replicates. The maximum likelihood tree reconstruction was carried out with RAxML 8.2.12 [68]. Three visualization was carried out with ETE3 [69].
Variable functional categories in Sulfitobacter species
To study the functional variation between the Sulfitobacter species, we searched for diagnostic orthologous groups. We additionally summarized the KEGG pathways and COG categories, comprising of orthologous groups which were missing from at least one of the genomes. To recover orthologous groups we first obtained the protein fasta files of the publically available genomes (Protector strains: S. brevis DSM11443, GCF_900112755.1; S. litoralis DSM17584, GCF_900103185.1; S. mediterraneus DSM12244, GCF_003054325.1; S. noctilucicola DSM101015, GCF_014197555.1; S. pontiacus DSM10014, GCF_900106935.1. Neutral strains: S. dubius DSM16472, GCF_900113435.1; S. indolifex DSM14862, GCF_022788655.1; S. marinus DSM23422, GCF_900116285.1) and predicted protein coding genes in the novel isolates genomes of the protector S. pontiacus i., https://doi.org/10.5281/zenodo.7520022, and neutral S. geojensis i., https://doi.org/10.5281/zenodo.7520006, using Prokka [70]. Orthologous groups were determined with OrthoFinder 2 [71] using the default inflation parameter value (1.5). Functional annotation of the protein sequences was carried out with eggNOG mapper 2 [72] using the eggNOG DB version 5 [73]. Orthologous groups which were missing from at least one of the genomes were listed. For each such orthologous group, each category or pathway was represented by the copy number of that orthologous groups, in each of the samples. Then, the occurrence of each category or pathway was summarized across orthologous groups. The resulting table was log transformed (supplementary file “functional_categories_log_counts.tsv”).
Environmental occurrence of the Sulfitobacter genus
Environmental data was obtained using the Ocean Barcode Atlas (OBA) server [39]. Data was retrieved from sampling points within the DCM and SRF and the filter size was limited to size fraction of 0.22–3 µm. The tool for community ecological analysis was used to search for the taxonomic family “Rhodobacteraceae” in the “Tara Oceans miTag 16S 18S version 2” dataset. Automatically generated datasets of the most abundant “Rhodobacteraceae” groups at the genus level were manually inspected for the presence and abundance of the Sulfitobacter genus.
Acknowledgements
We thank Dr. Amir Szitenberg from the Nancy and Stephen Grand Israel National Center for Personalized Medicine for the phylogenetic and functional analysis of Sulfitobacter genomes. We are thankful to Dr. Stephan Green from the Genomics and Microbiome Core Facility at Rush University, Chicago, IL for the 16S rRNA amplicon sequencing and bioinformatics analysis of algal associated bacterial community. We thank Dr. Hadas Keren-Shaul, Dr. Merav Kedmi and Dr. David Pilzer from the life sciences core facilities at the Weizmann institute for the help with the library preparation and sequencing using PacBio technology. Finally, we are grateful for valuable input from Dr. Martin Sperfeld and thank all members of the Segev lab for insightful comments and discussions.
Author contributions
RB and ES designed the study. RB and NO performed and analyzed experiments. RB and ES wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
The Weizmann SAERI program (RB). European Research Council, ERC StG 101075514 (ES). The Israeli Science Foundation, ISF 947/18 (ES). The Minerva Foundation with funding from the Federal German Ministry for Education and Research (ES). The de Botton Center for Marine Science (ES).
Data Availability
Strains used in this study can be found in Table 1. Isolated bacteria are available from the corresponding author on reasonable request. Genome sequences were deposited in zenodo.org, the accession numbers can be found in Table 1. Primers designed for species identification using qPCR are available in Table 3. Functional data used for Supplementary Fig. S4 is available in the supplementary file “functional_categories_log_counts.tsv”. Environmental data used for Supplementary Fig. S5 was retrieved from https://oba.mio.osupytheas.fr/ocean-atlas/ using the “community ecological analysis” function. Default criteria were used on the database “Tara Oceans miTAGs 16S and 18S version 2” to search for Rhodobacteraceae taxonomy.
Competing interests
The authors declare no competing interests.
References
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43705-023-00311-y.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}