





Abstract
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), is a class of severe and chronic diseases of the gastrointestinal (GI) tract with recurrent symptoms and significant morbidity. Long-term persistence of chronic inflammation in IBD is a major contributing factor to neoplastic transformation and the development of colitis-associated colorectal cancer. Conversely, persistence of transmural inflammation in CD is associated with formation of fibrosing strictures, resulting in substantial morbidity. The recent introduction of biological response modifiers as IBD therapies, such as antibodies neutralizing tumor necrosis factor (TNF)-α, have replaced nonselective anti-inflammatory corticosteroids in disease management. However, a large proportion (~40%) of patients with the treatment of anti-TNF-α antibodies are discontinued or withdrawn from therapy because of (1) primary nonresponse, (2) secondary loss of response, (3) opportunistic infection, or (4) onset of cancer. Therefore, the development of novel and effective therapeutics targeting specific signaling pathways in the pathogenesis of IBD is urgently needed. In this comprehensive review, we summarize the recent advances in drug discovery of new small molecules in preclinical or clinical development for treating IBD that target biologically relevant pathways in mucosal inflammation. These include intracellular enzymes (Janus kinases, receptor interacting protein, phosphodiesterase 4, IκB kinase), integrins, G protein-coupled receptors (S1P, CCR9, CXCR4, CB2) and inflammasome mediators (NLRP3), etc. We will also discuss emerging evidence of a distinct mechanism of action, bromodomain-containing protein 4, an epigenetic regulator of pathways involved in the activation, communication, and trafficking of immune cells. We highlight their chemotypes, mode of actions, structure-activity relationships, characterizations, and their in vitro/in vivo activities and therapeutic potential. The perspectives on the relevant challenges, new opportunities, and future directions in this field are also discussed.
Lay Summary
This comprehensive review summarizes recent advances in drug discovery of new small molecules that target biologically relevant pathways under preclinical and clinical development for the treatment of inflammatory bowel disease. The challenges, opportunities, and future directions are also discussed.
Introduction
Inflammatory bowel diseases (IBDs) include ulcerative colitis (UC) and Crohn’s disease (CD), which are chronic diseases with recurrent symptoms and significant morbidity afflicting over 1 million people in North America and 4 million worldwide.1–3 Long-term persistence of chronic inflammation in IBD contributes to neoplastic transformation and the development of colitis-associated colorectal cancer, responsible for the cause of death in 10% to 15% of IBD patients.4,5 Similarly, long-term persistence of CD is associated with symptomatic small bowel strictures in up to 15% of patients.6 Over the past decade, IBD has increased worldwide and has become an expanding global health problem.7,8
Inflammatory bowel disease is characterized by chronic inflammation and tissue destruction along the gastrointestinal (GI) tract with clinical symptoms of abdominal pain, persistent diarrhea, rectal bleeding, fatigue, and weight loss.9 Despite ongoing inflammation in the GI tract of both CD and UC patients, several important clinical and pathological features differ between them.10–12 As shown in Figure 1, CD can affect any part from the mouth to the anus, especially the portion of the small intestine besides the colon. The damaged areas in CD appear in patches that are next to areas of healthy tissue, and inflammation may reach through the multiple layers of the walls of the GI tract (Figure 1A). In contrast, UC selectively occurs in the large intestine. The damaged areas are continuous, beginning at the rectum and ascending into the colon, and inflammation is only present in the innermost layer of the colon lining (Figure 1B).
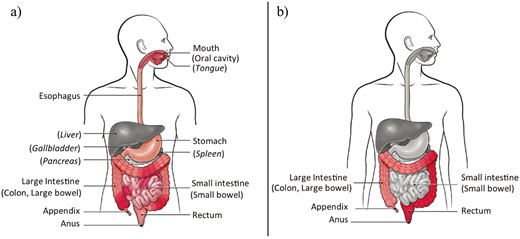
A, Pathological feature of Crohn’s disease (CD). B, Pathological feature of ulcerative colitis (UC). The 2 images were retrieved with permission from the Centers for Disease Control and Prevention (CDC) website: https://www.cdc.gov/ibd/what-is-IBD.htm.
Although the exact pathogenesis of IBD remains poorly understood, IBD is of multifactorial etiology due to interactions between genes, the immune system, gut microbiota, and other environmental aspects.13–15 A properly functioning immune system could effectively defend and kill foreign invaders, such as viruses, bacteria, fungi, and other microorganisms to protect the body. However, abnormal immune responses to environmental trigger(s) can occur in genetically susceptible patients with IBD, resulting in persistent intestinal inflammation and fibrosis. Additionally, there also appears to be a genetic component for IBD. Those who have inherited genes associated with cytokine regulation can develop inappropriate immune responses, leading to development of IBD.
No cure is currently available for IBD due to incomplete knowledge of its pathophysiology and a lack of effective medications.16–26 In addition to limited efficacy,16,27 some available agents (eg, corticosteroids) have been associated with serious side effects.28–30 Antibodies (eg, Remicade) against tumor necrosis factor-α (TNF-α) are more effective and have become the mainstay of long-term treatment for severe IBD.31,32 However, a large proportion (roughly 40%) of patients are initially unresponsive or lose their response to anti-TNF-α therapy. For those treated with anti-TNF-α for long durations, increased risks of infections, cancer, or death are seen.33–35 Therefore, the development of novel and effective therapeutics targeting IBD-specific signaling pathways has garnered a great deal of interest for maximizing the probability of treatment success. In this review, we aim to summarize the recent advances in drug discovery of new small molecules that regulate biologically relevant signaling pathways such as various intracellular enzymes, G protein-coupled receptors (GPCRs), integrins, inflammasomes, and epigenetic regulators under either preclinical or clinical development for the treatment of IBD. These representative drug targets are summarized and depicted in Figure 2. We highlight their chemotypes from hit-to-lead identifications and optimizations, mode of actions, structure-activity relationships (SARs), and pharmacological characterizations in vitro and in vivo, as well as their therapeutic potential for IBD. Perspectives on relevant challenges, new opportunities, and future directions in this field of drug discovery and development are also briefly discussed.
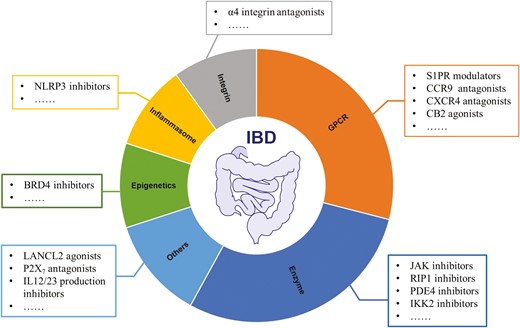
Summary on representative drug targets explored towards drug discovery and development of IBD therapeutics.
Targeting JAK-STAT Signaling Pathway
Cytokines are released by activated inflammatory cells in IBD. These proteins bind to specific receptors promoting inflammation, chemotaxis, and cell differentiation. Of these, the type 1 and 2 cytokine receptors are thought to play a key role in leukocyte activation and cellular inflammation in the gut. Binding the type 1/2 cytokine receptors activates key intracellular kinases known as the Janus kinases (JAKs). Janus kinases are intracellular nonreceptor tyrosine kinases that play a key role in cytokine-mediated signaling by activation of signal transducer and activator of transcription (STAT) proteins (Figure 3).36,37 The family of JAKs is composed of 4 members: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2).38,39 Janus kinase 1, JAK2, and TYK2 are ubiquitously expressed in all kinds of cells, whereas JAK3 is expressed mainly in hematopoietic, myeloid, and lymphoid cells.40 The 4 JAKs possess 2 kinase domains, JH1 and JH2.41 The domain JH1 is a true active kinase domain with adenosine triphosphate (ATP)–binding site, whereas JH2 is likely an inactive pseudo-kinase domain without catalytical functions.42 Blockade of the JAK/STAT signaling pathway leads to inhibition of several pro-inflammatory cytokines, including interferon (IFN)-α, IFN-γ, interleukin (IL)-2, IL-6, IL-12, IL-23, and GM-CSF.43,44 Therefore, the JAKs have been recognized as very attractive targets for drug development against inflammatory and autoimmune diseases.45–51 Over the past decade, various classes of small molecule inhibitors targeting JAKs have been developed, and some of them (chemical structures of representative drugs or drug candidates 1–9 are shown in Figure 4) have been approved or enrolled into different phases of human clinical trials focused on IBD therapies.52–56
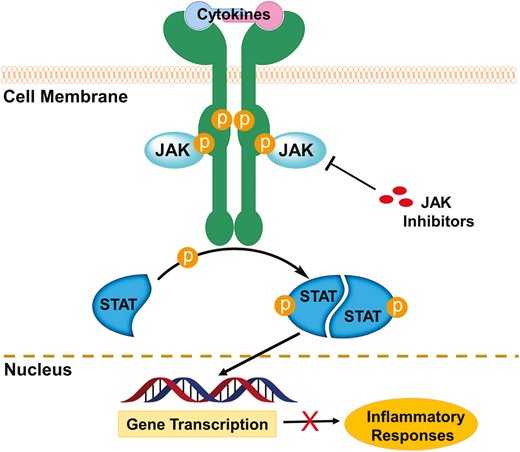
JAK inhibitors block chronic colonic inflammation of IBD.
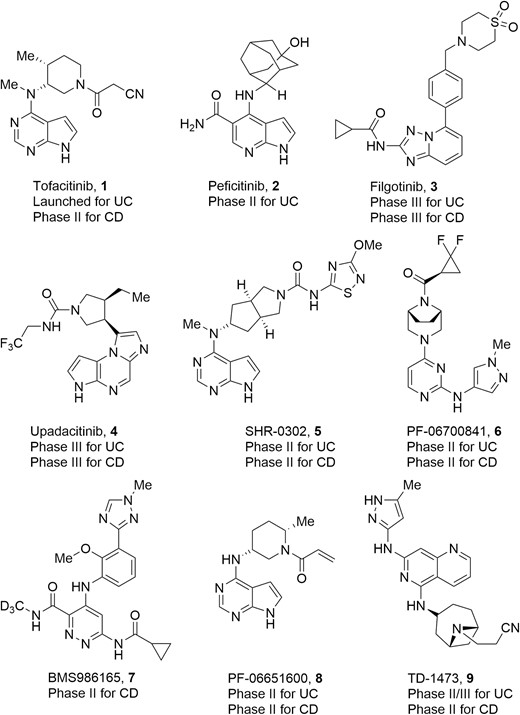
Representative drugs and drug candidates targeting JAKs for IBD therapies.
Pan-JAK Inhibitors
The high sequence homology of the ATP-binding site within the JAK family members (JH1 domains)57 makes the achievement of high selectivity for a specific JAK challenging. As a result, earlier efforts were focused on the discovery of nonselective JAK inhibitors. Tofacitinib (Figure 4, diagram 1) from Pfizer is the first-in-class pan-JAK inhibitor that exhibited potent inhibitory activity against JAK1, JAK2, JAK3 (IC50 = 3.2 nM, 4.1 nM, 1.6 nM, respectively), and, to a lesser extent, TYK2 (IC50 = 34.0 nM).58 The discovery of tofacitinib began with structural modifications on the hit 10 with a pyrrolo[2,3-d]pyrimidine scaffold that was identified by means of a high throughput screening method (Figure 5A). The binding mode of tofacitinib with JAK3 was confirmed by cocrystal analysis.59 As shown in Figure 5B, the pyrrolopyrimidine heterocycle of tofacitinib (magenta) forms key hydrogen bonds with the hinge region of the kinase. The cyanoacetamide group is directed into the glycine-rich loop (P-loop), and the methyl at the piperidine ring binds into a lipophilic pocket in the C-terminal lobe at the base of the active site. In preclinical studies, tofacitinib exhibited dose-dependent efficacy in multiple inflammatory models. In both rat and mouse models of arthritis, tofacitinib normalized inflammatory mediators implicated in disease and reversed bone resorption. In an oxazolone-induced rat model of chronic dermatitis, orally administered tofacitinib significantly inhibited ear swelling and epidermal thickening, and the effect at a dose of 10 mg/kg was comparable with that of cyclosporin A and etanercept.60 Tofacitinib has been approved by the United States Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis (RA) and psoriatic arthritis (PsA). Recently, a phase 3 OCTAVE program on the efficacy of tofacitinib in UC patients for 52 weeks demonstrated that 34.3% of the patients in the treatment group receiving 5 mg twice a day and 40.6% of the patients in the treatment group receiving 10 mg twice a day were in remission compared with 11.1% in the placebo group (P < .001).61 Furthermore, mucosal healing occurred more significantly in patients of tofacitinib groups (37.4% and 45.7% in the 5-mg and 10-mg treatment groups, respectively) compared with the placebo group (13.1%; P < .001).61 Notably, a post hoc analysis revealed that patients in tofacitinib groups had significant decrease from baseline in stool frequency subscore and rectal bleeding subscore vs placebo starting from the third day of the treatment.62 Based on these results, tofacitinib has also been approved by FDA and European Medicines Agency (EMA) for the treatment of patients with moderately to severely active UC after failure or intolerance to either conventional therapy or biologics. However, further development on tofacitinib for treating CD was discontinued after failing to achieve significant clinical efficacy in phase 2 clinical trials.63,64
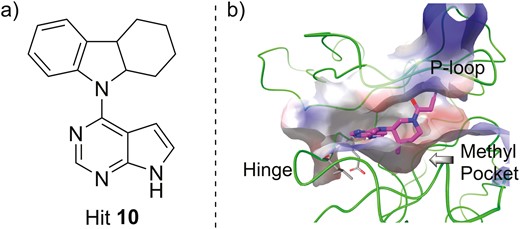
A, The chemical structure of hit 10 as the lead compound. B, The cocrystal structure of tofacitinib (magenta) with JAK3 (green, PDB ID: 3LXK).
Another pan-JAK inhibitor developed by Janssen was discovered by the optimization seen in Figure 6, diagram 11, which contains a 1H-pyrrolo[2,3-b]pyridine core with micromolar activities against JAK1 to JAK3 (Figure 6).65 Chemical modification of C4-substituent directed to the hydrophobic cavity and the introduction of a carbamoyl group at the C5 position led to diagram 12 in Figure 6, which had significant improvement of in vitro activities. However, it had poor metabolic stability in liver microsomes. To address this issue, the C4-cyclohexane ring was replaced by 2-adamantyl moiety, and a polar hydroxyl group was assembled, resulting in peficitinib (Figure 6, diagram 2). Peficitinib exhibited highly potent JAK1, JAK2, JAK3, and TYK2 inhibition. A slight selectivity for JAK3 over JAK1, JAK2, and TYK2 (IC50 = 0.71 nM, 3.9 nM, 5.0 nM, and 4.8 nM, respectively) and improved metabolic stability were observed.66 Moreover, peficitinib also showed good pharmacokinetics (PK) profile, high oral bioavailability, and dose-dependent efficacy.67 In an adjuvant-induced rat model of arthritis, peficitinib dose-dependently inhibited bone destruction and paw swelling by prophylactic or therapeutic oral dosing regimens. Peficitinib also showed efficacy in the model via continuous intraperitoneal infusion. Currently, peficitinib has been approved in Japan for RA and is also under phase 2 study for UC. Of patients with moderate to severe UC receiving ≥75 mg peficitinib daily, a higher rate achieved clinical response, remission, and mucosal healing, even though no significant dose-response relationship was observed.68
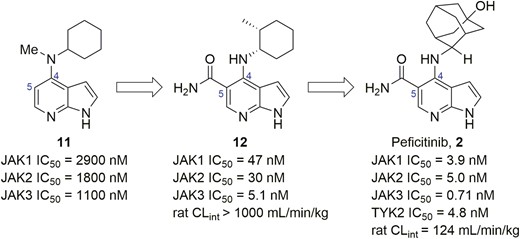
Evolution of compound structures from hit 11 to peficitinib (2).
Selective JAK Inhibitors
The first-generation nonselective JAK inhibitors have shown robust efficacy in various inflammatory and autoimmune diseases including UC and CD. However, dose-limiting tolerability and safety issues due to inhibition of erythropoietin signaling (EPO) mediated by JAK2 led to anemia and neutropenia being observed and thus limited the clinical application.69,70 Such safety concerns have resulted in the development of a second-generation of highly selective JAK inhibitors. Filgotinib (Figure 7, diagram 3) is the first inhibitor designed for improving selectivity for JAK1 over JAK2 to avoid undesired side effects.71–73 It was discovered and developed by using triazolo[1,5-a]pyridine (Figure 7, diagram 13) as a starting point for integrated lead optimization. The study of the SAR revealed that the cyclopropyl moiety appeared to be a preferred fragment for JAK1 selectivity over JAK2. The phenyl group at the 5-position of the triazolo[1,5-a]pyridine core placed a hydrophobic subpocket of JAK1, forming hydrophobic interaction with Leu881 (Leu855 in JAK2), Val938 (Val863 in JAK2), Ser963 (Ser936 in JAK2), Leu1010 (Leu983 in JAK2), and Gly1020 (Gly993 in JAK2). Modifications of the phenyl moiety further improved JAK1 selectivity and absorption, distribution, metabolism, and excretion (ADME) properties, eventually leading to filgotinib (Figure 7, diagram 3). In a head-to-head enzymatic assay performed at 1-mM ATP, filgotinib exhibited a greater selectivity for JAK1 over the other JAKs.52 In human whole blood (hWB), filgotinib potently inhibited JAK1 with an IC50 value of 629 nM, exhibiting >27-fold selectivity for JAK1 over JAK2. Filgotinib had a good PK profile and showed excellent in vivo efficacy in the dextran sulfate sodium (DSS)-induced mouse model of colitis. After treatment with filgotinib at 10 mg and 30 mg/kg/day, macroscopic disease scoring indicated moderate and strong protection against experimental colitis, respectively. Moreover, histological scoring also revealed 19% and 43% lower disease severity, respectively, compared with controls. Very recently, regulatory submissions of filgotinib for the treatment of RA have been approved by EMA and Ministry of Health, Labor and Welfare (MHLW) of Japan. Furthermore, a just-completed phase 2b/3 SELECTION trial (NCT02914522) evaluating the efficacy and safety of filgotinib in patients with moderately to severely active UC demonstrated that a statistically significantly higher proportion of patients achieved inducing clinical remission at week 10 and maintained clinical remission at week 58 compared with placebo when treated with 200 mg of filgotinib daily. The incidence of adverse events was low and comparable across treatment groups.74 Other Phase 3 studies on UC (NCT02914535) and CD (NCT02914600, NCT02914561) are currently underway.
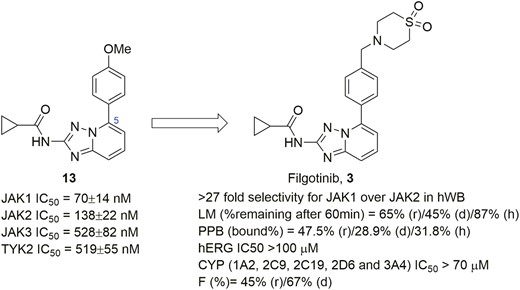
Evolution of compound structures from hit 13 to a selective JAK1 inhibitor filgotinib (3).
Upadacitinib (Figure 8, diagram 4), another JAK1 selective inhibitor developed by AbbVie, has a cellular IC50 value of 14 nM against JAK1 and displayed high selectivity over JAK2 (>40-fold), JAK3 (>130-fold), and TYK2 (>190-fold) in the Ba/F3 cellular assay. Moreover, in physiologically relevant cellular systems, upadacitinib was found to selectively inhibit the JAK1 dependent IL-2, cytokines IL-6, IFNγ, and oncostatin M (OSM), with weak potency against reticulocytes and natural killer (NK) cells related to the tolerability issues of tofacitinib, which was translated into a more favorable benefit-to-risk profile.75 As shown in Figure 8, the identification of upadacitinib began with the chemical modification of a novel tricyclic pyrrolopyrazine (Figure 8, diagram 14), which possessed an IC50 value of 290 nM against JAK1 with a 40-fold selectivity for JAK1 over JAK2.76 Investigation on both the size and nature of rings attached to tricyclic core led to what is seen in Figure 8, diagram 15, with an improved JAK1 cellular activity (IC50 = 80 nM) and JAK1/2 selectivity (227-fold). Subsequently, given that the P-loop in JAK1 can exist in a “closed” conformation compared with JAK2 because of amino acid differences within the loop, a key trifluoroethyl chain was designed to occupy van der Waals interaction space in the loop to offer an induced fit into JAK1, thereby conferring selectivity and increased activity. In preclinical models of RA, upadacitinib alleviated loss of bone volume and improved synovial hypertrophy, inflammation, cartilage damage, and bone erosion in a dose-dependent manner. Upadacitinib recently was approved for treating moderate to severe RA, and a phase 2 development for CD and UC has also achieved positive efficacy and favorable safety profiles.77,78 Further phase 3 trials in CD patients (NCT03345823, NCT03345836, NCT03345849) and UC patients (NCT03006068, NCT02819635, NCT03653026) are ongoing. Notably, the current phase 3 induction study (U-ACHIEVE) shows that upadacitinib (45 mg, daily) meets the primary end point of clinical remission after week 8 and all ranked secondary end points in adult patients with moderate to severe UC.79
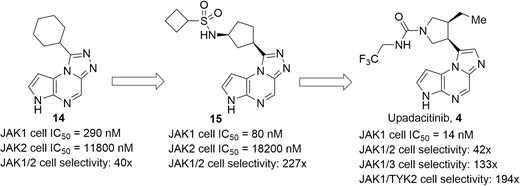
Evolution of compound structures from hit 14 to the JAK1 selective inhibitor upadacitinib (4).
In addition, SHR0302 (Figure 4, diagram 5) is also a highly selective JAK inhibitor with stronger JAK1-binding activity than others JAKs (>10-fold over JAK2, 77-fold over JAK3, 420-fold selectivity for TYK2). A preclinical study of SHR0302 demonstrated in vivo anti-inflammatory effects in a rat model of inflammation by inhibiting JAK1-STAT3 phosphorylation and then significantly suppressing T helper type 17 (Th17) cell function and total B cell proportion, including immunoglobulin G1 (IgG1) and IgG2a levels.80 Currently, SHR0302 is under phase 2 clinical trials in the patients with moderate to severe active CD (NCT03677648) and UC (NCT03675477).
Inspired by the success in the development of selective JAK1 inhibitors, other isoforms of selective JAKs inhibitors have also been explored. For example, PF-06700841 (Figure 9, diagram 6) is a dual JAK1/TYK2 inhibitor with IC50 values of 17 nM and 23 nM against JAK1 and TYK2, respectively, displaying a moderate selectivity over JAK2 (IC50 = 77 nM) and high selectivity over JAK3 (IC50 = 6494 nM).81 The program began with the optimization of a conformationally constrained piperazinyl-pyrimidine inhibitor (JAK1 IC50 = 23 nM, TYK2 IC50 > 4950 nM) for improvement of potency targeting TYK2 (Figure 9, diagram 16). The cocrystal of this in complex with TYK2 protein (Figure 10) indicated the conformationally rigid bridged piperazine played a key role in anchoring the cyanoacetamide to the P-loop of TYK2, making the nitrile further interact with Gly-906. In addition, it could offer an extra lipophilic interaction via filling a pocket at the bottom of the binding site. Subsequent modification on bridged diamines resulted in what we see in Figure 9, diagram 17, with a significant increase in TYK2 inhibition (TYK2 IC50 = 32 nM). Changing the benzene ring into a heteroaromatic ring gave the structural formula in Figure 9, diagram 18, which displayed little effect on the enzymatic potency but improved selectivity over EPO inhibition in the JAK2-driven hWB assay. Further efforts on exploring the amide moiety produced PF-06700841, with a particularly effective and balanced profile. In the preclinical rat model of adjuvant-induced arthritis, the increase in paw volume was significantly reduced and dose-dependent in PF-06700841 (3, 10, and 30 mg/kg, once daily) treated groups. In addition, PF-06700841 is currently being evaluated in clinical phase 2 development for CD (NCT03395184) and UC (NCT02958865).
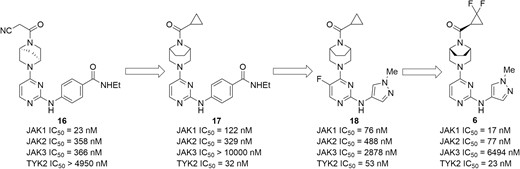
Evolution of compounds from lead 16 to a dual JAK1/TYK2 inhibitor PF-06700841 (6).
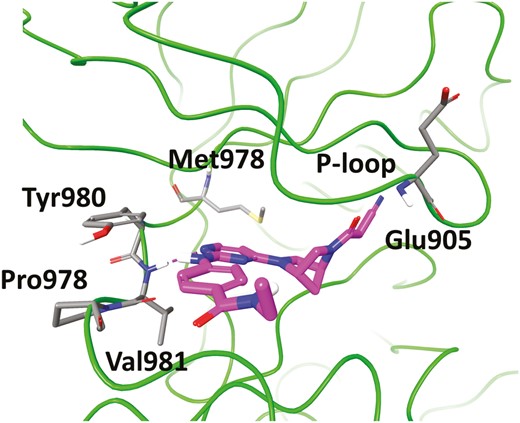
Cocrystal structure of 16 (magenta) with TYK2 (green, PDB ID: 6DBK). The key residues at the binding site are shown in stick representation.
Recently, Bristol Myers Squibb advanced the first selective and allosteric inhibitor of TYK2, BMS986165 (Figure 11, diagram 7), into phase 2 clinical trial for treating moderate to severe CD (NCT03599622). As shown in Figure 11, BMS986165 was discovered by the optimization of the nicotinamide (Figure 11, diagram 19), which was identified by a scintillation proximity assay (SPA)–based high throughput screen targeting the TYK2 pseudokinase domain.82 Extensive SAR efforts were explored, leading to the structural formula in Figure 11, diagram 20, which showed nearly complete selectivity against TYK2 JH2 inhibition by introducing a key deuteromethyl amide group. Notably, the structural feature occupied the unique alanine pocket in the JH2 ligand-binding domain of TYK2 to achieve high selectivity. Deuteration was used to suppress an N-demethylation metabolic pathway, blocking the generation of nonselective primary amide metabolite, thus accessing superior exposure. To further improve potency in hWB and mitigate the human Ether-à-go-go-Related Gene (hERG) issue, the effect of the C4 and C6 substituents of the structural formula in Figure 11, diagram 20, was carefully examined, eventually leading to the discovery of BMS986165 as a potent and allosteric inhibitor of TYK2; this acts by binding to the JH2 domain of TYK2 with high affinity (IC50 = 0.2 nM) and achieves remarkable selectivity over the JH1 domains of JAKs family.83 Of note, BMS986165 displayed IC50 values of 5 nM and 13 nM in the IFN-α cellular assay and hWB, respectively, and TYK2 selectivity of >300-fold and >5000-fold in JAK1/3-dependent IL-2 stimulated T cells and in JAK2-dependent EPO-induced TF-1 cells, respectively. Furthermore, BMS986165 also showed excellent pharmacokinetic properties and demonstrated significant efficacy in preclinical models of IBD.84 In the anti-CD40-induced colitis mouse, BMS986165 was highly efficacious at protecting against both weight loss and histologically evident colitis and being an effective anti-p40 antibody.
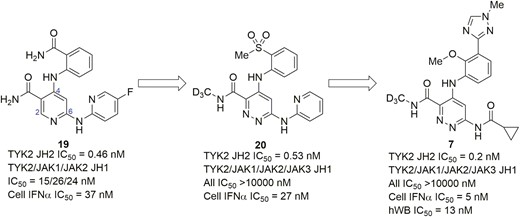
Evolution of compounds from lead 19 to a TYK2-selective allosteric inhibitor BMS986165 (7).
Considerable efforts have also been devoted to the discovery of highly selective JAK3 inhibitors. Analysis of crystal structures for the 4 JAK isoforms indicates that unique residue Cys-909 in the ATP-binding pocket of JAK3 can be used to impart selectivity for JAK3 over other family members in a covalent manner.52 By employing this mechanism, the highly selective irreversible inhibitor of JAK3, PF-06651600 (Figure 12, diagram 8), has been developed by Pfizer. As presented in Figure 12, the primary design used an acrylamide warhead to replace the cyanoacetamide in the tofacitinib core, which led to the structural model in Figure 12, diagram 21, with micromolar inhibition of JAK3 and poor JAK family selectivity.85 Modeling studies illustrated that the pyrolopyrimidine scaffold seen in Figure 12, diagram 21, was well-ordered in the active site, but the piperidine-containing acrylamide group was highly disordered due to a raised energy conformation. To address this issue, the N-methyl and the ring methyl groups were removed to afford the analog in Figure 12, diagram 22, which showed a robust increase in potency against JAK3 (IC50 = 56 nM), with no activity against JAK1, JAK2, or TYK2 (IC50 > 10 000 nM). Further optimization on substituents in the piperidine ring improved the cellular activity and ADME properties, resulting in giving PF-06651600. In a biochemical assay at 1 mM ATP concentration, PF-06651600 inhibited JAK3 kinase activity with an IC50 of 33 nM and exhibited high selectivity over JAK1, JAK2, and TYK2. The cocrystal structure (PDB code: 5TOZ) validated that the excellent selectivity for JAK3 resulted from a covalent interaction with the unique residue Cys-909 in the ATP-binding cavity of JAK3.86 The preclinical in vivo study demonstrated that JAK3-selective inhibition with PF-06651600 was efficacious, reducing disease pathology in rat adjuvant-induced arthritis and mouse experimental autoimmune encephalomyelitis models. Additionally, PF-06651600 is currently under a clinical phase 2 trial in UC and CD patients (NCT02958865, NCT03395184).
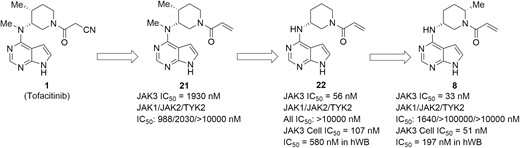
Drug design and optimization efforts culminated in the discovery of a JAK3-selective irreversible inhibitor PF-06651600 (8).
Intestinal-restricted JAK Inhibitors
An alternative strategy for alleviating undesirable side effects of nonselective JAK inhibition was focused on the development of an intestinal-restricted inhibitor of JAKs.87 An oral gut-selective pan-JAK inhibitor, TD-1473 (Figure 4, diagram 9), was designed to act locally in the gastrointestinal tract while minimizing the potential for adverse effects associated with systemic exposure.88,89 In vitro, TD-1473 potently inhibited JAK1, JAK2, JAK3, and TYK2 (IC50 values of 10.0 nM, 10.0 nM, 8.8 nM, and 9.5 nM, respectively) and reduced JAK-dependent responses in both mouse and human cells. The pharmacokinetic profile illustrated that oral dosing of TD-1473 at 10 mg/kg in mice could achieve high colonic tissue concentrations but with low plasma exposure and bioavailability (colon/plasma ratios of Cmax, and areas under the curve [AUC] were 2636 and 3883, respectively). In the oxazolone-induced colitis model, orally administered TD-1473 (0-1 mg/kg, twice daily) was found to dose-dependently attenuate colitis disease activity index and achieve maximum inhibition at 1 mg/kg without reduction in leukocyte subset counts. The phase 1 studies in healthy volunteers and UC patients revealed that TD-1473 was well tolerated in healthy participants and UC patients for up to 4 weeks without clinically significant changes in hematological or lipid parameters.90–92 Furthermore, higher rates of clinical response and mucosal healing were observed at week 4 in patients treated with all doses of TD-1473 vs placebo. These results demonstrated the potential of oral gut-selective pan-JAK inhibition for IBD therapy. Further clinical trials are underway to evaluate the safety and efficacy of TD-1473 for the treatment of CD (NCT03635112) and UC (NCT03758443).
Targeting RIP1 Kinase
Receptor-interacting protein 1 (RIP1) kinase is an important upstream serine/threonine kinase that plays a central role in regulating inflammatory responses.93–95 As a critical regulator, activated RIP1 can induce inflammation by directly regulating programmed necrosis. It can also lead to the production of pro-inflammatory cytokines, inflammasome assembly, and some forms of pathogenic apoptosis. Furthermore, RIP1 has also been proven to be a vital driver of inflammation in a variety of other pathways downstream of death receptors, such as Fas ligand (FasL), tumor necrosis factor receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL), and toll-like receptors.96–98 Hence, the blockade of this pathway has shown broad anti-inflammatory therapeutic potential.99
GlaxoSmithKline created GSK2982772 (Figure 13, diagram 24), which was the first-in-class RIP1 inhibitor that has been advanced into clinical trials for the treatment of inflammatory diseases. It was discovered through the optimization of a benzoxazepinone hit GSK481 (Figure 13, diagram 23) from a DNA-encoded library with high in vitro RIP1 potency (IC50 = 1.6 nM) and kinase selectivity.100 As shown in Figure 13, changing the isoxazole of GSK481 into a triazole was key to improve lipophilicity, solubility, and rat oral exposure, leading to the identification of clinical candidate GSK2982772, which exerted excellent activity and had highly favorable pharmacokinetic and physicochemical properties; GSK2982772 reduced spontaneous production of cytokines (such as IL-1β and IL-6) in biopsies from UC patients in overnight incubations. In addition, GSK2982772 had a weak inhibition of hERG in human embryonic kidney (HEK-293) cells with an estimated IC50 of 195 μM and showed weak activation of the human Pregnane X receptor (EC50 = 13 μM). It has also been shown to be RIPK1-dependent in an acute mouse model evaluating protection from TNF-induced lethal shock.101 Currently, GSK2982772 is under phase 2 studies in UC patients (NCT02903966).
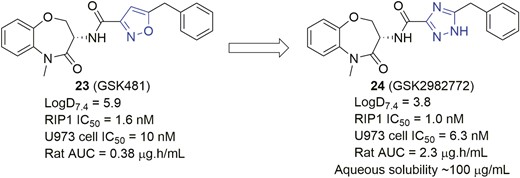
Drug design and optimization efforts culminated in the discovery of a RIP1 inhibitor GSK2982772 (24).
Targeting S1P Pathway
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid metabolite produced from the phosphorylation of sphingosine through sphingosine kinases (SphKs). sphingosine kinase regulates several physiological processes including vascular development, cardiac function, lymphocyte trafficking, and inflammation through interaction with 5 specific G protein-coupled receptors termed S1P1−5.102 The binding of S1P to related S1P receptors guides the migration of lymphocytes from the thymus and secondary lymph nodes into lymph in a S1P gradient-dependent manner, leading to an increase in circulating blood lymphocytes and inflammatory response.103 The blockage of this axis has been shown to induce the sequestration of lymphocytes preventing lymphocyte trafficking, thus resulting in inflammatory suppression. In this context, S1P receptors emerged as a novel and promising therapy for inflammatory and autoimmune diseases.104,105
To date, one S1P receptor modulator, known as fingolimod (Figure 14, diagram 25), has been approved by FDA for the treatment of relapsing-remitting multiple sclerosis.106 Several S1P receptor modulators are in various stages of clinical studies, and 4 of them are being evaluated in IBD, shown in Figure 14, diagram 25. Fingolimod was the first oral nonselective S1P receptor modulator discovered by chemical modification of the natural product myriocin. Fingolimod could be administered as a prodrug and subsequently phosphorylated in vivo to the bioactive monophosphate ester (Figure 14, diagram 26). P-Fingolimod activated S1P1,3,4,5 receptors (S1P1,4,5 EC50 = 0.3-0.6 nM, S1P3 EC50 = 3 nM) in the bioassays, but it had no activity on S1P2 receptors.107 Fingolimod has been proven to be clinically efficacious as an immunosuppressant; however, some adverse events (cardiovascular and liver) were observed in clinical studies, which were thought to be caused by agonism of the S1P3 receptor.108–110 These undesired adverse effects have prompted the investigation for next-generation S1P receptor modulators with greater selectivity against S1P3.
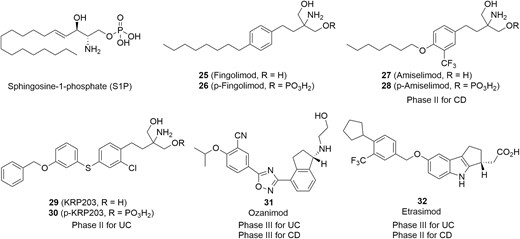
Drug candidates targeting S1P receptors for IBD therapies.
Amiselimod (Figure 14, diagram 27) is a second-generation oral selective S1P1,5 receptor modulator that was designed to lower the bradycardia risk associated with Fingolimod and other S1P receptor modulators.111 It acted by metabolizing the prodrug to its active phosphate (Figure 14, diagram 28) in vivo. In an intracellular Ca2+ mobilization assay, Amiselimod exhibited high potent S1P1 and S1P5 receptors agonism (S1P1 EC50 = .075 nM, S1P5 EC50 = 0.47 nM, respectively), with lower agonist activity for S1P4 (EC50 = 122.3 nM) and no distinct agonist activity for S1P2 and S1P3 receptors (EC50 > 10 000 nM). In a chronic colitis mouse model, Amiselimod effectively alleviated the severity of the disease.112 Further evidence showed the concentration of p-Amiselimod was lower than that of p-Fingolimod in rat heart tissue after oral dosing at 1 mg/kg, potentially leading to the minimal cardiac effects of Amiselimod. A telemetry study in monkeys demonstrated that Amiselimod did not affect heart rate or electrocardiogram parameters.111 A phase 1 study demonstrated that Amiselimod has a more favorable cardiac safety profile than Fingolimod (NCT02193217).113 Currently, Amiselimod is under phase 2 studies in CD patients (NCT02378688 and NCT02389790).
In a similar fashion, KRP-203 (Figure 14, diagram 29) from Kyorin and Novartis also acts as a selective S1P receptor modulator through its active metabolite (Figure 14, diagram 30). It showed significant agonistic activities on S1P1 (EC50 = 0.84 nM), with single-digit nanomolar activity on S1P4 (EC50 = 9.61 nM) but almost none on S1P3 (EC50 > 1000 nM). In an IL-10 gene-deficient (IL-10-/-) mouse model of colitis, after 4 weeks of treatment with KRP-203 at a dose of 0.3 mg/kg/day, a remarkable decrease in the severity of colitis was observed without loss of body weight.114 Currently, KRP-203 is under phase 2 trials for treating moderately active refractory UC (NCT01375179).
Alternatively, Ozanimod (Figure 14, diagram 31) is a novel oral S1P1 and S1P5 selective modulator that acts by binding directly to S1P receptors rather than by converting to its active metabolite. In vitro bioassays for receptor binding and function revealed that Ozanimod has highly potent agonist activity on S1P1 (EC50 = 0.41 nM) and S1P5 (EC50 = 11 nM) with high selectivities over S1P2, S1P3, and S1P4 (EC50 > 10 000 nM, >10 000 nM, and >7865 nM, respectively).115,116 In preclinical studies, Ozanimod showed high oral bioavailability and a half-life that supported once-daily dosing. Further in vivo study demonstrated that Ozanimod reduced inflammation and disease parameters in 2 models of IBD.115 Currently, phase 3 clinical trials of Ozanimod for UC (NCT02531126) and CD (NCT03440385, NCT03464097, NCT03467958) are underway.
Etrasimod (Figure 14, diagram 32) is in clinical development for treatment of IBD and selectively targets S1P1 (EC50 = 6.1 nM), S1P4 (147 nM), and S1P5 (EC50 = 24.4 nM) with high selectivity over S1P2 and S1P3 (EC50 > 10 000 nM) in in vitro signaling assays.117 Etrasimod has a favorable PK/PD (pharmacokinetics/pharmacodynamics) profile and significantly suppressed weight loss and colon inflammation at a dose of 3 mg/kg/day in a T-cell adoptive transfer mouse model of colitis.118 A phase 2 OASIS study (NCT02447302) in patients with moderate to severe UC for 12 weeks demonstrated that a 2-mg dose of etrasimod, compared with placebo, was significantly efficacious in achieving dose-dependent endoscopic improvement (43.2% vs 16.3%, P = .003), histological improvement (31.7% vs 10.2%, P = .006), histological remission (19.5% vs 6.1%, P = .027), and mucosal healing (19.5% vs 4.1%, P = .01).119 Currently, further phase 3 clinical trials for UC (NCT03996369, NCT03945188, NCT03950232) and phase 2 clinical trial for CD (NCT04173273) are ongoing.
Targeting Integrins
Integrins are broadly expressed transmembrane glycoproteins on the cell surface that can bind with adhesion molecules to mediate cell trafficking into peripheral tissues.120–123 Integrins are obligate heterodimers that consist of α and β subunits, and around 24 unique integrins are generated through different combinations of the α and β subunits.124 Among them, α4β1 mainly mediates the egress of lymphocytes both in the gut and brain, whereas α4β7 is responsible for attracting T cells into the intestine.125,126 It has been shown that IBD is characterized by the persistent recruitment of large quantities of lymphocytes from the blood to the inflamed intestinal mucosa.13,127,128 Therefore, inhibition of lymphocyte trafficking by blocking the interaction of α4β1 and α4β7 integrins with their respective ligands vascular cell adhesion molecule-1 (VCAM-1) and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) has emerged as an attractive therapeutic approach for IBD.129,130
EA Pharma developed AJM300 (Figure 15, diagram 33), an orally active antagonist that targets both α4β7 and α4β1 integrins. This structural formula is an esterified prodrug with limited pharmacological action by itself but converts to the active metabolite HCA2969 (Figure 15, diagram 34) through hydrolysis by carboxylesterase in the liver. In in vitro cell binding assay, HCA2969 potently inhibited the binding of α4β1 expressing cells (Jurkat) to VCAM-1 (EC50 = 5.8 nM) and the binding of α4β7-expressing cells (RPMI-8866) to MAdCAM-1 (EC50 = 1.4 nM). After single oral dosing of AJM300 in mice, lymphocyte homing to Peyer’s patches was dose-dependently suppressed, and the peripheral lymphocyte count was increased in the same dose range. In a mouse model of colitis induced by adoptive transfer of IL-10–deficient CD4+ T cells, AJM300 resulted in a decrease in T lymphocytes within lamina propria and reduced histopathological inflammatory score.131 A phase 2 randomized, controlled trial in patients with moderately active UC has shown that AJM300 at doses of 960 mg 3 times daily for 8 weeks led to higher rates of clinical response (62.7% vs 25.5%), clinical remission (23.5% vs 3.9%), and mucosal healing (58.8% vs 29.4%) than placebo. Moreover, no cases of progressive multifocal leukoencephalopathy (PML), a proven risk caused by inhibition of α4β1-VCAM 1 interaction, were observed.132 Currently, AJM300 is being evaluated in a phase 3 study for UC (NCT03531892).

Small molecule α4 integrin antagonists for potential IBD therapies.
Targeting CCR9
The C-C chemokine receptor 9 (CCR9) is expressed mainly on the surface of circulating lymphocytes and is responsible for homing of T lymphocytes to the small intestine tissue through the interaction with chemokine ligand 25 (CCL25; ie, thymus-expressed chemokine TECK) on the intestinal epithelial cells.133–135 Therefore, the blockade of the CCR9-CCL25 axis has been proposed as a therapeutic approach for IBD.136,137
Vercirnon (Figure 16, diagram 35; ie, GSK-1605786, CCX-282), developed by GlaxoSmithKline under license from ChemoCentryx for IBD therapies, represents the first selective antagonist of the CCR9 in clinical development (Figure 16).138 Vercirnon selectively binds to the intracellular side of the receptor, exerting allosteric antagonism and preventing G-protein coupling. In in vitro assay systems, vercirnon effectively inhibited CCL25-induced function with nanomolar potency.138,139 In a TNFΔARE mouse model of CD, both doses of vercirnon (10 mg/kg and 50 mg/kg) showed robust inhibition of inflammation and resulted in histopathologic normalization. At the high dose, 70% of mice in the treatment group were free of any histological indication of inflammation, and the remaining 30% experienced only a mild degree of inflammation.139 Vercirnon has completed successful phase 2 clinical trials in CD137,140 but failed to meet the primary end point in a subsequent phase 3 trial, attributed to the patient population selected for the study and the relatively poor PK profile.141 To improve the PK properties of vercirnon, a series of CCR9 antagonists containing a 1,3-dioxoisoindoline skeleton was discovered.138 Among them, the structural formula MOLT-FLIPR K = 6 nM (Figure 16, diagram 36) exhibited high in vitro potency at CCR9 with excellent PK properties. In an acute DSS-induced colitis mouse model, this formula showed a significant reduction in disease activity index (DAI) in a dose-dependent manner and provided excellent protection against the induced colitis at the high dose of 100 mg/kg. In a chronic model lasting 30 days, the formula in Figure 16, diagram 36, also demonstrated substantial protection against the cycles of DSS-induced colitis, and highly significant improvements on DAI were observed. This preclinical data suggest that this molecular structure could be efficacious in treating IBD patients.
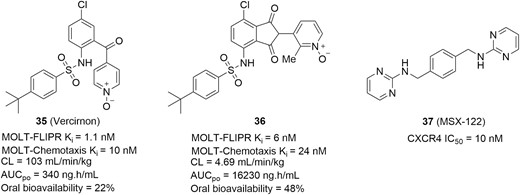
Small molecule chemokine receptor antagonists for IBD therapies.
Targeting CXCR4
The CXC chemokine receptor 4 (CXCR4) is broadly expressed in a variety of inflammatory cells and selectively binds to stromal cell-derived factor-1 (SDF-1, also called CXCL12) that attracts inflammatory cells to local tissue.142,143 Therefore, disruption of the CXCR4-CXCL12 interaction has also been proven to be an effective approach in preventing the inflammation, including for IBD. The compound MSX-122 (Figure 16, diagram 37) is an orally bioavailable, partial antagonist of CXCR4 with an IC50 value of 10 nM.144 In a DSS-induced mouse model of colitis, MSX-122 significantly inhibited epithelial erosion and crypt destruction at a dose of 10 mg/kg/day via intraperitoneal injection (i.p.). After treating for 7 days, MSX-122 attenuated the production of TNF-α, IL-6, and IL-1β. In addition, MSX-122 showed signs of recovery on colonic damage with crypt destruction, decreased cytokine concentration, and increased inflammatory cell infiltration at day 10 compared with the control group.
Targeting PDE4
Phosphodiesterase 4 (PDE4) belongs to intracellular nonreceptor enzymes that specifically catalyze the breakdown of cAMP (cyclic adenosine monophosphate) to inactive AMP in several types of cells, including inflammatory cells.145,146 The elevation of the intracellular level of cAMP caused by PDE4 inhibition has been demonstrated to downregulate the release of pro-inflammatory cytokines (eg, TNF-α, IFNγ, IL-12, IL-17, and IL-23) and upregulate the production of anti-inflammatory cytokine IL-10.147 Consequently, targeting PDE4 represents a valuable strategy for the treatment of inflammatory disorders, including IBD.148–150
Apremilast (Figure 17, diagram 38) is an orally available and PDE4-selective inhibitor that blocks the hydrolysis of cAMP by binding to the catalytic site of the PDE4 enzyme (IC50 = 74 nM; Figure 17). Apremilast has shown highly potent TNF-α inhibitory activity in lipopolysaccharide (LPS)-stimulated human peripheral blood mononuclear cells (hPBMCs, IC50 = 77 nM) and hWB (IC50 = 110 nM).151,152 Furthermore, apremilast has a good PK profile and demonstrated significant efficacy in LPS-induced models of inflammation. Apremilast has been approved for use in psoriasis and psoriatic arthritis and is currently under development for UC therapies (NCT02289417). A completed phase 2 study in patients with moderate to severe UC demonstrated that apremilast achieved clinical remission and ameliorated biomarker levels and histology at a dose of 30 mg twice daily relative to placebo (31.6% vs 13.8%, P < .05) after 12 weeks, and the safety profile was acceptable.153 A phase 3 clinical trial is expected to be launched soon.
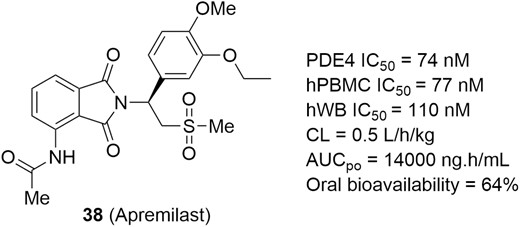
PDE4 inhibitor for potential IBD therapies.
Targeting LANCL2
Lanthionine synthetase C-like 2 (LANCL2), a member of the LANCL protein family, is broadly expressed in epithelial cells and the immune system including colon, spleen, thymus, and Peyer patches.154 Its natural ligand abscisic acid (ABA; Figure 18, diagram 39) was identified as a new endogenous mammalian hormone that exerted anti-inflammatory effects.155–157 Selective binding between LANCL2 and ABA increases the level of intracellular cAMP and activates protein kinase A (PKA), resulting in suppression of inflammation. Therefore, LANCL2 has been emerged as a new therapeutic target for treating inflammatory diseases.158
Additionally, BT-11 (Figure 18, diagram 41), developed by Landos Biopharma for the treatment of IBD, is an orally active and gut-restricted LANCL2 agonist with a Kd value of 7.7 µM for binding to LANCL2 (Figure 18).159–161 The discovery of BT-11 began with structural modification on the hit NSC61610 (Figure 18, diagram 40) that was identified by a virtual screen from public databases (eg, ZINC natural products, ChemBridge, NCI Diversity Set II, and FDA-approved drugs) based on the predicted structure of LANCL2. In a DSS-induced colitis mouse model, BT-11 (8 mg/kg/day) demonstrated a lower disease activity index, decreased colonic inflammatory lesions, and suppressed inflammatory cytokines (eg, IFN-γ, and TNF-α) within the gut. Further studies in LANCL2−/− mice (lacking LANCL2) abrogated beneficial actions of BT-11, indicating a high selectivity for the target. Moreover, BT-11 was tolerated in a preclinical study and has a favorable safety profile in healthy human subjects.162,163 Currently, a phase 2 trial in UC (NCT03861143) and CD (NCT03870334) patients is ongoing.
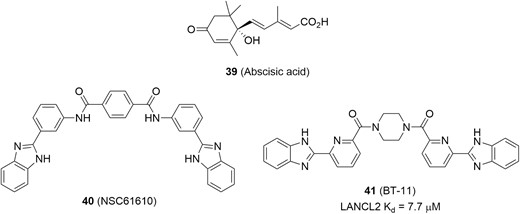
LANCL2 agonists for potential IBD therapies.
Targeting NLRP3
The nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3) inflammasome is a cytoplasmic multiprotein complex.164 This NLRP3 inflammasome is composed of a sensor (NLRP3), an adaptor (ASC), and an effector (caspase-1). Priming and activation of the NLRP3 inflammasome result in proteolytic activation of caspase-1, triggering cleavage and subsequent maturation of pro-inflammatory cytokines, such as IL-18 and IL-1β.165–167 Therefore, NLRP3 has been emerging as a promising target to develop novel and specific anti-inflammatory agents.168,169
In addition, MCC950 (Figure 19, diagram 42) is an orally potent, highly specific inhibitor of NLRP3 inflammasome, with IC50 values of 7.5 nM for mouse bone marrow–derived macrophages (BMDM) and 8.1 nM for human monocyte-derived macrophages (HMDM) cells (Figure 19). Subsequently, MCC950 has a favorable PK profile, showing high oral bioavailability (68%).170 In a spontaneous colitis murine model, MCC950 at a dose of 40 mg/kg significantly improved colon length, body weight gain, colon weight to body weight ratio, disease activity index, and histopathological scores. Moreover, MCC950 also potently inhibited the release of multiple pro-inflammatory cytokines, including IL-1β, IL-1α, IL-18, TNF-α, IFN-γ, IL6, chemokine MIP1-α, IL-17, and nitric oxide in colonic explants.171 Another specific small molecule inhibitor targeting NLRP3 inflammasome is INF39E (Figure 19, diagram 43), which inhibits NLRP3 activation via direct and irreversible interaction with NLRP3 and partial inhibition of LPS-driven pro-inflammatory gene expression. In vitro, INF39E concentration-dependently inhibited NLRP3 ATPase and prevented pyroptosis of human monocyte THP-1 cells. In a DNBS-induced rat model of colitis, INF39E displayed a dose-dependent reduction of the macroscopic damage score after treating with 12.5, 25, and 50 mg/kg/day and led to a significant reduction in IL-1β production in the intestine. Moreover, INF39E reduced colonic levels of myeloperoxidase and TNF. These results showcased the therapeutic potential of NLRP3 inflammasome inhibitor in IBD.172,173
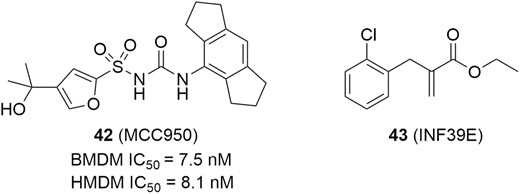
NLRP3 inhibitors for potential IBD therapies.
Targeting IKK2
The multisubunit IκB kinase (IKK) complex is the signal integration hub for NF-κB signaling pathway, which is composed of 2 the catalytic subunits IKK1 and IKK2 (ie, IKKα and IKKβ) and a regulatory subunit NEMO (ie, IKKγ).174 In the canonical NF-κB signaling pathway, the IKK complex integrates signals from NF-κB activating stimuli to phosphorylate IκB proteins, which triggers subsequent ubiquitination and proteolysis of IκB, thus making the NF-κB free to translocate to the nucleus and regulate specific gene expression to impact multiple physiological functions, particularly those with an inflammatory component.175 Even though both IKK1 and IKK2 are able to phosphorylate IκB proteins, evidence has shown that the IKK2 subunit plays the dominant role in NF-κB activation caused by pro-inflammatory stimuli.176 Therefore, the identification of selective IKK2 inhibitors as therapeutic agents in the treatment of IBD has received considerable attention.177
The structural formula in Figure 20, diagram 44 (R = Et), is a selective IKK2 inhibitor based on a tricyclic thiazole scaffold.178 It showed potent inhibitory activity against IKK2 (IC50 = 19 nM), with 22-fold selectivity against IKK1 (IC50 = 420 nM), and it had IC50 values of 220 nM and 3.5 µM in a human peripheral blood mononuclear cell assay (hPBMC) and a hWB assay, respectively. Moreover, this formula showed a reasonable profile, and twice-daily administration of this formula at 60 mg/kg was shown to be effective in a chronic intestinal inflammation model. Further investigation revealed that N-dealkylation of this formula was a major metabolic pathway in vivo, leading to the formation of the active metabolite found in Figure 20, diagram 45 (R = H) that was found to be Ames positive in mutagenicity assay.179 As a result, further optimization was mainly focused on the identification of a more metabolically stable heterocyclic core to avoid Ames genotoxicity, leading to the potent tricyclic azaindole in Figure 20, diagram 46; this compound was proven to be metabolically stable in vivo with a favorable half-life, low clearance, and acceptable bioavailability. Notably, the study of metabolites revealed that very little N-demethylation occurred in vivo, and the unsubstituted amino metabolite of the compound in diagram 46 was determined to be negative in the Ames assay. Unfortunately, this compound was unable to demonstrate efficacy in a chronic model of inflammation due to the lack of essential oral exposure.180 To address this issue, a polar nitrogen atom was assembled into the benzene ring, lowering the LogP and improving both cellular potency and pharmacokinetic parameters, eventually leading to compound BMS-066 (Figure 20, diagram 47). In the preclinical study, BMS-066 demonstrated high potency and selectivity in vitro, acceptable physicochemical and pharmacokinetic properties, and in vivo efficacy when orally administered in a mouse model of colitis, indicating its therapeutic potential for IBD.181,182

IKK2 inhibitors for potential IBD therapies.
Targeting Cannabinoid Receptor 2
Cannabinoid receptors (CBs) belong to the G protein-coupled receptor (GPCR) superfamily, which are a part of the endocannabinoid system.183 There are currently 2 known subtypes of cannabinoid receptors: CB1 and CB2. The CB1 receptor is mainly located in the brain (central nervous system) but also in the liver, lungs, and kidneys. The CB2 receptor is widely expressed in immune cells (eg, B cell, T cell, and macrophages) that are involved in various inflammatory responses, including modulating cytokine release and immune cell migration, which emphasized their important role in the control of inflammation. Selective targeting CB2 receptors to avoid CB1 receptor-mediated central nervous system (CNS) side effects has been proposed as an attractive strategy for the treatment of inflammatory diseases, including IBD.184
The compound in Figure 21, diagram 48, contains a novel 4-oxo-1,4-dihydropyridine scaffold and is a selective CB2 receptor agonist with high affinity at the CB2 receptor (Ki = 29 nM), exhibiting a >103-fold selectivity over CB1 receptor (Figure 21). In a functional assay at the CB2 receptor, this compound increased the [35S]-GTPγS binding to 212% (Emax), with an EC50 value of 12.5 nM. Interestingly, the replacement of tert-butyl with a phenyl group resulted in the compound seen in Figure 21, diagram 49, showing a shift of the functionality from agonist to inverse agonist (EC50 = 3.2 nM, Emax = 39%).185 In a 2,4,6-Trinitrobenzenesulfonic acid (TNBS)-induced colitis mouse model, intraperitoneal dosing of the compound in Figure 21, diagram 48 (10 mg/kg/day) once daily was capable of decreasing colitis macroscopic scores by 19.3% compared with the control group.186 To improve PK and in vivo efficacy, further optimizations were accessed, leading to the identification of the structural formula in Figure 21, diagram 50. It displayed high potency (Ki = 36.5 nM, EC50 = 114 nM, Emax = 213%) at the CB2 receptor, with low affinity at CB1 receptor (Ki > 1000 nM), and had an improved in vitro PK profile. The in vivo study demonstrated that this new compound drastically decreased macroscopic scores (61.4%) and significantly attenuated both histological scores (3.1) and myeloperoxidase (MPO) activity (34.1%) vs vehicle control (4.9 of histological scores; 73.7% of MPO activity).186
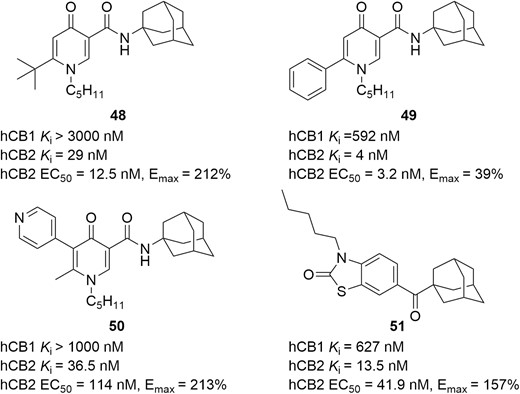
CB2 modulators for potential IBD therapies.
Another selective CB2 agonist seen in Figure 21, diagram 51, was identified through a lead optimization of the benzo[d]thiazol-2(3H)-one scaffold. It displayed higher CB2 receptor affinity (Ki = 13.5 nM) than CB1 receptor (Ki = 627 nM) and exhibited potent function activity for CB2 receptor (EC50 = 41.9 nM, Emax = 157%). Moreover, it showed no cytotoxicity, acceptable ADME-toxicity profile, a strong protective effect against experimental colitis, improved body weight (85.9 ± 2.0% vs 79.5 ± 1.8%; P = .04), a lower colon weight/size ratio (0.044 ± 0.0024 vs 0.050 ± 0.0014; P = .004), and a decrease of MPO activity (1.3 ± 0.20 vs 2.2 ± 0.31; P = .04) in the DSS-induced colitis mouse model.187
Targeting P2X7 Receptor
The P2X receptors (P2XRs) are a family of ionotropic ATP-gated receptors.188 They are cation-selective channels, equally permeable to Na+ and K+, with significant Ca2+ permeability. The family of P2XRs consists of 7 subunits (P2X1-7) that form either functional homo- or heterotrimers. Of them, the P2X7 receptor is a ligand-dependent ionic channel that opens in response to ATP binding.189 The binding of ATP induces the brief opening of a channel for small cations, permitting K+ efflux and Ca2+ and Na+ influx, which is a key step in the inflammatory process. Another well-known feature of P2X7 is its ability to form a larger pore in the case of a prolonged binding to ATP, resulting in the release of pro-inflammatory cytokines.190 The important role of the P2X7 receptor in regulating inflammation has made it a therapeutic target in numerous inflammation-related diseases such as IBD.191,192 The compound in Figure 22, diagram 52, is a pyroglutamide-based P2X7 receptor antagonist; it exhibited potent inhibitory activity against P2X7 (IC50 = 229 nM) in the biochemical assay and significantly reduced the production of pro-inflammatory cytokine IL-1β and reactive oxygen species (ROSs) in the human THP-1 macrophage systems. In the DSS-induced colitis mouse model, the DAI score and histologic score were decreased by 31% and 42%, respectively, after treated with this compound in Figure 22 (2.5 mg/kg, i.p.). Moreover, the ADME-Tox profile and selectivity for P2X7 were also encouraging for further development as a candidate towards potential IBD therapies.193
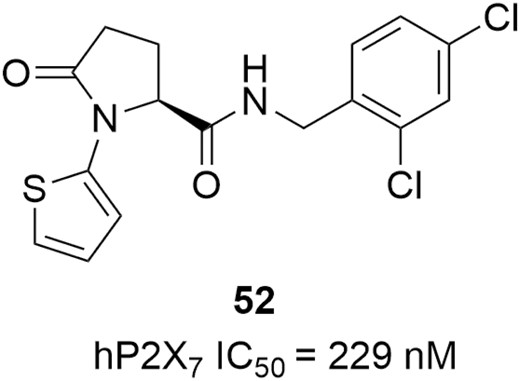
P2X7 receptor antagonist for potential IBD therapies.
IL-12/23 Production Inhibitors
Interleukin-12 and IL-23 are heterodimeric pro-inflammatory cytokines that share a common p40 protein subunit and induce T helper type 1 (Th1) cells and Th17 cells differentiation, respectively, which have been shown to induce inflammatory autoimmune diseases, including IBD.194–196 Inhibiting IL-12/23 to alleviate the excessive Th1/Th17 immune responses has been identified as a therapeutic strategy for diverse inflammatory and autoimmune diseases.197,198 In 2016, the approval of a monoclonal antibody (ustekinemab) against IL-12/23 for CD has validated the therapeutic utility of this approach and has stimulated efforts to develop small-molecule IL-12/23 inhibitors.199 In addition, APY0201 (Figure 23, diagram 53) is a unique IL-12/23 production inhibitor (Figure 23); it acts on PIKfyve kinase at the ATP-binding site to interrupt the conversion of phosphatidylinositol 3-phosphate (PtdIns3P) to phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2), resulting in strong suppression of IL-12/23 production in vitro. In the mouse colitis model induced by IL-10-/- adoptive transfer, APY0201 effectively alleviated inflammation in a dose-dependent manner, and a significant reduction (73%) in colon weight was observed after oral dosing at 30 mg/kg, showing promising potential for the treatment of IBD.200
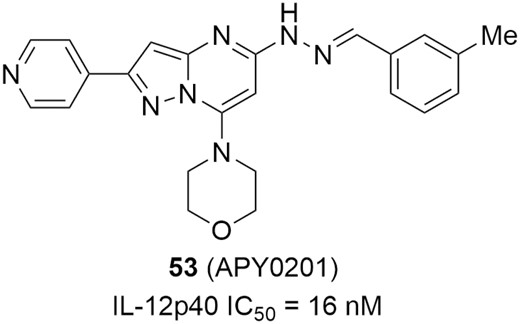
IL-12/23 production inhibitors for potential IBD therapies.
Targeting BRD4
Bromodomain-containing protein 4 (BRD4) is a family member of bromodomain and extraterminal domain (BET) proteins, which also includes BRD2, BRD3, and bromodomain testis-specific protein (BRDT).201,202 All BET members share 2 bromodomains, termed BD1 and BD2, exhibiting a high level of sequence conservation. As epigenetic readers, BET proteins play a key role in the regulation of gene transcription by binding to acetylated lysine residues on the histone tails. Therefore, BET family proteins, particularly BRD4, have emerged as promising epigenetic targets for various human diseases.203–209 As depicted in Figure 24, BRD4 plays an essential role in the signaling of IBD through the activation of RNA polymerase 2, and histone H3 Lys122 acetylation (H3K122Ac) to regulate the inflammatory gene expressions such as IL-1, IL-6, TNF-α, IL-13, and IFN-γ etc.210–220 Moreover, our compelling data (unpublished) demonstrates that the abundance of BRD4 activation biomarker H3K122Ac is significantly increased in human IBD patient samples (including both UC and CD patients) compared with that in healthy controls, suggesting that BRD4 activation is vital to the chronic inflammatory responses in IBD. Hence, inhibiting the BRD4 activation provides an attractive approach to the development of superior epigenetic therapeutics for IBD.
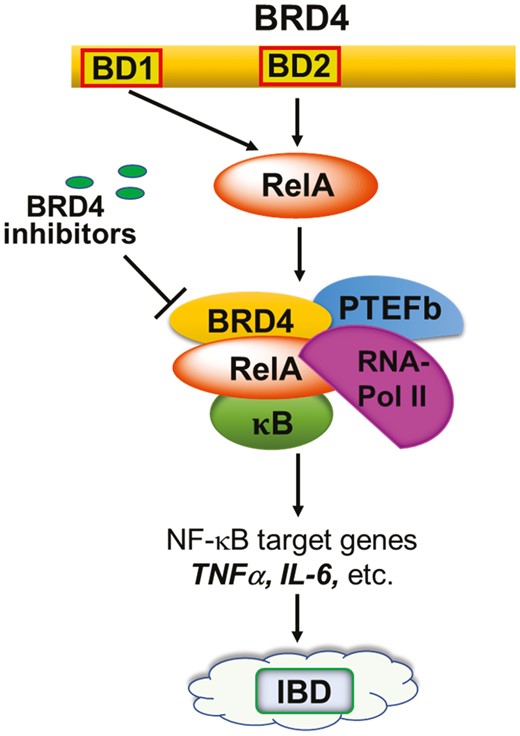
Essential role of BRD4 in the signaling of IBD. BRD4, as an epigenetic reader, is an essential transcriptional coactivator of the NFκB activity, regulating pro-inflammatory cytokine expression as a component of the positive transcriptional elongation factor (P-TEFb) complex. BRD4 serves to phosphorylate RNA polymerase 2 (RNA-Pol II) and modifies regulatory histones via its atypical histone acetyltransferase (HAT) activity, thereby regulating the production of key inflammatory cytokines that are implicated in both CD and UC such as TNF-α and the type 1, 2 and 17 cell inflammatory responses (IL-1, IL-6, IFN-γ, IL-13, etc.).
Bromodomain-containing protein 4 is a unique target because its inhibition simultaneously targets inflammatory responses induced by both innate and adaptive immune cells. Our team demonstrated that BRD4 is required for stabilization of NFκB-binding on the promoters of inflammatory genes, activation of RNA-Pol 2, and H3K122Ac formation to permit high levels of inflammatory gene expression by sentinel immune cells, epithelial cells, and myofibroblasts.214–219 Inhibition of BRD4 has been shown to inhibit the production of inflammatory IFN-γ by Th1, NK, and NKT cells in culture.221 Bromodomain-containing protein 4 was also found to positively regulate mouse Th17 cell differentiation, and its inhibition dramatically reduced Th17 cell activity.222,223 A similar finding was observed concerning the Th2 cytokine IL-13.218,222 Oncostatin M (OSM) is a member of the IL-6 cytokine family produced by several innate and adaptive immune cells in IBD and is shown to be critical to the pathogenesis of IBD.224 High expression of OSM is strongly associated with failure of anti-TNF-α therapy. We have revealed that BRD4 is needed for OSM signaling in the activation of immune cells and fibroblasts.225 Thus, BRD4 inhibition may open a new avenue to overcome the OSM-mediated anti-TNF-α resistance. Finally, pro-inflammatory activation of fibroblasts has been shown to play a critical role in extracellular matrix (ECM) remodeling during profibrotic changes in CD.226,227 Despite our knowledge on fibrosis, which is secondary to the intestinal inflammation in IBD, anti-fibrotic drugs are yet to be developed. Our recent findings indicate that inhibiting BRD4 is likely to provide a promising approach to inhibit profibrotic molecule production and, consequently, ECM remodeling in intestinal mucosa during chronic IBD.
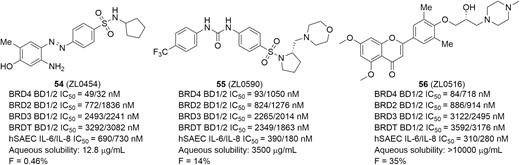
The compound ZL0454 (Figure 25, diagram 54) is our first-generation BRD4-selective inhibitor demonstrating strong in vivo anti-inflammatory activity for the therapeutic potential of IBD (Figure 25).228–233 It displayed highly potent BRD4-binding affinities, with greater BRD4 specificity over other bromodomains (eg, BRD2, BRD3, BRDT) in a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Moreover, ZL0454 also exhibited submicromolar cellular inhibitory activities against expressions of inflammatory cytokines (eg, IL-8 and IL-6) in poly(I:C)-stimulated human small airway epithelial cells (hSAECs). The cocrystal structural analysis of ZL0454 in complex with human BRD4 BD1 illustrated that the BRD4 inhibitor ZL0454 occupied the classical KAc binding pocket (Figure 26). The amine group in the benzene ring formed directly critical hydrogen bonds with the backbone carbonyl of Pro82, and the hydroxyl group interacted with Tyr97 via a water molecule. A network of water molecules connected the diazene moiety to the backbone carbonyl of Leu92. The phenyl ring of benzsulfonamide formed a π-π stacking with indole ring of Trp81. In a mouse model of DSS-induced colitis, ZL0454 restored body weight and reversed mucosal histopathology at a dose of 10 mg/kg/day (i.p.). In a Cbir1 T-cell transfer model that mimics the pathogenesis of CD, treatment with ZL0454 at a dose of 5 mg/kg significantly reduced intestinal inflammation. The results of quantitative real-time polymerase chain reaction (qRT-PCR) analysis of colon tissues demonstrated that ZL0454 significantly suppressed Th1 and Th17 cell responses (IFN-γ, IL-6, IL-1β, IL-17A, and TNF-α, respectively). In addition, treatment with ZL0454 also led to the dramatic reduction of the TNC (tenascin C) and COL1A2 (Collagen A) expression, the key profibrotic molecules upregulated in CD fibrosis (unpublished data). Pharmacokinetic profile indicated that ZL0454 had great drug exposure (AUC0-last = 11,400 ng ∙ h/mL) via injection, but with relatively short half-lives (T1/2 = 0.83 h) and low oral bioavailability (F = 0.46%), likely owing to the metabolic instability of the azo (N = N) linker in the molecular structure.
BRD4 inhibitors for potential IBD therapies.
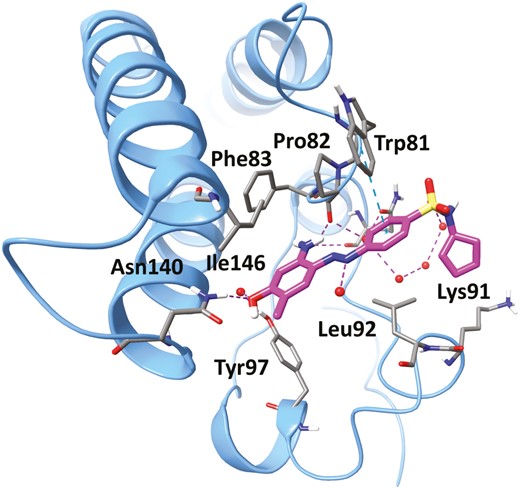
The cocrystal structure of ZL0454 (magenta) in complex with human BRD4 BD1 (blue). Key residues at the binding site are shown in stick representation. Water molecules are shown as red spheres. Hydrogen bonds are shown as purple dotted lines and π-cation is shown as blue dotted lines.
To improve PK properties, particularly oral bioavailability, optimization of ZL0454 was carried out. Replacing the azo linker with more metabolically stable urea followed by assembling a pyrrolidine moiety resulted in the advanced BRD4 inhibitor ZL0590 (Figure 25, diagram 55), which was found not only to improve oral bioavailability (F = 14%) and cell activities (IL-6/IL-8 IC50 = 390/180 nM in hSAECs) but also enhance the selectivity for BD1 over BD2 (>10-fold) while still maintaining specific BRD4-binding affinities (Figure 25). Excitingly, unlike ZL0454, the cocrystal structure revealed ZL0590 targeting a unique first-in-class non-KAc binding site located at the helix αB and αC surface with BRD4 residues that are not commonly shared among family members for high selectivity (unpublished data, manuscript in preparation). As shown in Figure 27A, the urea linker interacted via 2 water molecules with the backbone carbonyl of Tyr137 and the side-chain carboxylate group of Glu154. The basic amine moiety of the morpholine group was likely protonated and formed a salt bridge with the carboxylate moiety of Glu151. The sulfonamide directly formed a hydrogen bond with the backbone amide of Gly143. The other oxygen in the sulfonamide formed a hydrogen bond with a water molecule that mediated interaction with the backbone carbonyl of Asp144. Overlay of known crystal structure BRD4 BD1 and our structure clearly illustrated the different locations of the 2 nearby binding sites (Figure 27B). The in vivo study demonstrated that ZL0590 (10 mg/kg, i.p.) could safely reverse colonic histopathology and body weight loss in a DSS-induced mouse model of colitis. The expressions of inflammatory cytokines IL-6, TNF-α, and IL-17A in colonic mucosa were significantly downregulated. Also, the DSS-induced disruption of the epithelial crypts was significantly reversed. Furthermore, in the model of oxazolone (OXA)-induced chronic model of colitis, the survival rate of mice was increased, and colonic inflammation was reduced after treatment with ZL0590 (10 mg/kg, i.p.). Additionally, expressions of IL-6, TNF-α, IL-13, and IL-17A in colonic mucosa were also significantly downregulated by ZL0590 (unpublished data).
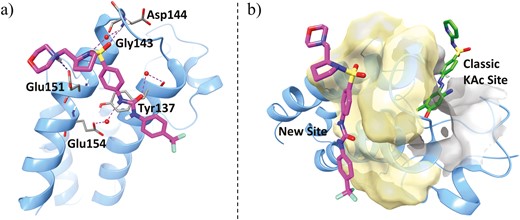
A, Ribbon representation of cocrystal structure (blue, PDB ID: 6U0D) of BRD4 BD1 in complex with ZL0590 (magenta). Key residues at binding site are shown in stick representation. Water molecules are shown as red spheres. Hydrogen bonds are shown as purple dotted lines, and salt bridge in blue dotted line. B, Overlay of cocrystal structures BRD4 BD1 (blue) in complex with ZL0590 (magenta) and MS436 (green, PBD ID: 4NUD). The traditional KAc pocket is shown in gray surface representation and the new binding site of ZL0590 is shown in yellow.
The compound ZL0516 (Figure 25, diagram 56) containing a chromone scaffold is another highly potent and specific BRD4 inhibitor identified by our team for the treatment of IBD.234 It possessed highly potent and specific BRD4-binding affinities with high BD1 selectivity over BD2. Moreover, the IC50 values of ZL0516 are potent in the range of 280 to 310 nM to inhibit IL-8 and IL-6 production in cellular assays. Similar to the binding mode of ZL0454, the cocrystal structure of ZL0516 in complex with BRD4 BD1 showed that the small molecular inhibitor occupied the classic KAc site, assisted by several water molecules. As shown in Figure 28, the carbonyl oxygen atom directly interacted with Asn140 and formed an indirect H-bond with Tyr97 with the aid of a water molecule. One methoxy group in the chromone ring was accommodated in the pocket by a water-mediated H-bond, whereas another methoxy group is extended to the Trp-Pro-Phe (WPF) shelf area. The 1-methylpiperazine was excluded from the pocket and directed to the solvent region. The chiral hydroxyl on the side chain was close to a water molecule where it formed a water-mediated H-bond with Asn93. The corresponding residue of Asn93 on BRD4 BD2 is glycine, and thus this interaction was losing for BRD4 BD2 with ZL0516. Similarly, the equivalent residues of Asn93 on BRD2 BD1 are glycine, on BRD2 BD2 are glycine, on BRD3 BD2 are glutamic acid, on BRDT BD1 are glutamine, and on BRDT BD2 are glycine. This may explain the selectivity of ZL0516 over other BET family members. Further study demonstrated that ZL0516 exhibited excellent physicochemical properties and PK profile and significantly reduced colonic inflammation and disease parameters in both DSS- and OXA-induced models of colitis (unpublished data), indicating its therapeutic potential for the treatment of IBD. The preclinical to clinical development of these selective BRD4 inhibitors toward epigenetic therapeutics of IBD medications are ongoing under the auspices of Crohn’s & Colitis Foundation Entrepreneurial Investing (EI) Initiative award and Litwin IBD Pioneers Program.

The cocrystal structures of ZL0516 (magenta) in complex with human BRD4 BD1 (blue, PDB ID: 6UWU), showing that the small molecular inhibitor ZL0516 occupies the classic KAc recognition site. Key residues at the binding site are shown in stick representation. Water molecules are shown as red spheres. Hydrogen bonds are shown as purple dotted lines.
Concluding Remarks And Future Perspectives
Significant progress has been made in the drug discovery and development of new small-molecule candidates that target biologically relevant pathways in the pathogenesis of IBD. The relevant drug targets include JAK, RIP1, S1P receptor, integrin, CCR9, CXCR4, PDE4, LANCL2, NLRP3, IKK2, CB2, P2X7 receptor, and emerging epigenetic target BRD4, which are associated with activation, communication, and trafficking of immune cells. The advances of representative small molecule agents described herein are summarized in Table 1. Notably, tofacitinib, a pan-JAK inhibitor that blocks intracellular signaling from multiple cytokines, has been approved by the FDA for clinical use in the treatment of moderate to severe UC in 2018 and is being widely prescribed in patients for whom conventional therapies or biologic drugs failed, highlighting the potential of target-based IBD therapies. Inspired by this success, the second-generation of highly selective JAK inhibitors (eg, filgotinib and upadacitinib) and gut-targeted JAK inhibitors (eg, TD-1437) is currently being evaluated at different clinical stages. Results indicate that significant clinical efficacy with favorable safety profiles was achieved. Other agents covering different mechanisms of action are also ongoing in the clinical and preclinical development. Among them, targeting immune cell trafficking has demonstrated promising treatment and minimal safety issues for both UC and CD. For example, S1PR modulators (eg, ozanimod and etrasimod) are efficacious at inducing and maintaining disease remission in phase 2 trials and might have fewer systemic side effects than JAK inhibitors. An α4 integrin subunit antagonist, AJM-300, showed significant improvement in all measured clinical criteria and found no patients had PML, which has previously been a concern associated with anti-integrins. Because increasing the level of intracellular cAMP can result in suppression of inflammation by regulating the release of inflammatory cytokines, some therapies blocking the hydrolysis of cAMP, including PDE4 inhibitor and LANCL2 agonist, have also been investigated in clinical trials. Apremilast, a PDE4 inhibitor approved for treating psoriasis and psoriatic arthritis, has completed the phase 2 study in UC patients, which demonstrated that 31.6% of the patients in the group receiving 30 mg twice daily were in clinical remission compared with 13.8% in the placebo group (P < .05). In addition, the NLRP3 inflammasome is also of great importance in the progression of inflammation.167 The inhibition of its activation by small molecules provides new hope for novel anti-inflammatory drug discovery. Currently, there are a few inflammasome inhibitors, spearheaded by MCC950, for use against IBD, and further cutting-edge research targeting the NLRP3 inflammasome will be promising. Another novel and attractive strategy was to target epigenetic regulators that play key roles in the regulation of inflammatory genes in IBD. It has been well documented that activation of BRD4, an epigenetic reader, is critical to the initiation of the human colonic inflammation during IBD, evidenced by the biomarker analysis using the IBD patient biopsies. Several novel BRD4 inhibitors such as ZL0454, ZL0516, and ZL0590 developed by our team have showcased promising therapeutic potential through the suppression and reversing of the colonic inflammation and fibrosis in IBD, and their future application as a novel epigenetic medication is anticipated through active ongoing preclinical and clinical development. Excitingly, this novel therapeutic strategy may offer effective treatment of IBD patients who are refractory to anti-TNF-α antibody therapy.
Representative small-molecule agents targeting biological pathways implicated in IBD.
| Agents | Target | Current Status in IBD | |
|---|---|---|---|
| UC | CD | ||
| Tofacitinib | pan-JAK | Launched | II, terminated |
| Peficitinib | pan-JAK/TYK2 | II | - |
| Filgotinib | JAK1 | III | III |
| Upadacitinib | JAK1 | III | III |
| SHR-0302 | JAK1 | II | II |
| PF-06700841 | JAK1/TYK2 | II | II |
| BMS986165 | TYK2 | - | II |
| PF-06651600 | JAK3 | II | II |
| TD-1473 | pan-JAK, gut-restricted | II/III | II |
| GSK2982772 | RIP1 | II | - |
| Amiselimod | S1P1,5 | - | II |
| KRP-203 | S1P1,4 | II | - |
| Ozanimod | S1P1,5 | III | III |
| Etrasimod | S1P1,4,5 | III | II |
| AMJ300 | Anti-integrins | III | |
| Vercirnon | CCR9 | - | III, terminated |
| Apremilast | PDE4 | II | - |
| BT-11 | LANCL2 | II | II |
| MSX-122 | CXCR4 | Discovery | Discovery |
| MCC950 | NLRP3 | Discovery | Discovery |
| INF39E | NLRP3 | Discovery | Discovery |
| BMS-066 | IKK2 | Discovery | Discovery |
| 50 | CB2 | Discovery | Discovery |
| 51 | CB2 | Discovery | Discovery |
| 52 | P2X7 | Discovery | Discovery |
| APY0201 | IL-12/23 production inhibitor | Discovery | Discovery |
| ZL0454 | BRD4 BD1/2 | Discovery | Discovery |
| ZL0590 | BRD4 BD1 | Discovery | Discovery |
| ZL0516 | BRD4 BD1 | Discovery | Discovery |
| Agents | Target | Current Status in IBD | |
|---|---|---|---|
| UC | CD | ||
| Tofacitinib | pan-JAK | Launched | II, terminated |
| Peficitinib | pan-JAK/TYK2 | II | - |
| Filgotinib | JAK1 | III | III |
| Upadacitinib | JAK1 | III | III |
| SHR-0302 | JAK1 | II | II |
| PF-06700841 | JAK1/TYK2 | II | II |
| BMS986165 | TYK2 | - | II |
| PF-06651600 | JAK3 | II | II |
| TD-1473 | pan-JAK, gut-restricted | II/III | II |
| GSK2982772 | RIP1 | II | - |
| Amiselimod | S1P1,5 | - | II |
| KRP-203 | S1P1,4 | II | - |
| Ozanimod | S1P1,5 | III | III |
| Etrasimod | S1P1,4,5 | III | II |
| AMJ300 | Anti-integrins | III | |
| Vercirnon | CCR9 | - | III, terminated |
| Apremilast | PDE4 | II | - |
| BT-11 | LANCL2 | II | II |
| MSX-122 | CXCR4 | Discovery | Discovery |
| MCC950 | NLRP3 | Discovery | Discovery |
| INF39E | NLRP3 | Discovery | Discovery |
| BMS-066 | IKK2 | Discovery | Discovery |
| 50 | CB2 | Discovery | Discovery |
| 51 | CB2 | Discovery | Discovery |
| 52 | P2X7 | Discovery | Discovery |
| APY0201 | IL-12/23 production inhibitor | Discovery | Discovery |
| ZL0454 | BRD4 BD1/2 | Discovery | Discovery |
| ZL0590 | BRD4 BD1 | Discovery | Discovery |
| ZL0516 | BRD4 BD1 | Discovery | Discovery |
The highest level of clinical status was obtained from https://www.clinicaltrials.gov (accessed Dec. 2020).
Representative small-molecule agents targeting biological pathways implicated in IBD.
| Agents | Target | Current Status in IBD | |
|---|---|---|---|
| UC | CD | ||
| Tofacitinib | pan-JAK | Launched | II, terminated |
| Peficitinib | pan-JAK/TYK2 | II | - |
| Filgotinib | JAK1 | III | III |
| Upadacitinib | JAK1 | III | III |
| SHR-0302 | JAK1 | II | II |
| PF-06700841 | JAK1/TYK2 | II | II |
| BMS986165 | TYK2 | - | II |
| PF-06651600 | JAK3 | II | II |
| TD-1473 | pan-JAK, gut-restricted | II/III | II |
| GSK2982772 | RIP1 | II | - |
| Amiselimod | S1P1,5 | - | II |
| KRP-203 | S1P1,4 | II | - |
| Ozanimod | S1P1,5 | III | III |
| Etrasimod | S1P1,4,5 | III | II |
| AMJ300 | Anti-integrins | III | |
| Vercirnon | CCR9 | - | III, terminated |
| Apremilast | PDE4 | II | - |
| BT-11 | LANCL2 | II | II |
| MSX-122 | CXCR4 | Discovery | Discovery |
| MCC950 | NLRP3 | Discovery | Discovery |
| INF39E | NLRP3 | Discovery | Discovery |
| BMS-066 | IKK2 | Discovery | Discovery |
| 50 | CB2 | Discovery | Discovery |
| 51 | CB2 | Discovery | Discovery |
| 52 | P2X7 | Discovery | Discovery |
| APY0201 | IL-12/23 production inhibitor | Discovery | Discovery |
| ZL0454 | BRD4 BD1/2 | Discovery | Discovery |
| ZL0590 | BRD4 BD1 | Discovery | Discovery |
| ZL0516 | BRD4 BD1 | Discovery | Discovery |
| Agents | Target | Current Status in IBD | |
|---|---|---|---|
| UC | CD | ||
| Tofacitinib | pan-JAK | Launched | II, terminated |
| Peficitinib | pan-JAK/TYK2 | II | - |
| Filgotinib | JAK1 | III | III |
| Upadacitinib | JAK1 | III | III |
| SHR-0302 | JAK1 | II | II |
| PF-06700841 | JAK1/TYK2 | II | II |
| BMS986165 | TYK2 | - | II |
| PF-06651600 | JAK3 | II | II |
| TD-1473 | pan-JAK, gut-restricted | II/III | II |
| GSK2982772 | RIP1 | II | - |
| Amiselimod | S1P1,5 | - | II |
| KRP-203 | S1P1,4 | II | - |
| Ozanimod | S1P1,5 | III | III |
| Etrasimod | S1P1,4,5 | III | II |
| AMJ300 | Anti-integrins | III | |
| Vercirnon | CCR9 | - | III, terminated |
| Apremilast | PDE4 | II | - |
| BT-11 | LANCL2 | II | II |
| MSX-122 | CXCR4 | Discovery | Discovery |
| MCC950 | NLRP3 | Discovery | Discovery |
| INF39E | NLRP3 | Discovery | Discovery |
| BMS-066 | IKK2 | Discovery | Discovery |
| 50 | CB2 | Discovery | Discovery |
| 51 | CB2 | Discovery | Discovery |
| 52 | P2X7 | Discovery | Discovery |
| APY0201 | IL-12/23 production inhibitor | Discovery | Discovery |
| ZL0454 | BRD4 BD1/2 | Discovery | Discovery |
| ZL0590 | BRD4 BD1 | Discovery | Discovery |
| ZL0516 | BRD4 BD1 | Discovery | Discovery |
The highest level of clinical status was obtained from https://www.clinicaltrials.gov (accessed Dec. 2020).
Great milestones in the development of drugs for the treatment of IBD have been made, especially the biological therapies using antibody-blocking TNF-α and the use of small-molecule agents that target specific pathways implicated in disease pathology. However, significant gaps in terms of efficacy, target specificity, gut selectivity, and drug safety still remain. Future research should focus on several directions. First, a deeper understanding of the complex biological mechanisms underlying IBD and the identification of relevant biomarkers to stratify patients. Prospectively identifying patients that are most likely to respond favorably to a specific therapy is critical for achieving clinical success. AstraZeneca’s 5Rs model (the right target, tissue, safety, patient, and commercial potential) should be considered and extensively applied to the drug development pipeline for better outcomes with a potential success.209,235 Secondly, because of the complexity and severity of this chronic disease, it is challenging to cure or control the course through targeting a single mechanism triggering the IBD. Therefore, combination therapy of 2 or more drugs or developing dual inhibitors with complementary mechanisms may offer alternative therapeutic approaches that should be explored. Furthermore, another aspect that should not be neglected is the drug delivery route. The focal zone of IBD is the GI tract, and the rational design of intestinal-restricted oral drug or specific local drug delivery can minimize the systemic side effects caused by the oral route.
In summary, the target-based drug development for the treatment of IBD represents a challenging but attractive research area. With the significant advances achieved in developing the new and effective therapeutic strategies and the exciting success with the FDA approval of increasing numbers of drugs targeting different factors and signaling pathways implicated in the disease pathogenesis, novel target-based small molecules are anticipated to have enormous potential to offer unique and safer treatment options of IBD to benefit patients that are suffering from this disease.
Abbreviations
- ATP
adenosine triphosphate
- ADME
absorption, distribution, metabolism, and excretion
- AMP
adenosine monophosphate
- ABA
abscisic acid
- ASC
apoptosis-associated speck-like protein containing a CARD
- BMDM
mouse bone marrow derived macrophages
- BRD4
Bromodomain-containing protein 4
- BET
bromodomain and extra-terminal domain
- BRDT
bromodomain testis-specific protein
- CD
Crohn’s disease
- CDC
Centers for Disease Control and Prevention
- CCR9
C-C chemokine receptor 9
- CCL25
chemokine ligand 25
- CXCR4
CXC chemokine receptor 4
- CXCL12
C-X-C motif chemokine 12
- cAMP
cyclic adenosine monophosphate
- CARD
caspase activation and recruitment domain
- CB
cannabinoid receptor
- CNS
central nervous system
- COL1A2
collagen A
- DSS
dextran sulfate sodium
- DAI
disease activity index
- DNBS
dinitrobenzene sulfonic acid
- EMA
European Medicines Agency
- EPO
erythropoietin signaling
- FDA
The United States Food and Drug Administration
- FasL
Fas ligand
- GI
gastrointestinal
- GPCR
G protein-coupled receptor
- GM-GSF
granulocyte-macrophage colony stimulating factor
- hWB
human whole blood
- hERG
the human Ether-à-go-go-Related Gene
- hPBMC
human peripheral blood mononuclear cell
- hSAECs
human small airway epithelial cells
- HEK-293 cell
human embryonic kidney 293 cell
- HMDM
human monocyte derived macrophages
- H3K122Ac
histone H3 Lys122 acetylation
- IBD
inflammatory bowel disease
- IL-1α
interleukin-1α
- IL-1β
interleukin-1β
- IL-2
interleukin-2
- IL-6
interleukin-6
- IL-10
interleukin-10
- IL-10-/-
IL-10 gene-deficient
- IL-12
interleukin-12
- IL-17
interleukin-17
- IL-23
interleukin-23
- IFN-α
interferon-α
- IFN-γ
interferon-γ
- IgG1
immunoglobulin G1
- IgG2a
immunoglobulin G2a
- i.p. injection
intraperitoneal injection
- IKK
IκB kinase
- JAK
Janus kinase
- LPS
lipopolysaccharide
- LANCL2
lanthionine synthetase C-like 2
- MHLW
Ministry of Health, Labor and Welfare of Japan
- MAdCAM-1
mucosal addressin cell adhesion molecule-1
- MIP1α
macrophage inflammatory protein 1α
- MPO
myeloperoxidase
- NK
natural killer
- NLRP3
nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing protein 3
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cell
- NEMO
NF-kappa-B essential modulator
- OSM
oncostatin M
- OXA
oxazolone
- P-loop
glycine-rich loop
- PsA
psoriatic arthritis
- PK
pharmacokinetics
- PD
pharmacodynamics
- PML
progressive multifocal leukoencephalopathy
- PDE4
phosphodiesterase 4
- PKA
protein kinase A
- P2XR
P2X receptor
- PtdIns3P
phosphatidylinositol 3-phosphate
- PtdIns(3,5)P2
phosphatidylinositol 3,5-bisphosphate
- QD
once a day
- qRT-PCR
quantitative real-time polymerase chain reaction
- RA
rheumatoid arthritis
- RIP1
receptor-interacting protein 1 kinase
- ROS
reactive oxygen species
- SAR
structure-activity relationship
- STAT
signal transducers and activators of transcription
- STAT3
signal transducers and activators of transcription 3
- S1P
sphingosine-1-phosphate
- S1P1−5
sphingosine-1-phosphate receptors 1–5
- SphKs
sphingosine by sphingosine kinases
- SDF-1
stromal cell-derived factor-1
- SPA
scintillation proximity assay
- TNF
tumor necrosis factor
- TYK2
tyrosine kinase 2
- TNFR1
tumor necrosis factor receptor 1
- TRAIL
TNF-related apoptosis-inducing ligand
- Th1
T helper 1
- Th17
T helper 17
- TECK
thymusexpressed chemokine
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- TNC
tenascin C
- UC
ulcerative colitis
- VCAM-1
vascular cell adhesion molecule-1
- WPF
Trp-Pro-Phe
Funding
This work was partially supported by Crohn’s & Colitis Foundation Entrepreneurial Investing (E.I.) Initiative award (J.Z., A.R.B., and B.T.), Litwin IBD Pioneers Program Award (B.T. and J.Z.), and a postdoctoral research fellowship award (Z.L.) from the Crohn’s & Colitis Foundation of America, UTMB Technology Commercialization Program, and the John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.).
Supplement Sponsorship
This supplement was sponsored by the Crohn’s & Colitis Foundation.
Conflicts of Interest
A.R.B. and J.Z. are cofounders of QuadraGenics, Inc. The authors declare no competing financial interest.
References
Author notes
These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}