





Abstract
We aimed to examine, for the first time, the effect of cannabidiol (CBD) and palmitoylethanolamide (PEA) on the permeability of the human gastrointestinal tract in vitro, ex vivo, and in vivo.
Flux measurements of fluorescein-labeled dextrans 10 (FD10) and fluorescein-labeled dextrans 4 (FD4) dextran across Caco-2 cultures treated for 24 hours with interferon gamma (IFNγ) and tumour necrosis factor alpha (TNFα) (10 ng·mL−1) were measured, with or without the presence of CBD and PEA. Mechanisms were investigated using cannabinoid receptor 1 (CB1), cannabinoid receptor 2 (CB2), transient receptor potential vanilloid 1 (TRPV1), and proliferator activated receptors (PPAR) antagonists and protein kinase A (PKA), nitric oxide synthase, phosphoinositide 3-kinases, extracellular signal–regulated kinases (MEK/ERK), adenylyl cyclase, and protein kinase C (PKC) inhibitors. Human colonic mucosal samples collected from bowel resections were treated as previously stated. The receptors TRPV1, PPARα, PPARδ, PPARγ, CB1, CB2, G-coupled protein receptor 55 (GPR55), G-coupled protein receptor 119 (GPR119), and claudins-1, -2, -3, -4, -5, -7, and -8 mRNA were measured using multiplex. Aquaporin 3 and 4 were measured using enzyme-linked immunosorbent assay (ELISA). A randomized, double-blind, controlled-trial assessed the effect of PEA or CBD on the absorption of lactulose and mannitol in humans taking 600 mg of aspirin. Urinary concentrations of these sugars were measured using liquid chromatography mass spectrometry.
In vitro, PEA, and CBD decreased the inflammation-induced flux of dextrans (P < 0.0001), sensitive to PPARα and CB1 antagonism, respectively. Both PEA and CBD were prevented by PKA, MEK/ERK, and adenylyl cyclase inhibition (P < 0.001). In human mucosa, inflammation decreased claudin-5 mRNA, which was prevented by CBD (P < 0.05). Palmitoylethanolamide and cannabidiol prevented an inflammation-induced fall in TRPV1 and increase in PPARα transcription (P < 0.0001). In vivo, aspirin caused an increase in the absorption of lactulose and mannitol, which were reduced by PEA or CBD (P < 0.001).
Cannabidiol and palmitoylethanolamide reduce permeability in the human colon. These findings have implications in disorders associated with increased gut permeability, such as inflammatory bowel disease.
INTRODUCTION
The gut provides a barrier between the external and internal environment. This selectively permeable barrier allows absorption of nutrients and water from gastrointestinal contents, while preventing the transfer of noxious material such as bacteria and lipopolysaccharide. During episodes of inflammation, the barrier becomes compromised, allowing transfer of noxious material into the systemic circulation, leading to disease states such as inflammatory bowel disease (IBD) and septic shock.1
The use of Cannabis sativa for its analgesic and anti-inflammatory effects have been described well.2 Interest in the psychoactive properties of Cannabis sativa leads to the characterization of its active major compounds Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD) and approximately 80 other compounds.3, 4 Discovery of the receptors for these ligands followed, with cloning of the cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2) receptors.5, 6 The endogenous ligands anandamide and 2-arachiodonoylglycerol that act at these receptors have been well described.7–9 Endocannabinoid-like compounds acting alongside these, such as palmitoylethanolamide (PEA), have emerged, with affinity for receptors such as the GPR55 receptor and the nuclear peroxisome proliferator activated receptors (PPARs).10–12 Palmitoylethanolamide is widely used as a food additive and is being assessed for clinical use in pain and inflammation.13, 14 Similarly, CBD is already in use as a treatment of multiple sclerosis (as part of the medicine Sativex) and by itself in childhood epilepsy.15, 16
We previously demonstrated that inflammation causes an increase in the permeability of fully confluent Caco-2 monolayers, measured by trans-epithelial electrical resistance (TEER).17 We identified that application of CBD or PEA rescued falls in TEER (ie, reduces inflammation-induced increases in permeability). The effects of CBD were blocked by antagonism of the CB1 receptor, and PEA was blocked by antagonism of the PPARα receptor.17, 18 Our recent meta-analysis demonstrated that preclinical studies with both CBD and PEA show promise in animal models.19
In light of this background, we hypothesized that CBD and PEA reduce inflammation-induced permeability in the human gut and examined the effects of PEA and CBD on the human colon using Caco-2 cells and ex vivo human colonic tissue, identifying their mechanisms of action. Based on these positive findings, we went on to examine the potential of CBD and PEA to modulate intestinal permeability in humans in vivo using a randomized controlled trial.
MATERIALS & METHODS
In Vitro Permeability Studies
Caco-2 cells were purchased from European Collection of Cell Culture (Wiltshire, UK; passages 21–42). Cells were cultured in Eagle’s minimum essential medium supplemented with L-glutamine, 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% nonessential amino acids mixture (all Sigma-Aldrich, St Louis, US). Cells were kept at 37°C in 5% CO2 and 95% humidity. For permeability experiments, cells were seeded at 5 × 104/mL per well in the apical compartment of 12 mm diameter 3.0 µm pore polyester membrane inserts within polyester 24-well plates (Corning Incorporated, ME, USA) and cultured for 14 to 16 days until fully confluent. Randomly assigned wells (n = 8) were treated with or interferon gamma (IFNγ) (basolateral, 10 ng.mL−1, Sigma-Aldrich) for 8 hours, followed by tumour necrosis factor alpha (TNFα) (basolateral, 10 ng.mL−1 [Sigma-Aldrich]) for 16 hours to induce inflammation. Palmitoylethanolamide (basolateral, 10 µM, Tocris Bioscience, Bristol UK), CBD (apical, 10 µM, Tocris Bioscience, Bristol UK), or vehicle (0.01% ethanol) were added with or without inflammation at the start of the 24-hour experimental period. Drug concentrations were based upon previous experiments.17
Confluent monolayers cultured on membrane inserts were washed 3 times with prewarmed phosphate buffered saline (PBS). Clear, nonfluorescent media (FluoroBrite DMEM, ThermoFisher Scientific, MA, USA), supplemented with 1% nonessential amino acids and 1% penicillin/streptomycin, was added to both the apical (225 µL) and basolateral compartment (700 µL) as per manufacturer’s instructions and left for 30 minutes to equilibrate. The fluorescein-labeled dextrans 4 (FD4) and fluorescein-labeled dextrans 10 (FD10) fluorescent dextrans (FDs; 25 mg.mL−1) were then added to the apical compartment. At 37°C baseline (0 hours), the fluorescence of 100-µL aliquot basolateral samples were determined (Fluoroskan Ascent FL2.5, ThermoFisher Scientific, Waltham, MA, USA; excitation and emission wavelengths of 485 and 520 nm, respectively). Fluorescence was determined at regular intervals for 36 hours.
Cannabidiol and palmitoylethanolamide were co-applied with antagonists under inflammatory conditions: AM251 100 nM (CB1 antagonist, Tocris Bioscience, Bristol, UK), AM630 100 nM (CB2 antagonist, Tocris Bioscience, Bristol, UK), GW6471 500 nm (PPARα antagonist, Tocris Bioscience, Bristol, UK), and SB366791 500 nM (transient receptor potential vanilloid 1 [TRPV1] antagonist, Tocris Bioscience, Bristol, UK). We co-applied various inhibitors with PEA or CBD under inflammatory conditions: KT5720 (protein kinase A [PKA] inhibitor, 1 µM, Tocris Bioscience, Bristol, UK), L-NAME (nitric oxide synthase inhibitor, 10 µM, Tocris Bioscience, Bristol, UK), PD98059 (extracellular signal–regulated kinases [MEK/ERK] inhibitor, 10 µM, Tocris Bioscience, Bristol, UK), SQ22536 (adenylyl cyclase inhibitor, 1 µM, Tocris Bioscience, Bristol, UK) and G06983 (protein kinase C [PKC] inhibitor, 1 µM, Tocris Bioscience, Bristol, UK).
mRNA and Protein Expression
Caco-2 cells were cultured in 6-well plates (Corning Incorporated, ME, USA) for 14 days. Following 24-hour treatments with CBD or PEA, cells were washed twice with ice-cold PBS and treated with radioimmunoprecipitation assay (RIPA) buffer containing phosphatase inhibitor and protease inhibitors (Sigma-Aldrich) at 4°C for 1 hour on a rocking platform to cause cell lysis. Cell lysates were then collected and stored at −80°C until analysis.
Experiments on ex vivo human colonic tissue were performed on healthy colonic tissue taken from colon removed at elective resection for bowel cancer at Royal Derby Teaching Hospital NHS Trust, Derbyshire (n = 8). After informed consent, samples of macroscopically normal colon at least 10 cm proximal or distal to any bowel tumor were obtained immediately after resection within the operating theater. Ischaemic times after pedicle clamping were not recorded. Sections of tissue 2 cm × 2 cm were excised and transferred on ice to the laboratory within 10 minutes in prechilled Eagle’s minimum essential medium supplemented with 1% FBS, 1% penicillin/streptomycin, and 1% nonessential amino acids mixture (Sigma-Aldrich). Mucosa with submucosa was dissected free from the underlying muscularis layer. Samples were further dissected into approximately 2 mm × 2 mm sections and placed in individual wells of 24-well polystyrene plates (Corning Incorporated, USA) at 37°C in 5% CO2 and 95% humidity and treated with PEA and CBD. After 24 h, the tissue was washed with ice cold PBS and stored frozen at −80°C until homogenization and analysis. Samples were cryohomogenized as previously described by von Zeigler.20 Collected homogenates were then dissolved in 100 µL of RIPA buffer containing phosphatase inhibitor and protease inhibitor (Sigma-Aldrich), incubated on an oscillating thermomixer for 30 minutes at 60°C and then centrifuged at 10,000G for 15 minutes.
The mRNA levels of PPARγ, PPARα, PPARδ, G-coupled protein receptor 55 (GPR55), G-coupled protein receptor 119 (GPR119), CB1, CB2, and TRPV1 were determined using Quantigene Plex kit QP1013; claudin-1, -2, -3, -4, -5, -7, and -8 were determined using kit QP101. Both kits were measured using Magpix (MAGPX 11326002, Luminex, Texas US). The mRNA levels were normalized according to levels of 3 housekeeping proteins β-actin, peptidyl-prolyl cis-trans isomerase B (PPIB), and hypoxanthine phosphoribosyltransferase 1 (HPRT1), which did not change in expression under the inflammatory protocol.
Membrane-bound aquaporin (AQP) cell membrane fractions were prepared with Mem-PER Plus kit (#89842, ThermoScientific). Enzyme-linked immunosorbent assay (ELISA) for aquaporin-3 (AQP3) and aquaporin-4 (AQP4) were performed as per manufacturer’s instructions (#LS-F13079 and #LS-F13078, Lifespan Biosciences, WA USA) and normalized by mean protein content or cell-membrane supernatants using a bicinchoninic acid assay (Sigma-Aldrich).
Randomized, Placebo Controlled, Double-Blind Controlled Trial
In healthy human subjects, we induced a state of increased gut permeability with aspirin, measuring the urinary concentrations of sugar probes. Participants were treated with oral PEA and CBD, and the change in gut permeability was measured in a randomized, double-blind, controlled trial. All experiments and procedures received prior approval of the University of Nottingham Ethics Committee (approval number J16122016); this trial was not registered on a trials registry. Healthy male participants between the ages of 18 and 50 were recruited after informed consent. Participants were screened to exclude any gastrointestinal medical conditions or symptoms, regular medications or recreational drugs, heavy alcohol use, previous abdominal surgery, or personal or family history of IBD. Participants were asked to refrain from the use of any pro or prebiotics the week before the study, and refrain from alcohol or nonsteroidal anti-inflammatory use or heavy exercise for 3 days before the study. Participants separately attended fasting at 8:00 am. Aspirin dose of 600 mg was administered orally with 400 mLs water with 600 mg of CBD, 600 mg of PEA, or placebo. At 9:00 am, 1 g of lactulose and 1 g of mannitol in 600 mL water were further administered; then a baseline urine sample was collected. Urine was collected hourly for 5 hours until 2:00 pm. A further 400 mL of water was administered at noon. Urinary samples were immediately centrifuged at 1000 G for 7 minutes at 3°C and then frozen at −80°C until analysis.
Based on previously published literature, a sample population under the same experimental conditions demonstrated a urinary lactulose-mannitol ratio (LMR) of 0.81 when treated with aspirin vs an LMR of 0.053 (SEM = 0.02).21 Based on α = 0.05 using a paired t test (effect size = 1.0), it was calculated that to detect a difference in LMR of 0.02 between treatment and placebo arms with a power of 80% and α of 0.05, a sample size of 10 patients was required—should a difference exist between the treatment and placebo groups. Using the block method in a 1:1:1 ratio, participants were assigned to receive CBD, PEA, or placebo. Both participants and investigators were blinded to the group assignments. Participants were numbered sequentially and were assigned to the treatment groups in the order of recruitment. Code assignment was sealed within an envelope until after the samples were analyzed. Participant assignment was only revealed at the end of the study.
Cannabidiol was gifted from Artelo Biosciences (CA, USA), extracted from Cannabis sativa, and analyzed at an independent laboratory to assure >99.65% purity. Palmitoylethanolamide was obtained from Russel Science (Nicosia, Cyprus). Placebo was manufactured and gifted by Artelo Biosciences using the base cellulose used in the preparation of CBD. Aspirin was obtained from Aspar Pharmaceuticals (London, UK). Lactulose was obtained from TEVA (Castleford, UK). D-mannitol was obtained from Sigma Aldridge (MO, USA). All products used in the test solution were intended for human use and tested safe for oral consumption. The purity of these sugar probes was reported to be >99.0%.
Concentrations of urinary lactulose and mannitol were determined using a liquid chromatography–mass spectrometry (LC-MS) method. Twenty µL aliquots of urine were thawed and diluted to precipitate any excess salt with 980 µL 90% acetonitrile, to which internal standards xylitol and raffinose were premixed at 0.5 µg/mL of final concentration. These were vortexed and incubated at −20°C for 4 hours. Previous work demonstrated that it is necessary to monitor sucrose to ensure that the assay is specific, correctly identifies lactulose, and therefore is included as an analyte.22 Calibration standards were made as a dilution series from 2.5 to 250 µg/mL of the analytes mannitol and lactulose. The method had been previously validated by creating 6 independently prepared dilutions of 5, 50, and 500 µg/mL as previously stated.22 The LC method was based on that of Kubica.23 The LC column was a Sequant ZIC-pHILIC pHILIC (150 × 4.6 mm, 5 µm) kept at 40°C. The mobile phases were a) acetonitrile and b) 5 mM ammonium acetate, adjusted drop-wise to pH6.85 with 5 mM ammonium hydroxide solution. The B Samples were kept in a chilled auto-sampler, and 2 µL volumes were injected for analysis. The detector was a Sciex 4000 QTrap operating in negative ion electrospray mode, with the source at 450ÅãC with curtain, nebuliser and auxiliary gases set to 10, 40, and 20 respectively. The ion-spray voltage was −4200 V. A minimum of 5 points were used for each analyte. Xylitol was used as the internal standard to normalize the lactulose signal, but the raw peak area was used to calculate the mannitol concentrations. Raffinose had been included as an internal standard as it has been used in previous studies.22, 24 A standard prepared at 50 µg/mL was injected every 20 samples and used to monitor precision and accuracy. The coefficient of variation were 14.1 and 7.3 for lactulose and mannitol respectively. The precisions were 107 and 89%. Calibration lines are linear over a range greater than 2.5 to 250 µg/mL, with R2 > 0.9995 for both analytes.
Statistical Analysis
For in vitro and ex vivo experiments, data are presented as mean (or mean percentage) change from baseline where indicated ± standard error of the mean (SEM). Permeability study results were compared using 2-way ANOVA. Caco-2 and human tissue group results were compared using 1-way ANOVA. The calculated concentration of each urinary sugar was assessed by repeated-measures ANOVA, with probabilities of post hoc comparisons subject to the Bonferroni correction. The differences in excretion of lactulose and D-Mannitol over the study period were compared using 2-way ANOVA. Normality was assessed using the D’Agostino & Pearson normality test. A P value of <0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 7.01 for Windows (GraphPad Software, San Diego, USA). All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
Dextran-Transfer Studies
Within Caco-2 cultures, inflammation caused an increase in the transfer of labeled FD4 and FD10 dextrans from 2 and 4 hours, respectively (P < 0.0001), until the end of the study period at 36 hours (Fig. 1A and B). Palmitoylethanolamide, in the presence of inflammation, reduced FD4 transfer compared with inflammation from 4 hours (P < 0.01, Fig. 2A), increasing with time, with the maximal difference seen at 12 hours (P < 0.0001). For FD10, PEA in the presence of inflammation caused a reduction in the transfer of dextrans compared with inflammation from 4 hours (P < 0.0001, Fig. 1B) until 36 hours. Antagonism of the PPARα receptor (but not TRPV1, CB1, and CB2) blocked the ability of PEA to reduce transfer of FD10 dextran over the study period (Fig. 2A). The effect of PEA in reducing the transfer of dextrans was also blocked by various intracellular signalling inhibitors, such as KT5720 (PKA inhibitor), PD98059 (MAPK inhibitor), and SQ22563 (adenylyl cyclase inhibitor, P < 0.0001, Fig. 3C and D) but not by L-NAME (nitric oxide synthase inhibitor), LY294002 (PI3 kinase inhibitor), or G06983 (PKC inhibitor, Fig. 3C and D).

The effects of PEA, applied basolaterally, and CBD, applied apically, on the permeability of Caco-2 monolayers in response to 24-hour exposure to TNFα and IFNγ measured by transfer of fluorescent dextrans (A:FD4 B:FD10 C:FD10 PEA & CBD). Raw data are expressed as the mean fluoescence per group ± SEM. N = 8 per condition. Data were analyzed by 2-way ANOVA using the Dunnett post hoc test comparing against the vehicle control (**** P < 0.0001).
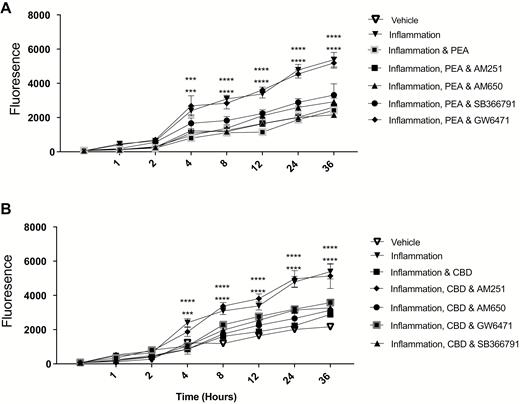
The effects of PEA (basolateral, A) and CBD (apical, B) on the permeability of Caco-2 monlayers in response to 24-hour exposure to TNFα and IFNγ in the presence of various receptor anatgonists, measured by transfer of fluorescent dextrans (FD10). Raw data are expressed as the mean fluoescence per group ± SEM. N = 8 per condition. Data were analyzed by 2-way ANOVA using the Dunnett post hoc test comparing against the vehicle control (*** P < 0.001, **** P < 0.0001).
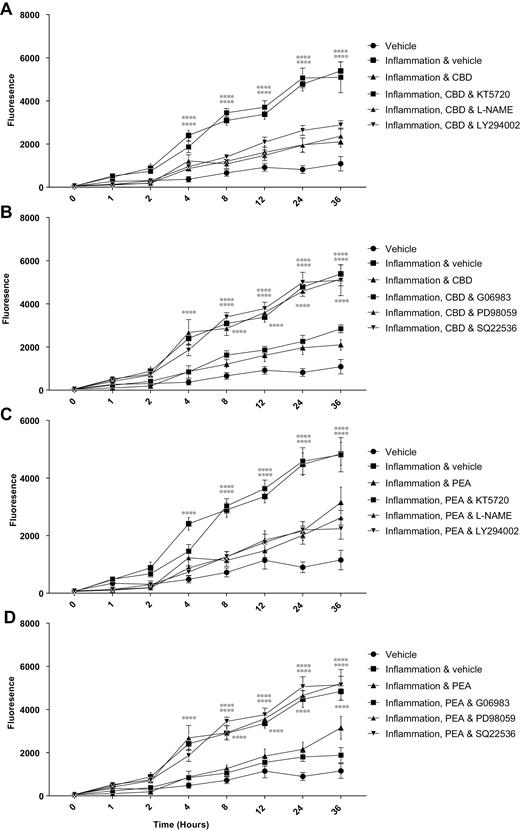
The effects of CBD (apical, A and C) and PEA (basolateral, B and D) on the permeability of Caco-2 monolayers in response to 24-hour exposure to TNFα and IFNγ in the presence of various protein inhibitors (KT5720, L-NAME and LY294002: A and C, and PD88059, SQ22536 and G06983: B and D), measured by transfer of fluorescent dextrans (FD10). Raw data are expressed as the mean fluoescence per group ± SEM. N = 8 per condition. Data were analyzed by two-way ANOVA using the Dunnett post hoc test comparing against the vehicle control (**** P < 0.0001).
Cannabidiol in the presence of inflammation caused a reduction in dextran transfer of FD4 compared with inflammation alone from 4 hours (P < 0.01, Fig. 1A). This difference became more pronounced with time, with maximal difference seen at 8 hours (P < 0.0001). With FD10, CBD in the presence of inflammation caused a reduction in the transfer of dextran from 4 hours (P < 0.0001, Fig. 1B) until 36 hours. Repeating in the presence of various antagonists revealed that only antagonism of the CB1 receptor blocked the effects of CBD (Fig. 2B). As with PEA, the effect of CBD on the transfer of dextrans was blocked by the presence of KT5720, PD98059, and SQ22563 but not L-NAME, LY294002, or G06983 (P < 0.0001, Fig. 3A and B). When applied together, PEA and CBD had no added effect on permeability in the presence of inflammation (Fig. 1C).
Effects of CBD and PEA on Claudin and Cannabinoid Receptor Expression
Compared with the vehicle, inflammation, PEA, and CBD did not change the mRNA levels of claudins-1, -2, -3, -4, -5, 7, or -8 in Caco-2 cells (Fig. 4A, C and Supplementary Fig. S1A–E). Inflammation, CBD, and PEA treatment did not affect the mRNA of claudins-1, -2, -4, -7, or -8 in human colonic tissue (Supplementary Fig. S1F–J).
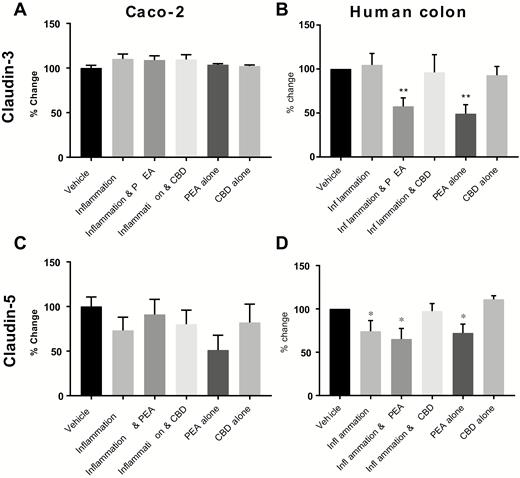
The effects of PEA and CBD on the expression of mRNA levels for tight junction proteins in response to a 24-hour inflammatory procotol in cultured Caco-2 cells (A = claudin-3, C = claudin-5) and human colonic tissue (B = claudin-3, D = claudin-5) measured by multiplex. Data are presented as percentage change from vehicle per plate ± SEM, n = 8 (A and C) and n = 7 (B and D) per condition. Data were analyzed by 1-way ANOVA comparing against vehicle (* P < 0.05, **P < 0.01).
In human colonic tissue, PEA decreased claudin-3 mRNA levels both in the presence of inflammation and alone (P < 0.01, Fig. 4B). Inflammation decreased claudin-5 mRNA levels compared with control tissue (P < 0.05, Fig. 4D), and this was prevented by treatment with CBD (P < 0.05) but not PEA. Palmitoylethanolamide treatment alone also decreased claudin-5 mRNA levels (P < 0.05, Fig. 4D).
In Caco-2 cells, inflammation decreased TRPV1 mRNA levels (Fig. 5A), which was prevented by PEA and CBD. In human colonic tissue, inflammation caused a fall in the mRNA of TRPV1, prevented by CBD (P < 0.05, Fig. 5B). Inflammation increased PPARα mRNA but not by treatment with PEA or CBD (P < 0.05, Fig. 5D).
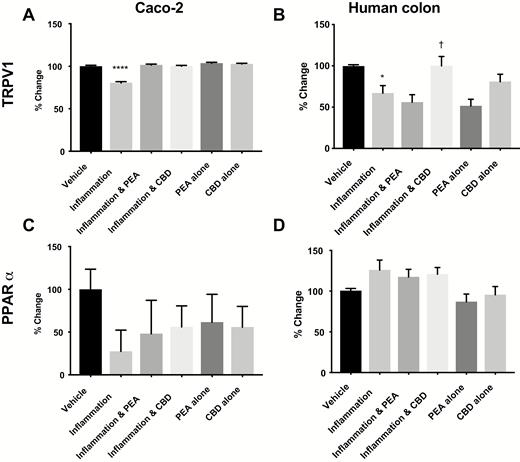
The effects of PEA and CBD on the expression of mRNA levels for receptor proteins in response to a 24-hour inflammatory procotol in cultured Caco-2 cells (A = TRPV1, C = PPARα) and human colonic tissue (B = TRPV1, D = PPARα) measured by multiplex. Data are presented as percentage change from vehicle per plate ± SEM, n = 8 (A and C) and n = 7 (B and D) per condition. Data was analyzed by 1-way ANOVA comparing against vehicle (* P < 0.05, ****P < 0.0001) or against inflammation († P < 0.05).
Inflammation did not affect mRNA levels of PPARδ, PPARγ, PPARα, GPR55, GPR119, CB1 or CB2 in Caco-2 cells or human colonic tissue (Fig. 5C and Supplementary Fig. 2A–L).
Effects of CBD and PEA on Membrane-bound AQP Channels
In Caco-2 cultures, IFNγ and TNFα increased the presence of AQP3 (Fig. 6A), and this was prevented by PEA (apical, P < 0.05) and CBD (apical, P < 0.05). Neither PEA nor CBD alone affected AQP3 compared with control. In human colonic tissue, inflammation did not change AQP3, though AQP3 levels were increased in the presence of inflammation and PEA and also in the presence of inflammation and CBD (P < 0.01 and <0.001, respectively, Fig. 6C). Neither PEA nor CBD alone affected the levels of AQP3 in human mucosal tissue compared with vehicle.
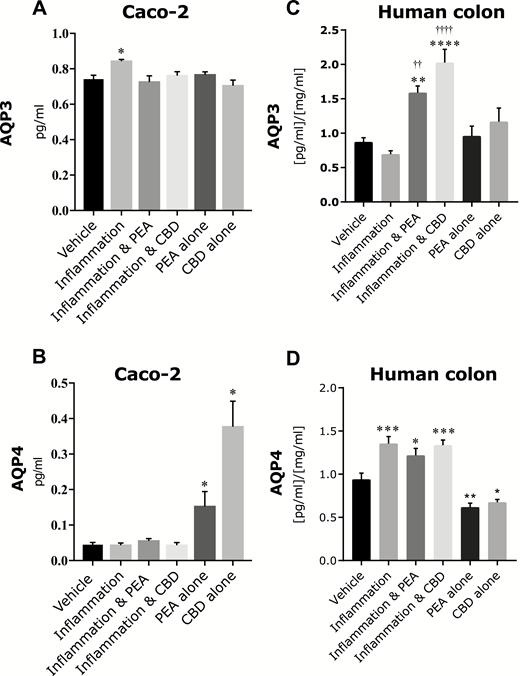
The effects of PEA and CBD on levels of membrane-bound AQP3 in Caco-2 cultures (A) and human colonic mucosa (C) and AQP4 in Caco-2 cultures (B) and human colonic mucosa (D) in response to an inflammatory protocol measured by ELISA. Raw data are expressed as mean protein levels per group ± SEM. N = 6–8 per condition. Data were analyzed by 1-way ANOVA comapring agianst vehicle (* P < 0.05, ** P < 0.01, ***P < 0.001, **** P < 0.0001) or inflammation (†† P < 0.05, ††††P < 0.0001).
In Caco-2 cells, the inflammatory protocol did not affect the presence of AQP4; although treatment with CBD or PEA alone did cause an increase in the presence of AQP4 (P < 0.05, Fig. 6B). Addition of IFNγ and TNFα increased the presence of AQP4 in human colonic mucosa (P < 0.001), which was not affected by treatment with PEA or CBD; however, treatment of colonic mucosa with PEA or CBD alone decreased the presence of these proteins compared with vehicle (P < 0.01 and <0.05, respectively, Fig. 6D).
Absorption and Excretion of Urinary Sugars In vivo
Thirty male participants between 22 and 51 years old (median age 28.7) successfully completed the study. No exclusions were made on the grounds of fitness. No participants reported any side effects in experimental sessions. Urinary concentrations of mannitol excreted over the 6-hour study period were normally distributed.
In participants receiving placebo only, aspirin administration caused an increase in the urinary concentration of mannitol and lactulose over the 6-hour study period (P < 0.0001 and P < 0.001, respectively, Table 1). Maximal increases in LMR were found at 2 hours after aspirin and placebo administration (P < 0.0001 compared with baseline).
Urinary Concentrations of Lactulose and Mannitol in Healthy Volunteers Receiving Aspirin and Placebo (n = 10), CBD (n = 10) and PEA (n = 4) Therapy. Results Are Expressed as Mean Concentrations ± Standard Deviation. Concentrations Were Compared With Baseline Using Repeated Measures ANOVA and the Dunnett Multiple Comparison Test (a<0.05, b<0.01, c<0.001 d<0.0001). LMR—Lactulose: Mannitol Ratio
| Placebo and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 7.1 | 2.0 | P > 0.05 | 3.2 | 3.8 | P > 0.05 | 2.2 |
| 1 hr | 139.1 | 76.5 | P > 0.05 | 4.8 | 5.0 | P > 0.05 | 28.9 |
| 2 hr | 313.4 | 196.4 | P < 0.0001 | 9.2 | 6.8 | P < 0.01 | 34.5 |
| 3 hr | 290.1 | 183.5 | P < 0.0001 | 7.0 | 3.0 | P < 0.001 | 41.4 |
| 4 hr | 194.8 | 134.2 | P < 0.05 | 6.2 | 2.5 | P < 0.05 | 31.2 |
| 6 hr | 182.3 | 129.5 | P < 0.05 | 5.9 | 2.1 | P > 0.05 | 30.8 |
| Placebo and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 7.1 | 2.0 | P > 0.05 | 3.2 | 3.8 | P > 0.05 | 2.2 |
| 1 hr | 139.1 | 76.5 | P > 0.05 | 4.8 | 5.0 | P > 0.05 | 28.9 |
| 2 hr | 313.4 | 196.4 | P < 0.0001 | 9.2 | 6.8 | P < 0.01 | 34.5 |
| 3 hr | 290.1 | 183.5 | P < 0.0001 | 7.0 | 3.0 | P < 0.001 | 41.4 |
| 4 hr | 194.8 | 134.2 | P < 0.05 | 6.2 | 2.5 | P < 0.05 | 31.2 |
| 6 hr | 182.3 | 129.5 | P < 0.05 | 5.9 | 2.1 | P > 0.05 | 30.8 |
| Cannabidiol and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 6.05 | 1.9 | P > 0.05 | 3.19 | 1.3 | P > 0.05 | 1.9 |
| 1 hr | 83.09 | 51.9 | P > 0.05 | 6.6 | 2.9 | P < 0.05 | 12.5 |
| 2 hr | 108.5 | 46.1 | P < 0.0001 | 8.73 | 3.8 | P < 0.01 | 12.4 |
| 3 hr | 98.59 | 57.4 | P < 0.01 | 7.56 | 4.1 | P < 0.05 | 13.0 |
| 4 hr | 82.84 | 42.5 | P < 0.05 | 9.68 | 5.8 | P < 0.01 | 9.2 |
| 6 hr | 77.74 | 32.9 | P > 0.05 | 8.76 | 4.2 | P < 0.01 | 8.8 |
| Cannabidiol and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 6.05 | 1.9 | P > 0.05 | 3.19 | 1.3 | P > 0.05 | 1.9 |
| 1 hr | 83.09 | 51.9 | P > 0.05 | 6.6 | 2.9 | P < 0.05 | 12.5 |
| 2 hr | 108.5 | 46.1 | P < 0.0001 | 8.73 | 3.8 | P < 0.01 | 12.4 |
| 3 hr | 98.59 | 57.4 | P < 0.01 | 7.56 | 4.1 | P < 0.05 | 13.0 |
| 4 hr | 82.84 | 42.5 | P < 0.05 | 9.68 | 5.8 | P < 0.01 | 9.2 |
| 6 hr | 77.74 | 32.9 | P > 0.05 | 8.76 | 4.2 | P < 0.01 | 8.8 |
| Palmitoylethanolamide and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 12.4 | 77.0 | P > 0.05 | 2.52 | 1.1 | P > 0.05 | 4.8 |
| 1 hr | 76.3 | 36.0 | P > 0.05 | 3.32 | 1.0 | P > 0.05 | 21.9 |
| 2 hr | 125.7 | 47.6 | P < 0.01 | 5.78 | 1.0 | P > 0.05 | 22.3 |
| 3 hr | 96.12 | 32.9 | P > 0.05 | 7.34 | 1.4 | P > 0.05 | 13.1 |
| 4 hr | 90.42 | 30.7 | P > 0.05 | 6.9 | 1.8 | P > 0.05 | 11.2 |
| 6 hr | 78.97 | 34.5 | P > 0.05 | 11.6 | 2.3 | P > 0.05 | 7.1 |
| Palmitoylethanolamide and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 12.4 | 77.0 | P > 0.05 | 2.52 | 1.1 | P > 0.05 | 4.8 |
| 1 hr | 76.3 | 36.0 | P > 0.05 | 3.32 | 1.0 | P > 0.05 | 21.9 |
| 2 hr | 125.7 | 47.6 | P < 0.01 | 5.78 | 1.0 | P > 0.05 | 22.3 |
| 3 hr | 96.12 | 32.9 | P > 0.05 | 7.34 | 1.4 | P > 0.05 | 13.1 |
| 4 hr | 90.42 | 30.7 | P > 0.05 | 6.9 | 1.8 | P > 0.05 | 11.2 |
| 6 hr | 78.97 | 34.5 | P > 0.05 | 11.6 | 2.3 | P > 0.05 | 7.1 |
Urinary Concentrations of Lactulose and Mannitol in Healthy Volunteers Receiving Aspirin and Placebo (n = 10), CBD (n = 10) and PEA (n = 4) Therapy. Results Are Expressed as Mean Concentrations ± Standard Deviation. Concentrations Were Compared With Baseline Using Repeated Measures ANOVA and the Dunnett Multiple Comparison Test (a<0.05, b<0.01, c<0.001 d<0.0001). LMR—Lactulose: Mannitol Ratio
| Placebo and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 7.1 | 2.0 | P > 0.05 | 3.2 | 3.8 | P > 0.05 | 2.2 |
| 1 hr | 139.1 | 76.5 | P > 0.05 | 4.8 | 5.0 | P > 0.05 | 28.9 |
| 2 hr | 313.4 | 196.4 | P < 0.0001 | 9.2 | 6.8 | P < 0.01 | 34.5 |
| 3 hr | 290.1 | 183.5 | P < 0.0001 | 7.0 | 3.0 | P < 0.001 | 41.4 |
| 4 hr | 194.8 | 134.2 | P < 0.05 | 6.2 | 2.5 | P < 0.05 | 31.2 |
| 6 hr | 182.3 | 129.5 | P < 0.05 | 5.9 | 2.1 | P > 0.05 | 30.8 |
| Placebo and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 7.1 | 2.0 | P > 0.05 | 3.2 | 3.8 | P > 0.05 | 2.2 |
| 1 hr | 139.1 | 76.5 | P > 0.05 | 4.8 | 5.0 | P > 0.05 | 28.9 |
| 2 hr | 313.4 | 196.4 | P < 0.0001 | 9.2 | 6.8 | P < 0.01 | 34.5 |
| 3 hr | 290.1 | 183.5 | P < 0.0001 | 7.0 | 3.0 | P < 0.001 | 41.4 |
| 4 hr | 194.8 | 134.2 | P < 0.05 | 6.2 | 2.5 | P < 0.05 | 31.2 |
| 6 hr | 182.3 | 129.5 | P < 0.05 | 5.9 | 2.1 | P > 0.05 | 30.8 |
| Cannabidiol and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 6.05 | 1.9 | P > 0.05 | 3.19 | 1.3 | P > 0.05 | 1.9 |
| 1 hr | 83.09 | 51.9 | P > 0.05 | 6.6 | 2.9 | P < 0.05 | 12.5 |
| 2 hr | 108.5 | 46.1 | P < 0.0001 | 8.73 | 3.8 | P < 0.01 | 12.4 |
| 3 hr | 98.59 | 57.4 | P < 0.01 | 7.56 | 4.1 | P < 0.05 | 13.0 |
| 4 hr | 82.84 | 42.5 | P < 0.05 | 9.68 | 5.8 | P < 0.01 | 9.2 |
| 6 hr | 77.74 | 32.9 | P > 0.05 | 8.76 | 4.2 | P < 0.01 | 8.8 |
| Cannabidiol and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 6.05 | 1.9 | P > 0.05 | 3.19 | 1.3 | P > 0.05 | 1.9 |
| 1 hr | 83.09 | 51.9 | P > 0.05 | 6.6 | 2.9 | P < 0.05 | 12.5 |
| 2 hr | 108.5 | 46.1 | P < 0.0001 | 8.73 | 3.8 | P < 0.01 | 12.4 |
| 3 hr | 98.59 | 57.4 | P < 0.01 | 7.56 | 4.1 | P < 0.05 | 13.0 |
| 4 hr | 82.84 | 42.5 | P < 0.05 | 9.68 | 5.8 | P < 0.01 | 9.2 |
| 6 hr | 77.74 | 32.9 | P > 0.05 | 8.76 | 4.2 | P < 0.01 | 8.8 |
| Palmitoylethanolamide and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 12.4 | 77.0 | P > 0.05 | 2.52 | 1.1 | P > 0.05 | 4.8 |
| 1 hr | 76.3 | 36.0 | P > 0.05 | 3.32 | 1.0 | P > 0.05 | 21.9 |
| 2 hr | 125.7 | 47.6 | P < 0.01 | 5.78 | 1.0 | P > 0.05 | 22.3 |
| 3 hr | 96.12 | 32.9 | P > 0.05 | 7.34 | 1.4 | P > 0.05 | 13.1 |
| 4 hr | 90.42 | 30.7 | P > 0.05 | 6.9 | 1.8 | P > 0.05 | 11.2 |
| 6 hr | 78.97 | 34.5 | P > 0.05 | 11.6 | 2.3 | P > 0.05 | 7.1 |
| Palmitoylethanolamide and aspirin | |||||||
|---|---|---|---|---|---|---|---|
| Lactulose | Mannitol | ||||||
| Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | Mean Concentration (ug/mL) | Standard Deviation | Significance from Baseline | LMR | |
| 0 hr | 12.4 | 77.0 | P > 0.05 | 2.52 | 1.1 | P > 0.05 | 4.8 |
| 1 hr | 76.3 | 36.0 | P > 0.05 | 3.32 | 1.0 | P > 0.05 | 21.9 |
| 2 hr | 125.7 | 47.6 | P < 0.01 | 5.78 | 1.0 | P > 0.05 | 22.3 |
| 3 hr | 96.12 | 32.9 | P > 0.05 | 7.34 | 1.4 | P > 0.05 | 13.1 |
| 4 hr | 90.42 | 30.7 | P > 0.05 | 6.9 | 1.8 | P > 0.05 | 11.2 |
| 6 hr | 78.97 | 34.5 | P > 0.05 | 11.6 | 2.3 | P > 0.05 | 7.1 |
In participants administered both CBD and aspirin, urinary lactulose and mannitol concentrations were also increased across the study period (P < 0.001 and P < 0.01, respectively, Table 1). The lactulose to mannitol ratio across the experimental period was also increased (P < 0.001 compared with baseline); however, compared with the placebo and asprin group, LMR was reduced, reaching maximal difference at 2 hours post-administration (P < 0.0001, Fig. 7).
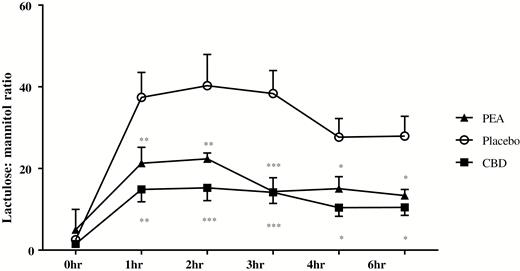
The concentration ratios of urinary lactulose and mannitol over time in healthy participants treated with aspirin and either placebo, CBD or PEA, measured by LC-MS. Results are expressed as mean ratios ± SEM. Time points between groups were compared using 2-way ANOVA using the Dunnett multiple comparisons test comparing to placebo at the same time point (*P < 0.05, **P < 0.01, ***P < 0.001).
In the PEA and aspirin group, urinary lactulose—but not mannitol concentrations—were also increased (P < 0.01, Table 1). In 6 patients in this group, mannitol levels were undetectable at baseline and subsequent timepoints, and hence, LMR was unable to be calculated. These patients were excluded from the results. The lactulose to mannitol ratio across the study period was increased (P < 0.01 compared with baseline). Compared with the placebo and aspirin group, LMR was reduced, reaching maximal difference at 3 hours post-administration (P < 0.001, Fig. 7). There was no difference in significance between PEA and CBD in reduction of LMR compared with placebo.
DISCUSSION
The aims of this study were to assess the effect of PEA and CBD on the permeability of the human gastrointestinal tract and identify underlying mechanisms of action. Within in vitro and ex vivo, PEA and CBD prevented inflammation-induced permeability, and these effects were mediated by different receptors but similar intracellular pathways and associated with changes in claudin, AQP, and receptor expression. We then measured the excretion of sugar probes in vivo using an aspirin-induced pro-inflammatory model, with or without 600 mg of PEA or CBD in healthy volunteers. Aspirin increased the LMR, suggesting an increase in intestinal permeability, and this was prevented by both CBD and PEA. Together, these findings suggest that CBD and PEA decrease intestinal permeability and may have future therapeutic use in IBD.
We chose to examine the effect of CBD and PEA on the transfer of FD4 and FD10 dextrans as these molecules are of a similar size to that of lipopolysaccharide from Escherichia coli and pseudomonas species (20–40 Å).25, 26 Palmitoylethanolamide and cannabidiol both reduced FD4 and FD10 transfer across Caco-2 membranes after an inflammatory protocol, blocked by PPARα and CB1 antagonism respectively, in line with previous reports.18, 27 We hypothesized that there may be additive effects if applied simultaneously but found this not to be the case. Looking at the intracellular mechanisms of action, we found that inhibition of PKA, ERK/MEK, and adenylyl cyclase prevented the actions of both drugs, suggesting that although the membrane receptors of CBD and PEA are different, they exert their actions through similar intracellular pathways and demonstrate no additive effects. This is the first report of the signalling pathways through which PEA acts in human colonic mucosa and is in line with those explored in murine colitis models where PEA acts through phosphorylation of ERK28 and in neuronal tissue where it acts through PKA.29 Similarly, our findings match previously described CBD actions at CB1 through adenylyl cyclase.5
We hypothesized that permeability changes caused by inflammation may be due to transcriptional changes in tight junction (TJ) proteins such as claudin, as changes in the presence of these paracellular proteins have been shown in active Crohn’s disease.30 Tight junction proteins are composed of 2 transmembrane proteins, occludin and claudin, with a third adjacent protein, the junctional adhesional molecule, within the inter-cellular space. These proteins are fused to identical molecules on neighboring epithelial cells, at which point the intercellular space is sealed around a charged pore. The flux of material through this paracellular pore is determined by TJ structure, dependent on claudin type.31 Claudin-2 increases the permeability of the TJ, whereas claudin-5 and claudin-8 decrease permeability.32 We found inflammation had no effect on mRNA for claudin proteins in Caco-2 cultures, and this was uninfluenced by PEA or CBD treatment. We compared these findings with experimentally inflamed human colonic tissue and found a decrease claudin-5 mRNA in response to inflammation and that this change was prevented by treatment with CBD. Claudin-5 is highly expressed in the human colon and acts to strengthen the mucosal barrier by decreasing permeability though cysteine residues.33 Inflammation is known to cause a decrease in the presence of claudin-5 and promotes increased permeability across mucosal types, and therefore, this may be a mechanism by which CBD affects permeability.30, 34 We also found PEA decreased the transcription of claudin-3, a protein providing a barrier function by decreasing permeability to charged ions in the healthy colon.35 The finding that PEA may decrease this transcription in human colon yet decrease hyperpermeability in Caco-2 cultures suggests that either the role of claudin-3 in the gut is incompletely understood or that PEA does not simply affect permeability in terms of pore formation. In support of our findings, Zeissig et al (2007) found that claudin-3 expression is unaffected by Crohn’s disease;30 however, in cell culture experiments, expression is reduced by TNFα treatment36 but not by IL-13.35 As no other reports have examined the effect of cannabinoids on claudin expression in human tissue, we are unable to determine if the effect of PEA on claudin-3 is a mechanism by which permeability may be affected; this requires further study.
Aquaporins have been found to have an increasingly important role in both permeability to water and the immune response; therefore, we hypothesized that changes in the expression of 2 AQPs (AQP3 and AQP4) may be a mechanism by which cannabinoids affect permeability. These proteins allow transport of water and solute through epithelial barriers via a transcellular route. Similarly to TJs, epithelial AQP populations may change dynamically in response to varying physiological environments.37 Aquaporin 3 expression in the ileum is reduced in IBD, which was suggested as a mechanism to reduce oxidative stress through limiting water loss.38 Although it has been shown that knockdown of AQP3 paradoxically impairs gut barrier function and increases permeability.39 We found that in Caco-2 cells, inflammation increased levels of membrane-bound AQP3, which was prevented both by PEA and CBD. Conversely, in human tissue, the inflammatory protocol alone had no effect on AQP3 levels, whereas in the presence of the inflammatory protocol and PEA or CBD, AQP3 levels were increased. Previous work demonstrated that PEA and CBD are not anti-inflammatory in Caco-2 cultures but do prevent the increased secretion of pro-inflammatory cytokines in human colonic tissue.40 A potential mechanism, therefore, for PEA and CBD on the inflammatory response may be through upregulation of functional membrane-bound AQP3 and glycerol uptake, although this may not be the direct mechanism through which permeability is affected in Caco-2 cultures.41 Further study is required examining the effect of CBD and PEA on glycerol uptake in conjunction with AQP3 expression in the human colon.
Aquaporin 4 is known not to contribute to water transfer in the gut, as knock-down of the channel does not affect permeability;42 however, there is evidence suggesting AQP4 plays a role in the immune response of the colonic mucosa as AQP4 is upregulated in colonic mucosa of IL-10 knockout mice.43 In Caco-2 cells, we found no change in membrane-bound AQP4 expression in response to inflammation but found upregulation in expression caused by PEA and CBD treatment. In comparison with experimentally inflamed human colonic tissue, inflammation increased the presence of AQP4, which was not affected by PEA or CBD, although in the absence of inflammation, these levels were reduced compared with vehicle alone. The absence of an immune cell–mediated response to inflammation in Caco-2 cells may explain the difference between the 2 culture models’ response to TNFα and IFNγ. As AQP4 expression was reduced in human tissue compared with vehicle, it may be possible that prophylactic administration of CBD and PEA may change the response to inflammation. We were unable to examine the effect of PEA and CBD on other AQP subtypes in this study; however, this does pose a future avenue of experimentation for cannabinoid and cannabinoid-like compounds.
It has been previously demonstrated that the expression of CB1 and CB2 receptors on the gut epithelium, immune cells, and enteric nervous system change with inflammation.44, 45 As PEA has been shown to alter the expression of these receptors in mice, we hypothesized that PEA and CBD might affect their expression (and of other molecular targets of cannabinoids) in experimentally inflamed human colon.11, 28 Surprisingly, we did not find any effect of PEA or CBD on the expression of these 2 receptors both in Caco-2 and human explant models. However, in both Caco-2 cultures and human colon, we found a significant decrease in the expression of TRPV1 in response to inflammation, in line with previous reports.11 In both cell culture and explant tissue models, CBD prevented these falls, whereas PEA only prevented falls in TRPV1 expression in Caco-2 cultures. We also found that PPARα expression was increased by inflammation, but this was not affected by PEA or CBD treatment. No other receptors were affected by inflammation. The absence of change in CB1 or CB2 transcription is interesting and contradicts existing evidence that CB1 and CB2 are upregulated in biopsies from IBD patients.45 One possible explanation for this difference is the role of the enteric nervous system. Peripherally restricted cannabinoid agonists have been shown not to prevent inflammatory changes in murine colitis, suggesting that cannabinoid action at the central nervous system is crucial to their effect on gut inflammation.46 This was supported by a study from Esposito et al, demonstrating that PEA may act directly on enteric glial cells rather than on mucosal immunocytes.28 We may suggest that our normal tissue, which is no longer innervated by the enteric nervous system, did not undergo any nerve-mediated changes in receptor expression. Alternatively, these differences could also be explained by the presence of a secondary immune response in ex vivo tissue compared with in vitro Caco-2 cultures, possessing no specialized immune response such as macrophages. This presents further evidence that the gut is dependent centrally on endocannabinoid tone for the immune response and barrier function.
Clinically, permeability in vivo is not measured; however experimentally, it may be estimated by the ingestion of probe molecules which undergo urinary excretion. Administering 2 sugars that are absorbed and excreted at differential rates avoids confounding factors such as delayed gastric emptying, differing volumes of distribution, intestinal transit time, and varying renal clearance.47 D-mannitol is passively absorbed by the small intestine at a steady rate in health. Lactulose, a larger molecule, is not normally absorbed in health but is passively absorbed during inflammatory intestinal episodes.48 As both compounds undergo similar degradation in the gut and regular excretion, the urinary lactulose to mannitol ratio (LMR) gives a measure of intestinal permeability. This ratio has previously been used to calculate intestinal permeability in Crohn’s disease, where lactulose absorption proportionally increases to the absorption of D-mannitol.49 We have shown that aspirin causes an approximate 20-fold increase in LMR, in line with previously published reports.48 It has been hypothesized that the mechanism by which increased permeability to sugars occurs by inhibition of cyclooxygenase production, which may lead to a decrease in mucosal prostaglandin production.50 We found that PEA and CBD both decreased the concentrations of these sugars and therefore may have achieved this through inhibiting cyclooxygenase production or by manipulation of membrane-bound proteins or receptors. Although we have not proven the mechanism in this study, we have shown for the first time that in humans, these compounds can reduce intestinal permeability in vivo and may potentially be of clinical use in IBD.
There are several limitations to this study. Although high-dose aspirin was used to increase in the absorption and excretion of ingested probes, this may not be a reliable simulation of gut inflammation. Therefore, the effects of PEA and CBD may not translate clinically. Alternative models have been used to induce a state of hyperpermeability which may resemble clinical disease more closely. One study administered lipopolysaccharide to healthy human subjects, successfully increasing the absorption of orally administered polyethylene glycol.51 However, all 14 participants developed the systemic inflammatory response syndrome, becoming hemodynamically unstable and requiring medical care. In light of these risks, small phase 2 clinical trials examining the effect of CBD and PEA in IBD may now be considered. Secondly, 6 of 10 samples in the PEA cohort were found to contain mannitol levels which were below the level of quantification using our LC-MS method. Therefore, ratios of lactulose to mannitol were not possible to calculate for these 6 participants and were excluded, which may exaggerate group differences. It is not clear if mannitol levels were undetectable because of PEA administration or due to an error in the administration of sugar probes or quantification of sugars at LC-MS. If this were due to a PEA effect, this would have implications for the clinical use of PEA, as this would mean that it is highly permeability-decreasing. This will be the subject of further study in our research group.
In conclusion, we have demonstrated for the first time in humans that PEA and CBD prevent increases in permeability in the inflamed gut and may do so through changes in AQP, TJ, and receptor expression. These data add to the growing body of data demonstrating the anti-inflammatory and permeability-reducing effects of PEA and CBD in the gastrointestinal tract.19 This holds significant promise for the development of future intestinal therapies treating disorders of increased intestinal permeability such as IBD. Their clinical effects should now be assessed in phase 1 and phase 2 clinical trials.
Abbreviations
- AQP3
Aquaporin-3
- AQP4
aquaporin-4
- CBD
cannabidiol
- FBS
fetal bovine serum
- IBD
inflammatory bowel disease
- LMR
lactulose:mannitol ratio
- PEA
palmitoylethanolamide
- PKA
protein kinase A
- PBS
phosphate buffered saline
- RIPA
radioimmunoprecipitation assay
- THC
Δ9-tetrahydrocannabinol
- TEER
trans-epithelial electrical resistance
- TRPV1
transient receptor potential vanilloid 1
Author Contribution: DGC, JNL, AND SEO contributed to the conception and design of the study. DGC, HC, CO, DB, JNL, SEO contributed to the generation, collection, assembly, analysis, and/or interpretation of data. All authors contributed to the drafting or revision of the manuscript and approval of the final version of the manuscript.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}