





Abstract
Patients suffering from inflammatory bowel diseases (IBDs) express increased mucosal levels of transforming growth factor (TGF)-β compared with non-IBD controls. SMAD7 negatively regulates TGF-β signaling. An earlier study aiming to target Smad7 showed a lack of clinical benefit. It remains unknown whether inhibition of SMAD7 is beneficial in specific settings of IBD. We evaluated the effect of Smad7 deficiency on inflammation, fibrogenesis, and wound healing.
For the initiation of fibrosis in Smad7-/- (Smad7Δex-I) CD-1 mice, the dextran sodium sulfate–induced chronic colitis model and the heterotopic transplantation model of fibrosis were used. Wound closure of fibroblasts from Smad7-/- mice was determined using culture inserts and electric cell-substrate impedance sensing in vitro.
In dextran sodium sulfate–induced chronic colitis, Smad7 deficiency was associated with ameliorated inflammation, as evidenced by decreased clinical score, histological score, and myeloperoxidase activity. Absence of SMAD7 decreased T-cell accumulation in colonic tissue and tumor necrosis factor (TNF) mRNA expression levels. Smad7-/- mice showed a significant increase in hydroxyproline and collagen content, as well as ColIVa1 mRNA expression. Wild type mice transplanted with terminal ileum from Smad7-/- mice in the heterotopic animal model for intestinal fibrosis showed a significant increase in collagen content and protein expression of α-smooth muscle actin.
Smad7 deficiency is associated with a decrease in intestinal inflammation and an increase in fibrosis. Targeting SMAD7 constitutes a potential new treatment option for IBD; progression of disease-associated fibrosis should be considered.
Lay Summary
We evaluated the effect of Smad7 deficiency on inflammation and fibrogenesis. Smad7 deficiency was associated with ameliorated inflammation and increased collagen deposition. When targeting Smad7 as therapeutic strategy in IBD, potential initiation or aggravation of fibrosis should be considered.
What is already known?
SMAD7 negatively regulates TGF-β signaling: furthermore, IBD patients express increased TGF-β mucosal levels compared with non-IBD controls.
What is new here?
Smad7 deficiency was associated with ameliorated inflammation and increased collagen deposition.
How can this study help patient care?
Targeting Smad7 as therapeutic strategy in IBD, however, potential initiation or aggravation of fibrosis should be considered.
Introduction
The prevalence of inflammatory bowel disease (IBD) in the industrialized world is approximately 0.5%. Inflammatory bowel disease comprises 2 main conditions, namely ulcerative colitis (UC) and Crohn’s disease (CD), and is characterized by a chronic inflammation of the intestinal wall. Crohn’s disease represents a chronic transmural inflammatory condition, although inflammation in UC only affects the mucosa. Not all IBD patients have severe intestinal inflammation. The clinical presentation at onset of IBD varies, and the disease course is highly variable, ranging from a quiescent disease with a long period of remission to a chronic refractory inflammation in patients suffering from CD1 and UC.2 Persistent mucosal tissue damage is one of the main features of a severe course of IBD. Excessive tissue repair promotes fibrosis, impairs gastrointestinal function, and is a common clinical problem in patients with CD. Fibrogenesis can also occur in long-standing UC, leading to the formation of strictures.3 Fibrosis is increasingly recognized as an important cause of morbidity and mortality in patients with IBD. Intestinal fibrosis leads to stricture formation due to thickening of the intestinal wall in 30% to 50% of patients with CD,4 and approximately 80% of patients with stricturing CD will eventually require surgery.4 Fibrogenesis is also associated with impaired absorption.5 Wound healing following tissue damage requires an exquisite balance between multiple pro- and antifibrotic stimuli on extracellular matrix (ECM)-producing cells (eg, activated myofibroblasts).6
Transforming growth factor (TGF)-β1 is a cytokine with immunoregulatory properties and is an important signaling molecule of mesenchymal cell activation. It is elevated in lamina propria lymphocytes in active IBD.7 Serum TGF-β levels is higher in naïve, active UC patients compared with healthy controls.8 Nonetheless, no differences have been determined in the expression of TGF-β1 in the colonic mucosa of healthy individuals and inactive UC and CD patients.7 Transforming growth factor β1 levels are also increased in the inflamed tissues of mice with colitis induced by either 2,4,6-trinitrobenzene sulfonic acid (TNBS) or oxazolone.9
Transforming growth factor β expression and active TGF-β level are elevated in the intestinal mucosa overlying intestinal strictures in CD patients.10,11 Transforming growth factor β production by myofibroblasts is elevated near intestinal strictures. Not surprisingly, the link between IBD and TGF-β, one of the main anti-inflammatory mediators in the gut, is the subject of intensive research.
Three different isoforms are expressed in mammals and designated as TGF-β1, TGF-β2, and TGF-β3. Transforming growth factor β can control the differentiation, proliferation, and state of activation of immune cells. Transforming growth factor β1, the most abundant isoform, can inhibit the differentiation of T cells naïve to CD4+ and CD8+ into effectors12 and block T helper (TH) 1 and 2 development.12 Transforming growth factor β transmits signals through Smad-dependent or Smad-independent pathways.13 Receptor-regulated Smad1, 2, 3, 5, and 8 (R-SMADs) are directly phosphorylated and activated; these form heteromeric complexes with the Co-SMAD (SMAD4).14 Activated SMAD complexes are translocated into the nucleus and regulate the transcription of target genes. The inhibitory Smad6 and Smad7 (I-Smads) are target genes of TGF-β and provide an autoregulatory negative feedback loop.15Smad7 expression is induced by pro-inflammatory cytokines like interferon γ, tumor necrosis factor (TNF)α, and interleukin (IL)-1β.16 SMAD7 negatively regulates TGF-β signaling by inhibiting phosphorylation of the 2 major transcription factors SMAD2 and SMAD3.14 Elevated colonic SMAD7 is associated with enhanced inflammation in IBD patients.17–19 It was hypothesized that TGF-β1 does not exert a regulatory effect, as TGF-β1 signaling is blocked by high levels of Smad7.9 As both anti-inflammatory TGF-β and inhibitory SMAD7 are increased in IBD patients, a strategy was suggested to knock down Smad7 with specific antisense oligonucleotides to restore and sustain TGF-β function.20 This led to the development of the pharmaceutical compound Mongersen, which was tested in phase 3 clinical trials; however, the study was prematurely discontinued as an interim analysis showed a lack of clinical benefit.20
In this study, we analyzed the role of Smad7 in 2 different animal models of intestinal fibrosis. Due to the pleiotropic properties of TGF-β, not only could the absence of inhibitory SMAD7 strengthen immunoregulatory function of TGF-β, but prolonged mesenchymal cell activation could also carry the risk of increased fibrosis. Our results show that Smad7 deficiency is associated with a decrease in intestinal inflammation and an increase in fibrosis development. Targeting the TGF-β signaling pathway may be a new and powerful option for an anti-inflammatory treatment but could bear the risk of IBD-associated fibrosis.
Materials and Methods
Animals
Animal experiments were performed according to the ARRIVE criteria. Smad7-/- mice (Smad7ΔexI, coding region of the first exon, including the starting ATG codon, and a part of intron 1 replaced with a neomycin selection cassette, Supplementary Figure 1A) have been previously described.21 Heterozygous mutant mice were backcrossed at least 10 generations onto CD-1 mice, which were obtained from Charles River Laboratories (MA, USA). The animal experiment protocol was approved by the Veterinary Authority of the canton of Zurich (registration number ZH242/2016). Littermates were used in all experiments involving transgenic mice. The animals were cohoused wherever possible, and bedding was exchanged among the cages to minimize potential effects of microbiota variation. All the animals were housed in a specific pathogen-free (SPF) facility. The animals were kept in type 2, long clear-transparent, individually ventilated cages (IVCs, 365 mm × 207 mm × 140 mm, Allentown, New Jersey, USA) with autoclaved dust-free bedding and tissue papers as nesting material. They were fed a pelleted and extruded mouse diet (R/M–H Extrudat, ssniff Spezialdiäten, Soest, Germany) ad libitum. The light/dark cycle in the room was given through natural daylight (sunrise 7:00 AM, sunset 6:00 PM). The mice were weighed at 10:00 AM every morning. The temperature was set to 21 ± 1°C, with a relative humidity of 55 ± 5% and 75 complete changes of filtered air per hour (filter: Megalam MD H14, Camfil, Zug, Switzerland).
Phenotyping Study
Postmortem gross and histological examination was conducted on 6 female mice, as part of a standard primary phenotyping screen, to investigate the effects of mutant Smad7ΔEx-1 genotype. A complete necropsy, including a thorough external and internal gross postmortem examination, was performed on each mouse by a blinded investigator from the Institute of Veterinary Pathology, Vetsuisse Faculty, University of Zurich. Representative samples of the following organs and tissues were fixed in 10% buffered formalin for histological examination: adrenal glands (cortex and medulla), bone and joint (femoral tibial joint), brain, colon, duodenum, gall bladder, gut-associated lymphoid tissue (GALT), heart, ileum, jejunum, kidneys, liver, lungs, lymph nodes (sub mandibular and mesenteric), vagina, esophagus, ovaries, pancreas, salivary gland (submandibular), skeletal muscle (quadriceps), skin, spinal cord (thoracic), spleen, stomach, thymus, thyroid glands, tongue, trachea, urinary bladder, uterus, and nasal cavities. Microscopic findings were classified with standard pathological nomenclature, and severities of findings were graded on a scale of 1 to 5 as slight, mild, moderate, marked, or severe.22 Grades of severity for microscopic findings were subjective; slight was the least discernible extent, and severe was the greatest extent possible.
Induction of Colitis With DSS
For chronic colitis, wildtype (WT) and Smad7-/- mice were administered with 3 cycles of 3% DSS (MP Biomedicals, LLC, Solon, OH, USA) in drinking water ad libitum for 7 days followed by 14 days of regular drinking water. After the last cycle, all animals were allowed to recover for 2 weeks and then killed for sample collection. Mice receiving regular drinking water served as controls. Data for the DSS model of chronic colitis originated from 3 experimental rounds using mice with an identical genetic background (littermates). Female mice were used, as male mice frequently show aggressive behavior upon DSS treatment. In age-matched experiments, WT 99 ± 9 days vs heterozygous 90 ± 16 days vs Smad7-/- 101 ± 11 days littermates were used. In weight-matched experiments, 50 ± 5 days-old WT and 138 ± 25 days-old Smad7-/-- CD-1 mice with an average weight of 30 to 31 g were used. Additional weight-matched experiments were performed because mice of the same age with the described genotypes are not of equal weight. Small mice are at risk of suffering more from chronic DSS-induced colitis compared with heavier animals. Heavier animals lose less weight percentagewise and recover more easily during remission. Lighter mice are at risk of not fully recovering in remission. To assess the clinical condition, a clinical score that includes appearance, behavior/activity, pain, stool consistency, blood in stool, and weight loss (5, 5, 4, 5, 4, 6 graduations, respectively) was used. The score for stool consistency included 5 graduations: solid, soft but formed pellets, soft with no pellets formed, very soft, or liquid. Liquid stool is a criterion for the termination of the experiment, and very soft stool is a criterion for the administration of wet food; neither severity level was reached in our experiments.
Heterotopic Intestinal Transplant Model
Female CD-1 littermates obtained from heterozygous Smad7 breeding pairs weighed 25 to 30 g at the start of the experiment. For the heterotopic mouse intestinal transplant model, 30 to 40 mm of small bowel proximal to the cecum was excised from a female donor and implanted into a subcutaneous pouch into the neck of a female recipient during the light cycle.23 Cefazolin (Kefzol) was administered by intraperitoneal injection. As previously described, fibrosis develops over time in the transplant model of fibrosis. Many profibrotic parameters are significantly regulated after a few days, whereas others require weeks.23 As the absence of inhibitory SMAD7 may increase the risk of fibrosis, grafts were explanted at day 6 during the light cycle.
Colonoscopy and Histological Score
Animals were anesthetized intraperitoneally with a mixture of 90 to 120 mg of ketamine (Narketan 10%, Vétoquinol AG, Bern Switzerland) and 8 mg of xylazine (Rompun 2%, Bayer, Switzerland) per kg body weight and were examined with the Tele Pack Pal 20043020 (Karl Storz Endoskope, Germany). Mice were evaluated using the murine endoscopic index of colitis severity (MEICS) criteria. The MEICS score also includes an assessment of the transparency of the intestinal wall (from transparent to partially nontransparent, largely nontransparent, and nontransparent). Upon DSS treatment, the colon wall of a healthy mouse thickens, and this loss of transparency is synonymous with increasing inflammation.24 One cm of the distal colon was scored histologically.
Sirius Red, Collagen Content and Thickness Measurement, Immunohistochemistry
Sections (3 µm) were stained with Sirius red according to a standard protocol. Staining was examined using the Imager Z2 microscope and the software ZEN. At least 3 representative areas from each section were taken using transmission light microscopy (20x). Collagen quantity was analyzed using the Fiji software (1.52a, NIH). The total tissue area was determined by converting the image into an 8-bit image type and adjusting the black and white threshold. The area covered with collagen was determined setting thresholds to select the red color (hue >220) and adjusting the saturation. Collagen was quantified as hue >220/total tissue area.
The thickness of the submucosa was defined as the distance between the muscularis mucosae and the muscularis propria and measured with the software ZEN. The average of 5 individual measurements from a representative area of each section was combined into one data point.
For immunohistochemistry, sections were stained with monoclonal rabbit antimouse FOXP3 (MAB8214, R&D Systems, 1:1000) and polyclonal rabbit antimouse CD3 (ab5690, abcam, 1:1000) according to a standard protocol for 3,3ʹ-diaminobenzidine (DAB). The relative quantity of FOXP3+; and CD3+ was analyzed as described for Sirius red. The area covered with DAB was determined by setting thresholds to select the brown color and divided by the total tissue area as described previously.
The absolute number of CD3+ was determined by counting DAB stained cells from at least 3 representative areas from each section. Number of CD3+ is shown per 103 of total cells in the tissue determined by their blue colored nucleus.
4-Hydroxyproline Assay
The 4-hydroxyproline (HYP) was quantified from the colon and ileum using an HYP assay (MAK008-1KT, Sigma-Aldrich, Buchs, Switzerland). In brief, tissues (10-30 mg) were homogenized using gentleMACS Octo Dissociator (130-096-427, Miltenyi Biotec, Bergisch Gladbach, Germany) and hydrolyzed in 12 M of HCl (10 µL/mg tissue). The hydrolysate was transferred in duplicates to a 96-well plate and dried at 60°C. Dried samples were incubated with 50 µL of chloramine T/oxidation buffer mixture (3 µL of the chloramine T concentrate and 47 µL of the oxidation buffer) at room temperature for 5 minutes. Fifty µL of the diluted DMAB reagent (25 µL dimethylaminobenzaldehyde, 25 µL perchloric acid/isopropanol) was added, and samples were incubated at 60°C for 90 minutes for chromophore formation. Absorbance was measured at 560 nm.
Myeloperoxidase Activity Assay
Both colon and small bowel specimens were homogenized in 50 mM phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide (H-5882, Sigma) with the gentleMACS octo dissociator (130-095-937, Miltenyi, program MPO). After 3 freeze and thaw cycles, homogenates were centrifuged for 10 minutes at maximum speed. Twenty mL of the supernatant were transferred to a 96-well plate in duplicate and mixed with 280 μL of 0.02% dianisidine (in 50 mM phosphate buffer and 0.0005% H2O2, D-3252, Sigma, St Gallen, Switzerland). Absorbance was measured at 460 nm after 20 minutes or 60 minutes for samples from Smad7 deficient and WT mice. Protein concentration of the supernatant was determined using the BCA Protein Assay Kit (23252, Thermo Fisher Scientific, Reinach, Switzerland). Myeloperoxidase (MPO) activity was calculated as mean absorbance (460 nm)/incubation time/protein content.
RNA Isolation, cDNA Synthesis, and Real-time Quantitative PCR
Total RNA was isolated from freshly isolated colon and ileum using the Maxwell RSC simplyRNA tissue kit (Promega, Madison, WI, AS1340). For mouse samples, lysis buffer from the kit was added to snap frozen resections, and samples were shredded in M tubes (Miltenyi Biotec, Bergisch Gladbach, Germany) in a gentleMACS tissue homogenizer (Miltenyi Biotec). For in vitro assays, lysis buffer from the kit was added to the cells, and a rubber policeman was used; DNase digestion was prepared. RNA was quantified at 260 and 280 nm with a NanoDrop (Thermo Fisher Scientific). Complementary DNA (cDNA) synthesis was performed (Applied Biosystems, Foster City, CA). Quantitative PCR (qPCR) was performed using the TaqMan Fast Universal Master Mix (Applied Biosystems) on a QuantStudio 6 Flex Real-Time PCR System, and results were analyzed with the SDS software (Applied Biosystems). Samples were measured in triplicates, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as endogenous control (∆∆CT method). Il-1β Mm00443259_g1, Il-6 Mm00446190_m1, TNF Mm00443259_g1, ColIVa1 Mm01210125_m1, TGF-β1 Mm01178820_m1, and GAPDH 4352339E (Life Technologies).
Murine Primary Cells
For hematological analyses, murine whole blood was collected from ethylenediaminetetraacetic acid-treated tubes (367525, BD, Franklin Lakes, NJ) and analyzed with an ADVIA 2120 flow cytometer (Siemens, Munich, Germany).
For FACS, lamina propria mononuclear cells were isolated; 5 cm of the colon was resected. Small colon pieces were incubated with 5 mL of collagenase V (1 mg/mL, C9263-1G, Sigma), collagenase D (1 mg/mL, 11088882001, Roche, Basel, Switzerland), dispase (0.3 U/mL, 17105-041, Gibco), DNase (0.3 mg/mL, 10104159001, Roche), and RPMI medium (21875-034, Gibco) with 10% fetal bovine serum (FCS) for 30 minutes at 37°C. Suspension was vortexed and filtered (70 μm mesh).
Flow Cytometry
Cells were incubated with primary antibodies in phosphate-buffered saline (PBS) for 30 minutes at 4°C and then washed with PBS. Cells were fixed with BD Cytofix/Cytoperm for 20 minutes at 4°C, followed by a final washing step with Perm buffer. Samples were analyzed on an LSR II Fortessa (405, 488, 561, and 640 nm laser lines; BD) using FACS Diva Software. The following murine antibodies were used: anti-CD45 PB, 1:800 (Biolegend, San Diego, CA), 103126; anti-CD8 PerCp Cy5.5, 1:400 (eBioscience San Diego, CA), 45-0081-82; anti-CD4 APC Cy7, 1:200 (eBioscience 47-0041-82); anti-CD3 APC, 1:200 (Biolegend) 100236; anti-B220 PE Cy7, 1:200 (BD Pharmingen) 552772, Aqua for dead cell staining, 1:1000 (Life Technologies) L34957. Data analysis was performed using FlowJo 10.0.x (BD) with applied compensation correction.
Western Blot
Tissue lysis was performed in M-PER cell lysis buffer (Thermo Fisher Scientific). Protein was quantified by NanoDrop, and equal amounts of protein were loaded onto SDS/PAGE gels. Twelve percent polyacrylamide gel with Tris/SDS running buffer was used, and protein was transferred onto nitrocellulose (Invitrogen); 5% milk, 3% bovine serum albumin, and 0.1% Tween 20 were used for blocking. Western blots (WBs) were performed using polyclonal rabbit antimouse IL-6 (ab6627, Lucerna, 1:1000), polyclonal goat antimouse IL-1β (AF-401-NA, R&D/ bio-techne, 1:1000), polyclonal rabbit antimouse TGF-β (D157926 3711S BioConcept/ CellSignaling, 1:1000), polyclonal rabbit antimouse ACTA2 (ab5694, abcam, 1 μg/mL), polyclonal rabbit antimouse β-actin antibodies (4970S, 13E5, Cell Signaling, Danvers, MA, 1:2000), and the horseradish peroxidase-conjugated secondary goat antirabbit (sc-2357, Santa Cruz, CA 1:2000) and donkey antigoat (sc-2020, Santa Cruz, CA 1:2000) antibodies. Densitometry was performed using Fiji software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (v5.0). Differences were considered significant at a P value of <0.05, highly significant at a P value of <0.01 and <0.001. Results are presented as mean ± standard deviation (SD) or ±standard error of the mean (SEM).
Results
Altered Body Weight Regulation in Smad7-deficient Mice
The generation of Smad7-/- mice was performed as previously described.21 Heterozygous mutants, backcrossed onto CD-1 mice, were used for breeding. Out of 39 litters and 242 live births, the distribution of genotypes from Smad7-/+ parents was 37% WT, 51% Smad7-/+, and 12% Smad7-/- (Figure 1A). All of these mice survived to weaning. Nonetheless, Smad7-/- mice had a survival disadvantage during embryogenesis. Macroscopically Smad7-/- mice exhibited a lower body weight (P < .001, Figure 1B) and appeared to be smaller than WT and Smad7-/+ mice. Although Smad7-/- mice gained weight over time, they never caught up with WT and Smad7-/+ mice.
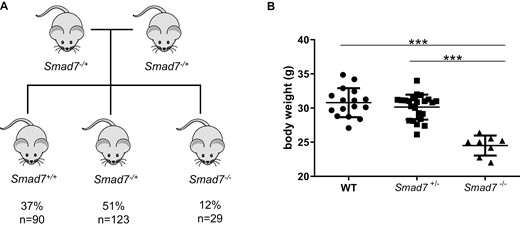
Body weight is decreased in Smad7-deficient mice. A, Out of 39 litters and 242 live births, the distribution of genotypes from Smad7-/+ parents was 37% WT, 51% Smad7-/+, and 12% Smad7-/-. B, Body weight, n = 16, 23, 8, male mice of the same age, ANOVA, Tukey’s multiple comparison test, ±SD, P < .0001 each.
A Smad7-/- postmortem phenotyping study was performed by a blinded investigator from the Institute of Veterinary Pathology. A complete necropsy, including a thorough external and internal gross postmortem examination, was performed on each mouse. Again, macroscopically Smad7-/- mice exhibited lower body weight compared with WT mice. However, this was not attributed to any particular lesion, as no macroscopic or histological findings were identified (Supplementary Figure 1B). In particular, the gastrointestinal tract did not show any evidence of underlying chronic inflammation or fibrosis (Supplementary Figure 1C and D).
Smad7-deficient Mice Showed Decreased Inflammation in DSS-Induced Chronic Colitis
To determine the relevance of SMAD7 in the development of inflammation and fibrosis, the DSS mouse model of chronic colitis was used, mice were cohoused wherever possible, and bedding was exchanged among the cages. To ensure that a different body weight at the start of the experiment was not affecting the course and the outcome of the experiment, mice with the same starting weight were used. Sibling animals from different litters were used. As cohousing was not possible for mice from different litters, bedding was exchanged among the cages to minimize potential effects of microbiota variation. Accordingly, mice had a different age at the start of the experiment (WT 50 ± 5 days vs Smad7-/- 138 ± 25 days). A total of 8 DSS-treated female WT mice were compared with 7 female Smad7-/- littermates in 3 independent experiments. The intake of drinking water containing DSS was not different between the 2 groups (WT 3.6 ± 0.2 mL/day vs Smad7-/- 3.5 ± 0.2 mL/day). Upon DSS-induced chronic colitis, Smad7-/- mice displayed a reduced weight loss when compared with WT mice (Supplementary Figure 2). The clinical condition of the mice was assessed using an appropriate score, as described in the Methods section. The DSS-treated mice developed clinical signs of colitis, as evidenced by a clinical score >1 as early as day 7 (Figure 2A). Upon DSS-induced chronic colitis, WT mice displayed a significantly increased clinical score compared with Smad7-/- mice, suggesting increased inflammation. Wild type mice also displayed a significantly and sustained increased clinical score during remission following the third and final cycle of DSS treatment (Figure 2A). Severity of inflammation in both groups was analyzed by coloscopy and MEICS. In the model of DSS-induced inflammation, a softening stool is indicative of more severe inflammation of the bowel. Smad7 deficiency was associated with lower inflammation, as evidenced by a significantly lower score for stool consistency (graduations described in Methods) when compared with WT mice (Figure 2B). In contrast, chronic colitis triggered an increased thickening of the colon in Smad7-/- mice when compared with WT mice (Figure 2B), suggesting increased fibrogenesis and ECM deposition in Smad7-/- mice. Thickening of the colon in DSS-treated Smad7-/- mice appeared to be increased, as shown during colonoscopy, as organs behind the bowel wall were no longer visible through the colon tissue. In the model of DSS-induced inflammation, shortening of the colon is indicative of more severe inflammation of the bowel. Smad7 deficiency was also linked to ameliorated colitis, as evidenced by a significantly increased colon length in DSS-treated Smad7-/- mice compared with WT (Figure 2C).
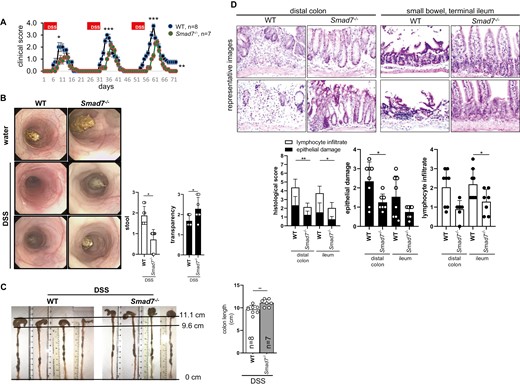
Absence of SMAD7 reduces inflammation upon DSS-induced chronic colitis. Mice of the same starting weight but different age were used. A, Clinical score, ±SEM, unpaired t tests, n as indicated, ***P < .001 and *P < .05. B, MEICS, ±SD, unpaired t test, n = 8, 7, 8, 7, **P = .0060 and *P = .0185. C, Colon length, ±SD, unpaired t test, n as indicated, **P = .0070. D, Histological score, ±SD, unpaired t test, **P < .01 and *P < .05.
Colonic specimens from both groups were analyzed for severity of inflammation by histological scoring. The histological score was significantly lower in Smad7-/- mice with DSS-induced chronic colitis compared with WT controls (Figure 2D). The DSS-treated Smad7-/- mice had significantly lower scores for both epithelial injury and leukocyte infiltration in the intestinal mucosa (Figure 2D).
Immunohistochemistry was performed to determine the number of CD3+ T cells in intestinal tissue upon DSS treatment. In Smad7-/- mice, the number of CD3+ cells was significantly decreased in the colon compared with WT mice (Figure 3A). Flow cytometry was performed to quantify the number of viable T cells in colonic tissue upon DSS. In Smad7-/- mice, the number of CD3+ cells was significantly decreased compared with WT mice (Figure 3B). The relative number of CD4+ cells within the CD3+ population was significantly decreased in Smad7-/- mice (WT 72.1 ± 5.1% vs Smad7-/- 62.6 ± 8.7%, Figure 3B). In Smad7-/- mice, the number of FOXP3+ cells was significantly increased in the colon compared with WT mice (Figure 3C).


Absence of SMAD7 reduces number of T cells upon DSS-induced chronic colitis. Mice of the same starting weight but different age were used. A, Immunohistochemical staining for CD3+ T cells. CD3+/ total tissue content, ±SD, unpaired t test, n = 5, 4, 6, 5, *P < .0229. Absolute number of CD3+ per 103 of total cells in the tissue, ±SD, unpaired t test, n = 6, 4, *P = .0497. B, Flow cytometry, ±SD, unpaired t test, n = 7, 6, ***P < .0001 and *P = .0311. C, Immunohistochemical staining for FOXP3+ T regs, ±SD, unpaired t test, n = 6, 5, *P = .0075.
Relative spleen weight was significantly decreased in DSS-treated Smad7-/- mice compared with WT mice, indicating less leukocyte accumulation (Figure 4A). Smad7-deficient mice showed a significant reduction of DSS-induced Il-1β, Il-6, TNF and higher TGF-β1 mRNA expression (Figure 4B) in the ileum indicating decreased inflammation. Smad7-/- mice also had both significantly lower IL-1β, IL-6 and higher TGF-β1 protein expression (Figure 4C) in the ileum. Myeloperoxidase activity was significantly decreased in colon from DSS-treated Smad7-/- mice (Figure 4D), suggesting decreased inflammation.
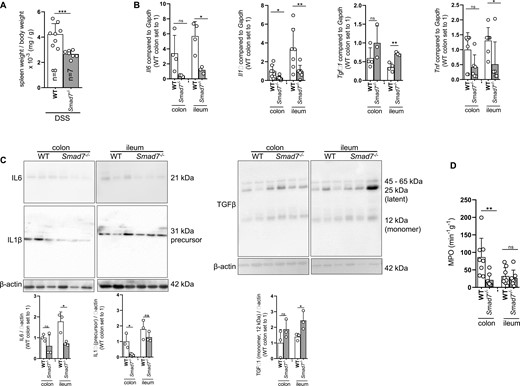
Absence of SMAD7 reduces spleen weight and inflammatory cytokine profile upon DSS-induced chronic colitis. Mice of the same starting weight but different age were used. A, Spleen weight, ±SD, unpaired t test, n as indicated, ***P = .0005. B, Il-6, Il-1β, TGF-β and TNF mRNA expression, ±SD, unpaired t tests, n = as indicated, *P = .0123, .0182, .0182, .0030, and .0367. C, IL-6, IL-1β and TGF-β protein expression, ±SD, unpaired t tests, n = as indicated, *P = .0182, .0422, and .0459. D, MPO, ±SD, unpaired t tests, n = 8, 7, 8, 7, **P = .0099.
Smad7-deficient Mice Showed Increased Fibrosis in DSS-Induced Chronic Colitis
We evaluated fibrogenesis parameters in Smad7-deficient mice. The amino acid HYP is a major component of collagen, and the HYP tissue content allows a reliable conclusion on the degree of fibrosis. The 4-hydroxyproline levels were significantly increased in colon, but not the ileum, from Smad7-/- mice compared with WT mice (Figure 5A), indicating increased collagen deposition. Additionally, collagen was determined by Sirius red staining (Figure 5B) and Masson’s trichrome staining (Figure 5C) after induction of fibrosis and quantified using transmission light microscopy—employed for the histological visualization of collagen fibers—and analyzed using the Fiji software. The collagen content was significantly increased in colon from Smad7-/- mice compared with WT mice. Additionally, ColIVa1 mRNA expression levels were significantly increased in colon tissue from Smad7-deficient mice (Figure 5D), suggesting increased fibrosis. Submucosal thickness between the muscularis mucosae and the muscularis propria was significantly increased in the colon from Smad7-/- mice compared with WT mice (Figure 5E).
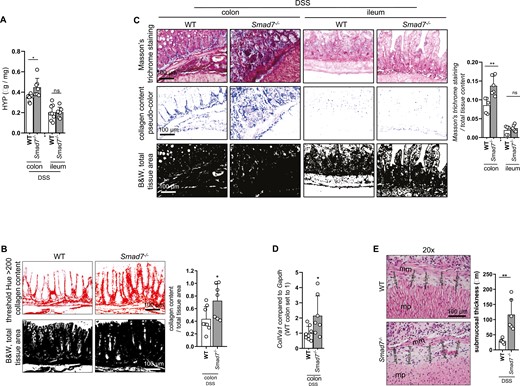
Absence of SMAD7 is increasing fibrosis upon DSS-induced chronic colitis. Mice of the same starting weight but different age were used. A, HYP content, ±SD, unpaired t tests, n = 8, 7, 8, 7, *P = .0103. B, Collagen content visualized by Sirius red staining was quantified with the Fiji software, ±SD, unpaired t test, n = 8, 7, *P = .0339. C, Collagen content visualized by Masson’s trichrome staining was quantified with the Fiji software, ±SD, unpaired t tests, n = 5, 6, 5, 6, *P = .0053. D, ColIVa1 mRNA expression, ±SD, unpaired t test, n = 8, 7, *P = .0366. E, Submucosal thickness, mm = muscularis mucosae, mp = muscularis propria, ±SD, each data point represents the average of five individual measurements from a representative area, unpaired t test, n = 5, 6, *P = .0045.
Smad7 Deficiency Is Not Protective Against DSS-induced Chronic Colitis When Using Animals of the Same Age
In the experiments described herein, mice with the same starting weight were used to ensure that differences in body weight did not affect the course of DSS-induced chronic colitis. At the beginning of the experiment, mice ranged from 50 ± 5 days of age to 138 ± 25 days of age; and therefore, they were neither juvenile nor senescent. However, as the immune system changes with age, we also determined inflammation and fibrogenesis in mice of the same age at the beginning of DSS treatment. In age-matched experiments, the following littermates and ages were compared: WT 99 ± 9 days vs heterozygous 90 ± 16 days vs Smad7-/- 101 ± 11.
A total of 9 DSS-treated female WT mice, 8 female heterozygous, and 6 female Smad7-/- littermates were used. Severity of inflammation was analyzed by coloscopy and MEICS. Upon DSS-induced chronic colitis, Smad7-/- mice displayed a clear trend towards an increased MEICS score compared with WT mice (Supplementary Figure 3A). An increased thickening of the colon was observed in Smad7-/- mice compared with WT mice (Figure 6A), suggesting increased fibrogenesis. Additionally, HYP levels were significantly increased in the colon from Smad7-/- mice compared with WT mice (Figure 6B), indicating increased collagen and ECM deposition. Submucosal thickness was significantly increased in the colon from Smad7-/- mice compared with WT mice (Figure 6C). However, absence of Smad7 did not ameliorate inflammation in mice of the same age during the initial phase of DSS treatment. In Smad7-/- mice, the number of FOXP3+ cells remained unchanged in the colon compared with WT mice (Supplementary Figure 3B). Smad7-deficient mice showed significantly higher DSS-induced Il-6 mRNA and protein expression in the colon (Supplementary Figure 3C). Il-1β and TNF remained unchanged (Supplementary Figure 3C). Inflammation parameters such as the histological score, colon length, relative spleen weight, MPO activity, and number of neutrophils in peripheral blood showed equal values in DSS-treated Smad7-/- and WT mice (Supplementary Figure 3D–H). Profibrotic TGF-β1 displayed a clear trend towards an increased mRNA expression level and significantly increased protein in DSS-treated Smad7-/- mice compared with WT mice (Supplementary Figure 3C).
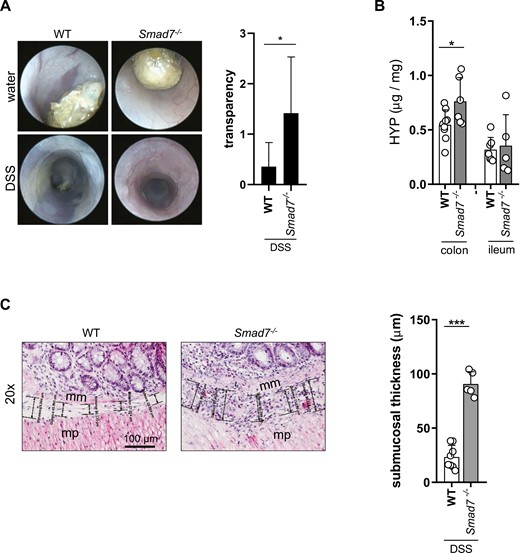
Absence of SMAD7 is increasing fibrosis in mice of the same age but different starting weight. A, Coloscopy and transparency score, ±SD, unpaired t test, n = 7, 6, *P = .0432. B, HYP content, ±SD, unpaired t tests, n = 9, 6, 7, 5, *P = .0341. C, Submucosal thickness, mm = muscularis mucosae, mp = muscularis propria, ±SD, each data point represents the average of five individual measurements from a representative area, unpaired t test, n = 8, 5, *P < .0001.
Fibrosis Is Increased in Smad7-deficient Mice in a Transplantation Model of Intestinal Fibrosis
We evaluated the development of fibrosis in an animal model that is unaffected by body weight. We used terminal ileum from Smad7-/- and WT mice as grafts in the heterotopic transplantation model of intestinal fibrosis. Important aspects of human intestinal fibrosis are reflected this model: exaggerated synthesis of matrix and an activation of intestinal myofibroblasts and obliteration associated with an increasing number of CD3+ lymphocytes. Grafts from 6 WT mice were compared with 6 Smad7-/- littermates in 2 independent experiments. Body weight remained unchanged in recipient groups receiving either Smad7-/- or WT grafts. From the 12 intestinal transplants, histologically evaluable tissue was obtained from all grafts. Collagen was visualized by Sirius red staining (Figure 7A) and Masson’s trichrome staining (Figure 7B) and quantified using bright light microscopy after induction of fibrosis. Microscopy was used for imaging, and quantification was performed using ImageJ/Fiji software. Collagen content was significantly increased in Smad7-/- mice, indicating a more severe fibrosis. Additionally, Smad7-/- mice showed significantly increased ACTA2 protein levels, indicating a higher number of myofibroblasts (Figure 7C). This result confirms that collagen deposition is increased upon Smad7 depletion in this murine model of intestinal fibrosis.
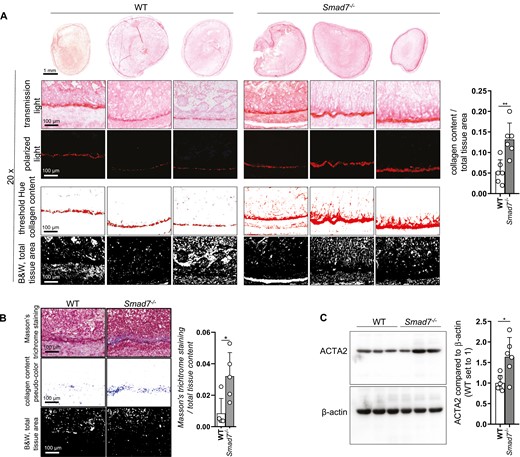
Absence of SMAD7 reduces deposition of collagen in the heterotopic transplantation model. A, Collagen content visualized by Sirius red staining was quantified with the Fiji software, ±SD, unpaired t tests, n = 6 each, **P = .0031. B, Collagen content visualized by Masson’s trichrome staining was quantified with the Fiji software, ±SD, unpaired t tests, n = 5 each, *P = .0177. C, ACTA2 protein expression and densitometry, n = 6 each, ±SD, unpaired t test, n = 6 each, *P = .0113.
Discussion
An orally available Smad7 antisense oligonucleotide, Mongersen, has been developed.20 Mongersen was reported safe and well tolerated by patients in a phase 1 study.25 Fourteen patients with active, inflammatory CD received a short-term treatment with Smad7 antisense oligonucleotide once daily for 7 days (3 cohorts with 40, 80, or 160 mg) and completed the 6-month phase 1 open-label study. None of the patients in the study developed small bowel stricture, and circulating levels of basic fibroblast growth factor, chitinase 3-like 1 (YKL-40), and matrix metalloproteases were not increased; thus it was concluded that short-term knockdown of Smad7 is not associated with intestinal fibrosis.26 This serves as a basis for the discussion on the safety of this drug when used at early time points. A phase 2 study was conducted to assess the efficacy of Mongersen in patients with active, steroid-dependent/resistant CD. Rates of clinical response were higher in patients receiving Mongersen compared with placebo.27 A phase 3 clinical trial showed the absence of clinical and endoscopic efficacy of Mongersen.28 The study was discontinued, as an interim evaluation showed a lack of clinical benefit.20 The specific antisense pharmacokinetics determine whether certain cells in the body are reached and to what extent. Moreover, formulation protects the antisense oligonucleotide from the gastric acid and facilitates the release of the active compound at the desired location in the body. In the present study, we analyzed the role of Smad7, a negative regulator of TGF-β signaling, in animal models of intestinal fibrosis. In our own work, we used mice with a systemic knockout of Smad7 in all cells. This allowed us to compare results from our chronic DSS-induced colitis experiments in animals constitutively lacking Smad7, with short-time treatments using antisense oligonucleotides. The results obtained with our experimental setup confirmed previous data that the absence of Smad7 has an anti-inflammatory effect. Furthermore, the systemic knockout also appears to cause increased fibrosis. The reason for this could be that the knockout of Smad7 is effective in all cells, whether in precursor or differentiated cells. Additionally, a systemic knockout could decrease the number of immune cells and reduce their activity. Upon DSS-induced colitis, increased TGF-β activity may on the one hand cause a dampened immune system, while on the other hand hyperactive myofibroblasts may develop and thereby lead to increased fibrosis.
Macroscopically, Smad7-/- mice exhibited a lower body weight but did not exhibit any histological change that could explain the small body size compared with WT mice. The gastrointestinal tract did not show any evidence of chronic inflammation and fibrosis. During the observation period, adult Smad7−/− mice never recovered their weight from the slow poor weight gain as newborns and maintained their body weight at a reduced percentage of normal weight compared with their wildtype littermates. No evidence of underlying chronic inflammation or fibrosis was identified in postmortem analyses. Others could show that loss of Smad7 (Smad7Δex-I) in mice on a mixed C57BL/6J/CD1 background impairs the cell cycle in chondrocytes, indicating that Smad7 is required for normal skeletal development.29 The C57BL/6 mice with a loss of Smad7 (Smad7Δex-IV) died within days after birth, whereas ICR mice developed to adulthood but were smaller than WT mice.30 The phenotype of the animals may also vary because animals with different genetic backgrounds were used in the studies mentioned. In our work, animals were used that were backcrossed to CD-1 mice for at least 10 generations.
Several studies have linked Smad7-deficiency and knockdown to the amelioration of intestinal inflammation in mice. Our data show that inflammation is decreased in Smad7-/- mice upon DSS-induced chronic colitis when animals with the same starting weight are used. This Smad7 deficiency is associated with a decrease in TNF mRNA expression and MPO. Changes in the gut microbiota were determined between young and aged mice.31 Therefore in our experiments, animals were cohoused wherever possible, and bedding material together with the stool was exchanged among the cages to minimize microbiota variation.
Work carried out by others has shown that a specific Smad7 deficiency in dendritic cells protected mice against DSS-induced acute colitis and upon adoptive T-cell transfer.32 Knockdown of Smad7 with oral antisense oligonucleotide, Mongersen, ameliorated intestinal inflammation in mice in different experimental approaches.9,33 The expression level of SMAD7 in the inflamed tissue in mice was increased upon TNBS- and oxazolone-induced colitis.9 Designed as a short-term treatment, knockdown with oral Smad7 antisense over a period of 7 or 4 days attenuated acute colitis in mice induced by TNBS or oxazolone, respectively.9 When Mongersen is applied as short-term treatment after the final fourth cycle of recurrent relapses of acute TNBS-induced colitis, persistent inflammation was also ameliorated.9 In contrast, severe combined immunodeficiency mice subjected to adoptive transfer of CD4+ CD45Rbhigh T cells show a lack of therapeutic effect upon knockdown of Smad7.9 Exposure to Mongersen over 3 and 4 weeks show that Smad7 antisense ameliorates persistent inflammation when applied at the end of an 11-week TNBS-induced colitis.33 Treatment with Mongersen was associated with decreased pro-inflammatory IL-6 and TNF mRNA expression. Mice upon Mongersen treatment show reduced TGF-β protein synthesis and decreased SMAD3 phosphorylation, as determined by ELISA and WB, respectively.33 Exposure to Mongersen show a trend towards increased profibrotic connective tissue growth factor, whereas, Il-13 and platelet-derived growth factor remained unchanged.33 Treatment was associated with reduced trichrome-positive and collagen 1–positive tissue.33 In addition, mice subjected to chronic DSS-induced colitis show decreased collagen 1–positive tissue upon Smad7 antisense treatment starting together with the fourth cycle of DSS, a time point at which mice had already developed fibrosis. Smad7-deficiency and knockdown in murine models of colitis suggest a therapeutic option for patients with IBD.
Altered cellular composition and cell responses could provide an explanation for Smad7-dependent effects on the course of inflammation. Mounting evidence indicates that Smad7 levels influence the quantity and differentiation of T cells in the gut. In our study, Smad7-/- mice showed a decreased number of CD4+ T cells in DSS-induced chronic colitis compared with WT mice. Work carried out by others, using a murine model of multiple sclerosis, showed that a specific Smad7 deficiency in CD4+ T cells causes a decreased number of intestinal T cells compared with an enhanced proliferative potential in T cells overexpressing Smad7 in the small intestine.34 Transforming growth factor β and inhibitory Smad7 is involved in the differentiation of naïve CD4+ T cells and their subtypes.35,36 The TGF-β signaling drives the induction of regulatory T (Treg) cells, characterized by low Smad7 expression.37 Additionally, Treg frequencies and numbers of anergic T cells are higher in the lamina propria of mice with Smad7-deficient CD4+ T cells.34 In contrast, overexpression of Smad7 in T cells favored the expansion of intestinal CD4+ T cells toward an inflammatory phenotype and causes autoreactive activation in the intestine in an animal mouse model of multiple sclerosis.34 Overexpression of Smad7 leads to an altered T lymphocyte composition of the intestinal mucosa and structural changes, including a thickened submucosa of the jejunum and colon compared with control mice.34 Inflammatory Th1 lymphocytes are increased in the lamina propria of mice overexpressing Smad7, whereas Smad7-deficient mice show fewer Th1 cells.34 Increased expression of Smad7 in T cells increases T-cell activation and differentiation toward Th1 cells in a murine model of experimental autoimmune encephalomyelitis, whereas Treg differentiation is decreased.35Smad7-deficient B cells are characterized by increased TGF-β signaling measured as increase of pSMAD2-positive B cells compared with WT.21Smad7-deficient B cells exhibited increased Ig class switch recombination to IgA, significantly enhanced spontaneous apoptosis, and decreased proliferation in the presence of lipopolysaccharide.21 These data suggest that Smad7 is an inhibitor of tolerance at the intestinal phalanx.34
Using 2 murine fibrosis models, we showed that absence of Smad7 leads to a significant increase in the transcriptional expression of fibrosis markers and an increase in collagen layer thickness and HYP when compared with WT mice. Confirmative to our findings, work carried out by others has shown increased fibrogenesis in Smad7-/- mice and decreased fibrosis upon Smad7 overexpression in specific animal models for various diseases. Smad7-deficient mice show an increased ECM deposition in a model of chronic pancreatitis.38 Available data suggest that fibrogenesis involves increased collagen type 1 and a higher number of mesenchymal cells.38Smad7 is directly regulated by microRNA miR-877-3p.39 Mice upon bleomycin-induced pulmonary fibrosis show increased silencer miR-877-3p and, accordingly, downregulation of Smad7. Treatment with miR-877-3p-inhibitor leads to ameliorated pulmonary fibrosis.39 When Smad7 overexpression was induced in renal tubular epithelial cells, TGF-β-induced transition into myofibroblasts was inhibited, and production of ECM and collagen protein was decreased.40 These data have shown that mice lacking Smad7 displayed a tendency towards fibrosis in a variety of tissues in animal models, especially concerning long-term, chronic inflammation. However, when Mongersen was applied in the last 3 weeks of an 11-week TNBS-induced colitis, fibrosis microscopic score and soluble collagen were decreased in colon samples, suggesting that Smad7 antisense is effective in reducing intestinal fibrogenesis in this model, whereas other profibrotic parameters remained unchanged.33 Mice subjected to chronic DSS-induced colitis also showed decreased collagen 1–positive tissue upon Smad7 antisense. In contrast to the mentioned study, where the TNBS-induced (with BALB/c mice) and DSS-induced colitis models were used, we employed CD-1 mice. Strain-specific differences may be relevant for different outcome for fibrogenesis. Western blots for SMAD7 and pSMAD3 suggest that the antisense molecules were highly effective.33 On the other hand, we used Smad7Δex-I knockout mice in our own study. In our experimental animals, SMAD7 is permanently absent from all tissues at all stages of development. Therefore, SMAD7 is not only decreased after the onset of inflammation, but may be completely switched off. This may also have an influence on the development, presence, and stimulability of cells because the developing mutant organism needs to adapt to the lack of Smad7.
Transforming growth factor β originates from damaged tissue, fibroblasts, and M2 macrophages41 and modulates cell growth, migration, apoptosis, differentiation, and collagen production. This results in long-term overactivation of fibroblasts, which leads to increased collagen in aberrant scars.42 Sustained TGF-β signaling is also caused by decreased Smad7.
In summary, our data show that Smad7 deficiency is associated with ameliorated inflammation, as evidenced by macroscopic and microscopic improvement, a decreased T cell accumulation in colonic tissue, and reduced pro-inflammatory molecular parameters. Several studies have linked Smad7 deficiency and knockdown to the amelioration of inflammation in mice.7,31,32 Further, we showed that absence of SMAD7 in a long-term model of DSS-induced chronic colitis is associated with an increase in HYP and collagen content. In line with our findings, Smad7Δex-I knockout mice displayed increased fibrogenesis in a model of chronic pancreatitis.38 Our results from Smad7-deficient mice showing increased intestinal fibrosis are in contrast to the results from the previously mentioned experiments, where animals exposed to Mongersen for a limited time period of 3 and 4 weeks revealed either no change or a decrease in profibrotic parameters and fibrogenesis.33 Likewise, in the aforementioned phase 1 study,25 none of the patients included experienced obstructive symptoms upon a short-term treatment with Smad7 antisense oligonucleotide.26 Short-term downregulation of SMAD7 by antisense RNA might promote resolution of inflammation without affecting fibrogenesis.26 Continuous absence of Smad7 in both immune cells and stromal cells might affect immune responses and developmental processes in a different way21,34 (eg, by a decreased number of intestinal T cells, Treg frequencies, and numbers of anergic T cells).34Smad7 deficient B cells are characterized by an overactive TGF-beta signaling and increase of pSMAD2.21 Further, orally administered antisense RNA is mainly taken up by epithelial cells, leaving the function of stromal cells unaltered.26
Thus, the notion of silencing Smad7 in an anti-inflammatory or antifibrotic manner as a novel therapy for human fibrotic disorders warrants further investigation. Long-term elimination of SMAD7 might have unfavorable consequences and should therefore also be judiciously investigated.
Abbreviations
- Acta2
α-smooth muscle actin
- Actb
beta-actin
- CD
Crohn’s disease
- Col4a1
pro-collagen type IV alpha 1
- ECIS
electric cell-substrate impedance sensing
- ECM
extracellular matrix
- FCS
fetal bovine serum
- GALT
gut associated lymphoid tissue
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HYP
4-Hydroxyproline
- IBD
inflammatory bowel disease
- IL
interleukin
- MEICS
murine endoscopic index of colitis severity
- mRNA
messenger RNA
- qPCR
real-time quantitative polymerase chain reaction
- TGF-β1
transforming growth factor β1
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- TNF
tumor necrosis factor
- UC
ulcerative colitis
- WB
Western blot
Acknowledgments
Authors would like to thank Josep Monné Rodríguez from the Institute of Veterinary Pathology, Vetsuisse Faculty, University of Zurich for his assistance with the postmortem gross and histological examination.
Author Contributions
Conceptualization: G.G.
Data curation and writing of original draft: C.S. and M.H.
Data curation: C.M., B.W., M.S., C.S., and F.F
Writing, review and editing: L.P., C.d.V., R.H., P.A.R., G.R., M.H.
All authors approved the final submitted version of the manuscript.
Funding
This work was supported by research grants from the Swiss National Science Foundation (grant 314730_152895/ 1) and FreeNovation from Novartis to M.H. and the Swiss National Science Foundation (grant numbers 310030_172870 and 314730_153380) to G.R.
Conflicts of Interest
M.H. discloses grant support from AbbVie and Novartis. G.R. discloses grant support from AbbVie, Ardeypharm, MSD, FALK, Flamentera, Novartis, Roche, Tillots, UCB and Zeller. All other authors have nothing to disclose.
Data Availability
The data underlying this article are available in a repository provided by the University of Zurich.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}