





Abstract
Do methylation changes in sperm DNA correlate with infertility?
Loss of spermatogenesis and fertility was correlated with 1680 differentially-methylated CpGs (DMCs) across 1052 genes.
Methylation changes in a number of genes have been correlated with reduced sperm count and motility.
This case-control study used spermatozoal DNA from 38 oligo-/oligoastheno-zoospermic infertile patients and 26 normozoospermic fertile men.
Genome-wide methylation analysis was undertaken using 450 K BeadChip on spermatozoal DNA from six infertile and six fertile men to identify DMCs. This was followed by deep sequencing of spermatozoal DNA from 32 infertile patients and 20 fertile controls.
A total of 1680 DMCs were identified, out of which 1436 were hypermethylated and 244 were hypomethylated. Classification of DMCs according to the genes identified BCAN, CTNNA3, DLGAP2, GATA3, MAGI2 and TP73 among imprinted genes, SPATA5, SPATA7, SPATA16 and SPATA22 among spermatogenesis-associated genes, KDM4C and JMJD1C, EZH2 and HDAC4 among genes which regulate methylation and gene expression, HLA-C, HLA-DRB6 and HLA-DQA1 among complementation and immune response genes, and CRISPLD1, LPHN3 and CPEB2 among other genes. Genes showing significant differential methylation in deep sequencing, i.e. HOXB1, GATA3, EBF3, BCAN and TCERG1L, are strong candidates for further investigations. The role of chance was ruled out by deep sequencing of select genes.
N/A.
Genome-wide analyses are fairly accurate, but may not be exactly validated in replication studies across all DMCs. We used the ‘t’ test in the genome-wide methylation analysis, whereas other tests could provide a more robust and powerful analysis.
DMCs can serve as markers for inclusion in infertility screening panels, particularly those in the genes showing differential methylation consistent with previous studies. The genes validated by deep sequencing are strong candidates for investigations of their roles in spermatogenesis.
The study was funded by the Council of Scientific and Industrial Research (CSIR), Govt. of India with grant number BSC0101 awarded to Rajender Singh. None of the authors has any competing interest to declare.
Introduction
Infertility affects ~10%–15% of reproductively active couples worldwide (Jarow et al., 2002). In about half of them, male factor infertility is the cause. Genetic factors remain the most complex and least understood contributors to variations in fertility (Ferlin et al., 2006; Zorrilla and Yatsenko, 2013). Epigenetics, which constitutes another layer of gene regulation, strongly affects gene expression and function. DNA methylation is one of the best-studied epigenetic modifications that affect gene expression (Lande-Diner et al., 2007) and chromatin packaging (Hammoud et al., 2011) during spermatogenesis. In germ cells, DNA methylation is critically involved in various processes, including paternal genomic imprinting (Houshdaran et al., 2007), silencing of transposable elements (Hollister and Gaut, 2009), silencing and DNA compaction, and numerous aspects of mitosis, meiosis and spermiogenesis (Gaysinskaya et al. 2018). The mammalian germ-line undergoes broad epigenetic reprogramming during germ cell maturation and gametogenesis. Widespread erasure of DNA methylation occurs in male primordial germ cells (Seisenberger et al., 2012) and de novo DNA methylation occurs during germ cell maturation and spermatogenesis before meiosis (Oakes et al., 2007a). Consequently, the pattern of sperm DNA methylation is unique in comparison with any somatic cell (Reik et al., 2001; Oakes et al., 2007b). In mature sperm, hypomethylated promoters are found in developmental genes and genes encoding signaling factors (Hammoud et al., 2009). As a result, the specific sperm DNA methylation in mammals is suggested to be essential for spermatogenesis, fertilization and early embryonic development (Li et al., 1992; Eckhardt et al., 2006).
Earlier studies on candidate genes have shown a strong association between abnormal semen parameters and aberrant DNA methylation in imprinted, testes-specific and other genes (Houshdaran et al., 2007; Kobayashi et al., 2007; Boissonnas et al., 2010; Hammoud et al., 2010; Marques et al., 2010; Poplinski et al., 2010; Pacheco et al., 2011; Rajender et al., 2011; Sato et al., 2011; Rotondo et al., 2012; Tian et al., 2014). These reports were followed by genome-wide studies utilizing 27 K array, which suggested that methylation could differentiate between high and low sperm count groups (Aston et al., 2012; Schutte et al., 2013; Montjean et al., 2015). Subsequent studies have employed 450 K BeadChip methylation to compare the methylation profile between normozoospermic fertile and normozoospermic infertile patients, finding that in addition to select targets, methylation at several repetitive sequences is lower in sperm than in somatic cells (Urdinguio et al., 2015). Two other similar studies based on 450 K arrays found that differential methylation correlates with sperm motility (Pacheco et al., 2011; Du et al., 2016). Similarly, in a mixed population of infertility patients attending an IVF clinic, Camprubi et al. (2016) found that methylation analysis could differentiate between fertile and infertile individuals. A recent 450 K-based study identified KCNJ5, MLPH and SMC1B as differentially-methylated genes between sub-fertile and fertile males (Laqqan and Hammadeh, 2017). Most of the above studies provided sufficient strength to the hypothesis that the sperm methylome is associated with sperm count and infertility. Inspite of these reports, the methylation differences between infertile and fertile men remain poorly characterized, particularly due to the lack of replication and a thorough deep analysis around the regions identified by BeadChip analyses.
In the present study, we undertook genome-wide DNA methylation analysis on sperm DNA from infertile individuals and compared them with fertile controls, then performed a validation of the differences in a larger sample size by employing massive parallel sequencing. We found that infertile men differed from fertile controls in 1680 CpG sites, with 1436 CpG sites showing hypermethylation and 244 CpG sites showing hypomethylation in the infertile group. Deep sequencing identified significant differential methylation in a number of genes known to participate in spermatogenesis.
Materials and Methods
Ethical approval
The Institutional Review Board and Ethics Committee of the Central Drug Research Institute (CDRI) approved this study. All subjects were informed about the purpose of sample collection and informed written consents were obtained. Study subjects were recruited at the Department of Endocrinology, Central Drug Research Institute (CDRI), Lucknow and the Institute of Medical Sciences, Banaras Hindu University (BHU), Varanasi.
Semen analysis and sample preparation
For genome-wide DNA methylation, we used samples from normozoospermic fertile controls (N = 6) and oligozoospermic infertile patients (N = 6). For validation by deep sequencing, 52 semen samples were collected and classified into two groups: normozoospermic fertile controls (N = 20) and oligo-/oligoastheno-zoospermic infertile patients (N = 32) (Supplementary Tables S1 and SII). Inclusion criteria for patients consisted of infertility persisting for longer than an year and the absence of other diseases known to affect fertility, such as mumps, mycoplasma infection, varicosity, prostatitis, epididymitis and orchitis. Individuals with abnormal karyotypes and those with Y-chromosome microdeletions were excluded. All of the participants showed endocrine profiles (serum FSH, inhibin B and testosterone) and testicular volumes within normal range, ruling out congenital hypogonadotropic hypogonadism.
Semen samples were collected after 3–5 days of abstinence and allowed to liquefy at 37°C. Semen parameters were analyzed according to the WHO 2010 recommendations (Cooper et al., 2010). Treatment with somatic cell lysis buffer (SCLB: 0.1% SDS, 0.5% Triton X-100) was used to remove leukocytes (Goodrich et al., 2007), followed by confirmation using light microscopic analysis. In case of controls, swim up, immediately upon liquefaction, was performed to prepare high-quality sperm (Jayaraman et al., 2012), as low quality sperm have been shown to be more inconsistent with respect to methylation (Jenkins et al., 2015).
DNA isolation and quality analysis
High-quality DNA was prepared using MasterPure™ kit (Illumina, San Diego, CA, USA). Samples having minimal shearing were taken for further study and low quality samples were prepared on a second occasion. Isolated DNA samples were quantified by spectrophotometry and fluorometry and 1 μg of DNA was used for bisulphite conversion using EZ DNA methylation-GoldTM Kit (Zymo Research).
Genome-wide (450 K) DNA methylation analysis
Genome-wide methylation analysis was carried out using the Illumina Infinium Human Methylation 450 K BeadChip (Illumina). Genomic DNA (500 ng) from each sample was bisulfite converted using the EZ DNA methylation kit (Zymo Research, California, USA) and 4 μl of it was used for Chip hybridization. Technical control samples of different methylation percentages (0%, 50%, 100%) were spiked-in and repeated across plates to ensure high-quality data.
Gene ontology and pathway enrichment
For gene ontology, the enrichment analysis was performed using the Panther (Panther GO slim) software. Gene names were uploaded and analysis was performed against a human reference genome. Pathway analysis was performed using the KEGG tool and statistical analysis was performed against the same using a Bonferroni multiple test adjustment threshold of P < 0.05.
Deep sequencing of select genes
A total of nine genes were selected on the basis of (i) their roles in development, immune response and reproduction from the GO and KEGG analyses (HOXB1, GPR123, CRISPLD1 and KDM4C), (ii) having more than three differentially-methylated CpG (DMC) spots (HLA-C, HLA-DRB6, HLA-DQA1, TCERG1L and EBF3) and (iii) being known to participate in spermatogenesis and male fertility (TP73, GATA3, CPEB2, LPHN2, SMYD3 and BCAN). In total, a set of 15 genes was finalized for validation through deep sequencing, which included sequencing of the CpGs adjacent to the target DMCs. PCR conditions for each primer set are detailed in Supplementary Table SIII.
For deep sequencing, we undertook targeted bisulfite amplicon sequencing using MiSeq (Illumina, San Diego, CA, USA) on an independent set of semen samples from oligozoospermic/oligoastheno-zoospermic infertile patients (n = 32) and fertile control (n = 20) individuals.
The meth primer tool was used to design primer pairs specific for bisulfite converted DNA (Li and Dahiya, 2002), producing PCR amplicons of 300–320 base pairs. PCR amplification was carried out using platinum Taq DNA polymerase (Thermo scientific) and 1 μl of converted DNA template per 25 μl reaction. PCR conditions for each primer set are detailed in Supplementary Table SIII. Amplicons were purified using QIA quick PCR purification kit (Qiagen) as per the manufacturer’s suggested protocol. Purified reaction products were run on a 2% agarose gel for confirmation, followed by quantification using the Qubit dsDNA high sensitivity fluorometric assay (Invitrogen). A 200-ng equimolar mix of the 13 amplicons was used as input for library preparation using the TruSeq DNA PCR-free library preparation kit. TruSeq DNA HT adapters (Illumina) were used for indexing. Library quantification was carried out using Kapa library quantification kit (Kapa Biosystems). Equimolar library pools were mixed and diluted to 8 pM for denaturation. PhiX control v3 (Illumina) was spiked in at a 5.0% final concentration, and subsequent cluster generation/sequencing was performed on the MiSeq using MiSeq Reagent Nano Kit (Illumina). A total of 500 cycles of 2 × 250 paired-end sequencing generated over 1820,000 reads.
For the genes where deep sequencing did not produce good or full coverage of the amplicons (TP73 and SMYD3), Sanger sequencing was employed to analyze the methylation level. For this, PCR products were purified and subjected to Sanger sequencing using standard protocols.
Methylation data analysis
For 450 K data analysis, mean ∆beta was calculated for a given CpG site by comparing methylation level in infertile with fertile individuals. Mean ∆beta is referred to as ∆beta across the manuscript. A positive ∆beta indicates relative hypermethylation and negative ∆beta indicates hypomethylation in the cases (Supplementary Table SIV). The T-test was used for testing statistical significance (P < 0.05). Corrections for multiple testing were made using a Bonferroni’s adjustment for all CpG sites passing quality filtering (Dunn, 1961).
In case of deep sequencing, de-multiplexed raw reads were quality assessed using the FastQC tool. Good quality reads were sorted by keeping the Phred score threshold of 20 and adapter trimming was performed with Illumina universal adapter sequence ‘GAGATCGGAAGAGC’ using trim galore application. QC reports were generated using FastQC for trimming. Good quality processed reads were then aligned using Bismark tool using human genome version hg38 reference sequence. Bismark genome preparation was used for in silico DNA bisulphite conversion. Mapping was done using ‘-non_directional’, ‘-sam-no-hd’ and ‘-sam’ commands to create.sam files for further use in downstream processes. The sam file thus created were used for secondary data processing in an R based tool. This alignment was executed in Ubuntu version 16.04 operating system on an one-core/128 Gb RAM work-station.
The methylation percentage at a locus was determined using R based tool called Methylkit by counting the number of ‘C’ nucleotides aligning to cytosine at a particular location. Bismark generated. sam files were used for obtaining percentage methylation scores in the CpG. This process was run by using ‘processBismarkAln’ command in the console of R studio. This command generates a tab separated text per Bismark.sam file containing percentages of C and T along with the information of chromosome number and position. In case of Sanger sequencing, the methylation level was estimated by the ratio of two alternate nucleotides at the target loci.
mRNA analysis of differentially-methylated genes
We selected nine genes (GPR123, HLA-C, EBF3, KDM4C, LPHN3, TCERG1L, BCAN, HOXB1 and TP73) for estimation of transcript levels in sperm. RNA was isolated from sperm of oligozoospermic infertile and fertile control individuals using Illumina MasterPure complete DNA/RNA purification kit. Equal amount of RNA was converted into cDNA, which was used for estimation of transcript levels using a LightCycler 480 instrument (Roche Life Sciences).
Results
Global differential DNA methylation
Global hypermethylation and hypomethylation were significantly different between cases and controls (Fig. 1A–C). A total of 1680 DMCs were observed, of which 1436 were hypermethylated and 244 were hypomethylated (Fig. 1D). Of these, 1662 DMCs were annotated with a unique CpG identifier number, 12 were single polymorphic sites with rs id and six were denoted only by chromosome number (Supplementary Table SIV). Region-wise distribution of the DMCs showed that methylation differences were more frequent in the gene body (43.13%) and intergenic regions (26.91%) and less frequent around transcriptional start sites (TSS), e.g. TSS1500 (6.63%) and TSS200 (2.18%), untranslated regions, e.g. 5′UTR (7.04%) and 3′UTR (12.8%), and in the first exon (1.31%) (Fig. 1E). Most of the DMCs belonged to open sea regions (71.89%) with a very little frequency in the CpG island (5.19%), N-shelf (7.76%), N-shore (3.88%), S-shelf (7.76%) and S-shore (3.52%) regions (Fig. 1F)
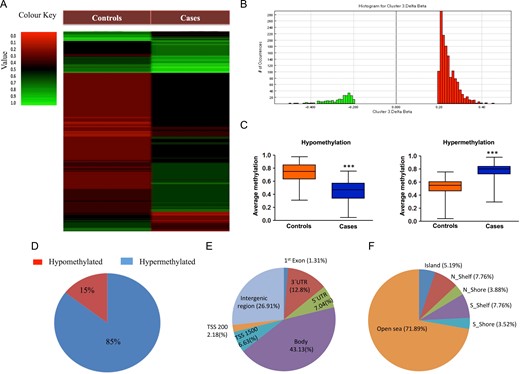
(A) Heat map of genome-wide DNA methylation. (B) Histogram showing the number of differentially-methylated CpGs (DMCs) across various β-values. (C) Box plot comparing mean β-values for hypermethylated and hypomethylated DMCs between cases and controls. (D) Percentage of DMCs showing hypermethylation and hypomethylation. (E) Distribution of DMCs on the basis of organization of gene transcript structure. (F) Distribution of DMCs on the basis of CpG island and neighborhood regions.
Out of 1662 annotated DMCs, 1230 spots were referred to genes, and other spots were either in the intergenic regions or placed distantly enough with respect to any gene. These 1230 DMCs represented 1052 genes, with four genes (CACNA2P1, HOXB1, MED12L and RICTOR) having four, three genes (HLA-DQA1, LMF1, RPH3AL) having five, two genes (HLA-C, TCERG1L) having six and one gene (EBF3) having seven DMCs (Supplementary Table SIV). A number of the genes possessing DMCs have been shown to play important roles in spermatogenesis and fertility (Table I). We have also undertaken an analysis on the regulatory regions to which the DMCs affiliate and found that seven hypermethylated DMCs belonged to the GTDC1, ABCA7, FHIT, TMEM87A, COL11 A1, DDX43 and ZNF492 genes and four hypomethylated DMCs belonged to the ATP1A4, HDAC4, TEKT5 and TSNAX-DISC1 genes. An additional analysis on DNase hypersensitive sites and enhancers identified 50 and 532 CpGs, respectively (Supplementary Table SIV). Out of 50 DNase hypersensitivity sites, 20 were hypomethylated and 30 were hypermethylated, and out of 532 enhancer sites, 67 were hypomethylated and 465 were hypermethylated.
DMC harboring genes involved in spermatogenesis or fertility or those with predominant testicular expression.
| Gene | Protein | Genomic localization | Methylation change | Function summary | β-value |
|---|---|---|---|---|---|
| CPEB2 | Cytoplasmic polyadenylation element binding protein | 3′UTR | Hypermethylation | Required for spermatogenesis | 0.2067 |
| HOXB1 | Homeobox B1 | Promoter | Hypermethylation | Highest expression in testis | 0.2164 |
| RECQL4 | RecQ Like Helicase 4 | Body | Hypomethylation | Abundantly expressed during spermatogenesis | −0.2698 |
| SOD3 | Superoxide Dismutase 3 | Promoter | Hypomethylation | Function in fertilization | −0.2698 |
| VRK1 | Vaccine Related Kinase 1 | Body | Hypermethylation | Spermatogonial proliferation and differentiation | 0.2589 |
| SPATA16 | Spermatogenesis Associated 16 | Promoter | Hypermethylation | Specifically expressed in testis and involved in acrosome formation and sperm–egg fusion. | 0.2443 |
| SPATA22 | Spermatogenesis Associated 22 | Body | Hypermethylation | Predominantly expressed in germ cells and essential for early meiotic prophase | 0.2443 |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | Body | Hypermethylation | Involved in spermatogonial differentiation and apoptosis | 0.2106 |
| TET2 | Tet Methylcytosine Dioxygenase 2 | Promoter | Hypermethylation | Required for spermatogenesis | 0.2480 |
| NPHP1 | Nephrocystin 1 | Body | Hypermethylation | Required for sperm morphogenesis | 0.2265 |
| JMJD1C | Jumonji Domain Containing 1C | Body | Hypermethylation | Involved in spermatogenesis and fertility | 0.2180 |
| PRDM1 | PR/SET Domain 1 | Body | Hypermethylation | Required for spermatogenesis | 0.2321 |
| AKAP9 | A-Kinase Anchoring Protein 9 | Body | Hypermethylation | Required for spermatogenesis and Sertoli cell maturation | 0.2008 |
| AKT3 | AKT Serine/Threonine Kinase 3 | Body | Hypermethylation | Dominantly expressed in testis and brain | 0.2008 |
| DDX4 | DEAD-Box Helicase 4 | 3′UTR | Hypermethylation | Required for germ cells development and spermatogenesis | 0.2077 |
| TBL1XR1 | Transducin Beta Like 1 X-Linked Receptor 1 | Body | Hypermethylation | Involved in chromatin remodeling and sperm differentiation | 0.2479 |
| SPATA5 | Spermatogenesis Associated 5 | Body | Hypermethylation | Required for mitochondrial function and integrity during spermatogenesis | 0.2444 |
| SPATA7 | Spermatogenesis Associated 7 | Body | Hypermethylation | Specifically expressed in testis and retina | 0.2444 |
| HLA-DQA1 | Major Histocompatibility Complex, Class II, DQ Alpha 1 | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2156 |
| HLA-DRB6 | Major Histocompatibility Complex, Class II, DR Beta 6 (Pseudogene) | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2160 |
| Gene | Protein | Genomic localization | Methylation change | Function summary | β-value |
|---|---|---|---|---|---|
| CPEB2 | Cytoplasmic polyadenylation element binding protein | 3′UTR | Hypermethylation | Required for spermatogenesis | 0.2067 |
| HOXB1 | Homeobox B1 | Promoter | Hypermethylation | Highest expression in testis | 0.2164 |
| RECQL4 | RecQ Like Helicase 4 | Body | Hypomethylation | Abundantly expressed during spermatogenesis | −0.2698 |
| SOD3 | Superoxide Dismutase 3 | Promoter | Hypomethylation | Function in fertilization | −0.2698 |
| VRK1 | Vaccine Related Kinase 1 | Body | Hypermethylation | Spermatogonial proliferation and differentiation | 0.2589 |
| SPATA16 | Spermatogenesis Associated 16 | Promoter | Hypermethylation | Specifically expressed in testis and involved in acrosome formation and sperm–egg fusion. | 0.2443 |
| SPATA22 | Spermatogenesis Associated 22 | Body | Hypermethylation | Predominantly expressed in germ cells and essential for early meiotic prophase | 0.2443 |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | Body | Hypermethylation | Involved in spermatogonial differentiation and apoptosis | 0.2106 |
| TET2 | Tet Methylcytosine Dioxygenase 2 | Promoter | Hypermethylation | Required for spermatogenesis | 0.2480 |
| NPHP1 | Nephrocystin 1 | Body | Hypermethylation | Required for sperm morphogenesis | 0.2265 |
| JMJD1C | Jumonji Domain Containing 1C | Body | Hypermethylation | Involved in spermatogenesis and fertility | 0.2180 |
| PRDM1 | PR/SET Domain 1 | Body | Hypermethylation | Required for spermatogenesis | 0.2321 |
| AKAP9 | A-Kinase Anchoring Protein 9 | Body | Hypermethylation | Required for spermatogenesis and Sertoli cell maturation | 0.2008 |
| AKT3 | AKT Serine/Threonine Kinase 3 | Body | Hypermethylation | Dominantly expressed in testis and brain | 0.2008 |
| DDX4 | DEAD-Box Helicase 4 | 3′UTR | Hypermethylation | Required for germ cells development and spermatogenesis | 0.2077 |
| TBL1XR1 | Transducin Beta Like 1 X-Linked Receptor 1 | Body | Hypermethylation | Involved in chromatin remodeling and sperm differentiation | 0.2479 |
| SPATA5 | Spermatogenesis Associated 5 | Body | Hypermethylation | Required for mitochondrial function and integrity during spermatogenesis | 0.2444 |
| SPATA7 | Spermatogenesis Associated 7 | Body | Hypermethylation | Specifically expressed in testis and retina | 0.2444 |
| HLA-DQA1 | Major Histocompatibility Complex, Class II, DQ Alpha 1 | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2156 |
| HLA-DRB6 | Major Histocompatibility Complex, Class II, DR Beta 6 (Pseudogene) | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2160 |
DMC harboring genes involved in spermatogenesis or fertility or those with predominant testicular expression.
| Gene | Protein | Genomic localization | Methylation change | Function summary | β-value |
|---|---|---|---|---|---|
| CPEB2 | Cytoplasmic polyadenylation element binding protein | 3′UTR | Hypermethylation | Required for spermatogenesis | 0.2067 |
| HOXB1 | Homeobox B1 | Promoter | Hypermethylation | Highest expression in testis | 0.2164 |
| RECQL4 | RecQ Like Helicase 4 | Body | Hypomethylation | Abundantly expressed during spermatogenesis | −0.2698 |
| SOD3 | Superoxide Dismutase 3 | Promoter | Hypomethylation | Function in fertilization | −0.2698 |
| VRK1 | Vaccine Related Kinase 1 | Body | Hypermethylation | Spermatogonial proliferation and differentiation | 0.2589 |
| SPATA16 | Spermatogenesis Associated 16 | Promoter | Hypermethylation | Specifically expressed in testis and involved in acrosome formation and sperm–egg fusion. | 0.2443 |
| SPATA22 | Spermatogenesis Associated 22 | Body | Hypermethylation | Predominantly expressed in germ cells and essential for early meiotic prophase | 0.2443 |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | Body | Hypermethylation | Involved in spermatogonial differentiation and apoptosis | 0.2106 |
| TET2 | Tet Methylcytosine Dioxygenase 2 | Promoter | Hypermethylation | Required for spermatogenesis | 0.2480 |
| NPHP1 | Nephrocystin 1 | Body | Hypermethylation | Required for sperm morphogenesis | 0.2265 |
| JMJD1C | Jumonji Domain Containing 1C | Body | Hypermethylation | Involved in spermatogenesis and fertility | 0.2180 |
| PRDM1 | PR/SET Domain 1 | Body | Hypermethylation | Required for spermatogenesis | 0.2321 |
| AKAP9 | A-Kinase Anchoring Protein 9 | Body | Hypermethylation | Required for spermatogenesis and Sertoli cell maturation | 0.2008 |
| AKT3 | AKT Serine/Threonine Kinase 3 | Body | Hypermethylation | Dominantly expressed in testis and brain | 0.2008 |
| DDX4 | DEAD-Box Helicase 4 | 3′UTR | Hypermethylation | Required for germ cells development and spermatogenesis | 0.2077 |
| TBL1XR1 | Transducin Beta Like 1 X-Linked Receptor 1 | Body | Hypermethylation | Involved in chromatin remodeling and sperm differentiation | 0.2479 |
| SPATA5 | Spermatogenesis Associated 5 | Body | Hypermethylation | Required for mitochondrial function and integrity during spermatogenesis | 0.2444 |
| SPATA7 | Spermatogenesis Associated 7 | Body | Hypermethylation | Specifically expressed in testis and retina | 0.2444 |
| HLA-DQA1 | Major Histocompatibility Complex, Class II, DQ Alpha 1 | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2156 |
| HLA-DRB6 | Major Histocompatibility Complex, Class II, DR Beta 6 (Pseudogene) | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2160 |
| Gene | Protein | Genomic localization | Methylation change | Function summary | β-value |
|---|---|---|---|---|---|
| CPEB2 | Cytoplasmic polyadenylation element binding protein | 3′UTR | Hypermethylation | Required for spermatogenesis | 0.2067 |
| HOXB1 | Homeobox B1 | Promoter | Hypermethylation | Highest expression in testis | 0.2164 |
| RECQL4 | RecQ Like Helicase 4 | Body | Hypomethylation | Abundantly expressed during spermatogenesis | −0.2698 |
| SOD3 | Superoxide Dismutase 3 | Promoter | Hypomethylation | Function in fertilization | −0.2698 |
| VRK1 | Vaccine Related Kinase 1 | Body | Hypermethylation | Spermatogonial proliferation and differentiation | 0.2589 |
| SPATA16 | Spermatogenesis Associated 16 | Promoter | Hypermethylation | Specifically expressed in testis and involved in acrosome formation and sperm–egg fusion. | 0.2443 |
| SPATA22 | Spermatogenesis Associated 22 | Body | Hypermethylation | Predominantly expressed in germ cells and essential for early meiotic prophase | 0.2443 |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | Body | Hypermethylation | Involved in spermatogonial differentiation and apoptosis | 0.2106 |
| TET2 | Tet Methylcytosine Dioxygenase 2 | Promoter | Hypermethylation | Required for spermatogenesis | 0.2480 |
| NPHP1 | Nephrocystin 1 | Body | Hypermethylation | Required for sperm morphogenesis | 0.2265 |
| JMJD1C | Jumonji Domain Containing 1C | Body | Hypermethylation | Involved in spermatogenesis and fertility | 0.2180 |
| PRDM1 | PR/SET Domain 1 | Body | Hypermethylation | Required for spermatogenesis | 0.2321 |
| AKAP9 | A-Kinase Anchoring Protein 9 | Body | Hypermethylation | Required for spermatogenesis and Sertoli cell maturation | 0.2008 |
| AKT3 | AKT Serine/Threonine Kinase 3 | Body | Hypermethylation | Dominantly expressed in testis and brain | 0.2008 |
| DDX4 | DEAD-Box Helicase 4 | 3′UTR | Hypermethylation | Required for germ cells development and spermatogenesis | 0.2077 |
| TBL1XR1 | Transducin Beta Like 1 X-Linked Receptor 1 | Body | Hypermethylation | Involved in chromatin remodeling and sperm differentiation | 0.2479 |
| SPATA5 | Spermatogenesis Associated 5 | Body | Hypermethylation | Required for mitochondrial function and integrity during spermatogenesis | 0.2444 |
| SPATA7 | Spermatogenesis Associated 7 | Body | Hypermethylation | Specifically expressed in testis and retina | 0.2444 |
| HLA-DQA1 | Major Histocompatibility Complex, Class II, DQ Alpha 1 | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2156 |
| HLA-DRB6 | Major Histocompatibility Complex, Class II, DR Beta 6 (Pseudogene) | Body | Hypermethylation | Involved in spermatozoa and oocyte interactions | 0.2160 |
Gene ontology and KEGG analysis of genes
We took the above 1052 genes for ontology and KEGG pathway analysis to classify them based on their functions and biological pathways. A total of 971 gene IDs were mapped and used for gene ontology (GO). GO showed 702 hits in molecular function, 589 hits in cellular component and 1391 hits in biological processes (Supplementary Fig. 1SA–C). We took reproduction as our target biological process for gene selection and delve down to the lowest possible level to spermatogenesis (Supplementary Fig. S1C).
KEGG pathway analysis suggested their participation in various pathways such as phagosome (15 genes, P = 2.87 × 10−06), axon guidance (13 genes, P = 9.62 × 10−06), cell adhesion molecule (12 genes, P = 6.29 × 10−05), MAP kinase signaling pathway (17 genes, P = 2 × 10−04), viral myocardidities (10 genes, P = 4.59 × 10−06), type 1 diabetes mellitus (8 genes, P = 5.49 × 10−06), alograft rejection (7 genes, P = 1.90 × 10−05), pathways in cancer (21 genes, P = 2.74 × 10−05) and graft versus host disease (7 genes, P = 3.84 × 10−05) (Supplementary Fig. S1D). Pathway analysis for genes specific to reproduction is presented in Supplementary Fig. S1E.
Deep sequencing of selected genes in infertile and fertile samples
To validate genome-wide differential methylation data, a targeted bisulphite deep sequencing of 15 genes was undertaken on sperm DNA from 32 oligo-/oligoastheno-zoospermic infertile and 20 normozoospermic fertile controls. Along with target DMCs identified by 450 K analysis, deep sequencing analyzed additional proximal CpGs, which helped us in assessing region-wise methylation pattern. Deep sequencing of the amplicons yielded on an average 60,000 pairs of reads with mapping efficiency close to an average of 60%. The qualities of reads were exceptionally good with almost 98% of reads with a Q score above 30 (Supplementary Fig. S2). The quality improved slightly after adapter trimming as adapter contamination in the reads was negligible.
In deep/Sanger sequencing, six genes (HOXB1, CRISPLD1, HLA-DQA1, HLA-DRB6, GATA3 and EBF3) showed hypermethylation in the cases (Figs 2 and 3), seven genes (BCAN, CPEB2, LPHN3, SMYD3, KDM4C, HLA-C and TCERG1L) showed hypomethylation in the cases (Figs 4 and 5) and two genes (TP73, GPR123) showed no difference in methylation between cases and controls (Figs 5 and 6). With regard to the above, the hypermethylation in HOXB1, GATA3 and EBF3 and hypomethylation in the BCAN and TCERG1L genes were statistically significant.
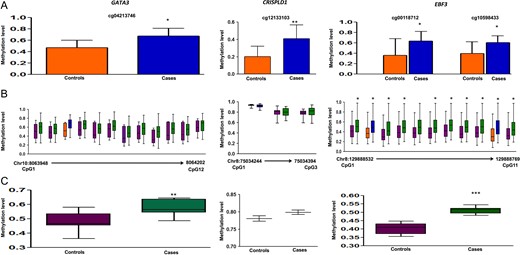
Methylation data for GATA3, CRISPLD1 and EBF3 for 450 K DMC (A), deep sequencing of neighboring CpGs in the target region (B), average methylation in the target region compared between cases and controls (C). 450 K DMC in deep sequencing (B) has been denoted using the same colors as in graphical representation of the DMC in panel A. In panel B, chromosome and nucleotide positions of the CpGs analyzed have been shown by numbers on the X-axis of the box plot.
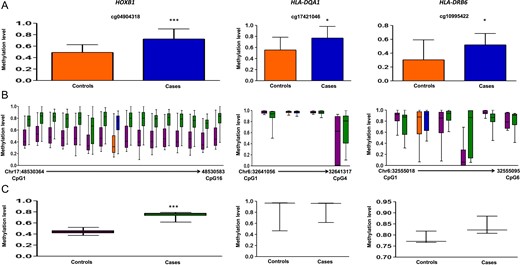
Methylation data for HOXB1, HLA-DQA1 and HLA-DRB6 for 450 K DMC (A), deep sequencing of neighboring CpGs in the target region (B), average methylation in the target region compared between cases and controls (C). 450 K DMC in deep sequencing (B) has been denoted using the same colors as in graphical representation of the DMC in panel A. In panel B, chromosome and nucleotide positions of the CpGs analyzed have been shown by numbers on the X-axis of the box plot.
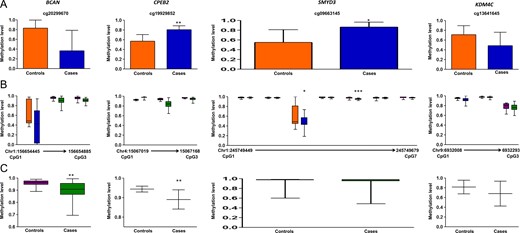
Methylation data for BCAN, CPEB2, SMYD3 and KDM4C for 450 K DMC (A), deep sequencing of neighboring CpGs in the target region (B), SMYD3 data was generated using Sanger sequencing. Average methylation in the target region compared between cases and controls (C). 450 K DMC in deep sequencing (B) has been denoted using the same colors as in graphical representation of the DMC in panel A. In panel B, chromosome and nucleotide positions of the CpGs analyzed have been shown by numbers on the X-axis of the box plot.
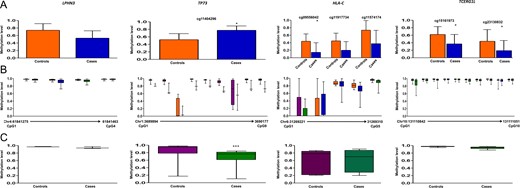
Methylation data for LPHN3, TP73, HLA-C and TCERG1L for 450 K DMC (A), deep sequencing of neighboring CpGs in the target region (B), TP73 data was generated using Sanger sequencing. Average methylation in the target region compared between cases and controls (C). 450 K DMC in deep sequencing (B) has been denoted using the same colors as in graphical representation of the DMC in panel A. In panel B, chromosome and nucleotide positions of the CpGs analyzed have been shown by numbers on the X-axis of the box plot.
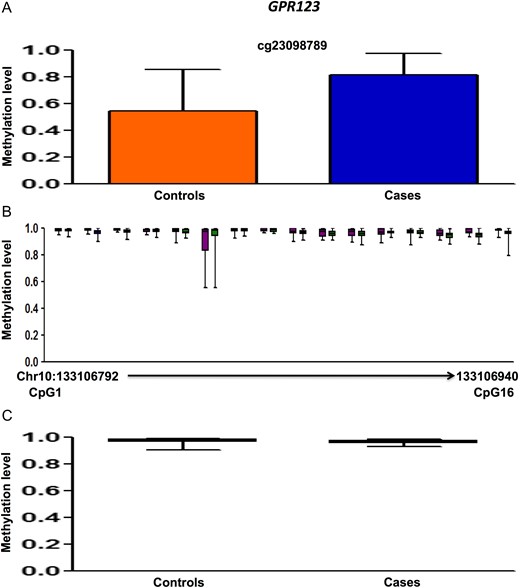
Methylation data for GPR123 for 450 K DMC (A), deep sequencing of neighboring CpGs in the target region (B), average methylation in the target region compared between cases and controls (C). 450 K DMC in deep sequencing (B) has been denoted using the same colors as in graphical representation of the DMC in panel A. In panel B, chromosome and nucleotide positions of the CpGs analyzed have been shown by numbers on the X-axis of the box plot.
Transcript levels in sperm
We compared the expression data with methylation output from the deep sequencing experiment. Out of nine genes tested for expression analysis, six genes (GPR123, HLA-C, KDM4C, LPHN3, TCERG1L, BCAN) showed expression concordant with methylation data; however, the expression of three genes (HOXB1, TP73 and EBF3) was not concordant with their methylation level (Fig. 7).
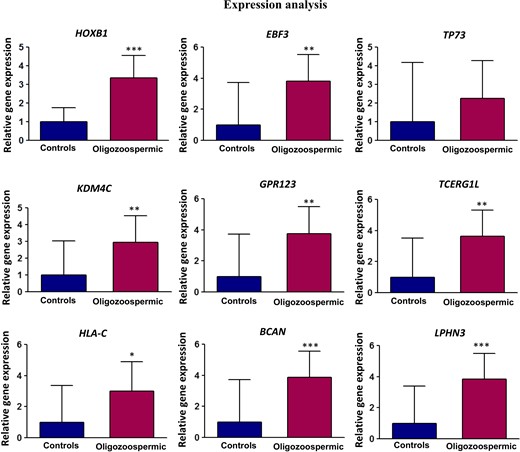
Expression analysis of selected genes. mRNA from sperm was converted into cDNA and subjected to analysis of transcript level by real time PCR.
Discussion
We found that out of 1680 DMCs, 1436 were hypermethylated and 244 were hypomethylated in the patients group. However, one limitation of our study could be the use of the ‘t’ test in the genome-wide methylation analysis, whereas other tests could provide a more robust and powerful analysis.
A majority of the DMCs were in the non-island regions, which is consistent with previous reports stating that the key mark of methylation in germ cells are non-CpG island sequences (Hajkova et al., 2002). Out of the differentially-methylated DNase hypersensitive sites, a majority showed hypermethylation in the patients, which is consistent with overall hypermethylation in this group. Similarly, Camprubi et al. (2016) found 696 DMCs, a majority of which were hypermethylated (74%). Another similar study found hypermethylation in sperm with poor morphology in comparison to sperm with good morphology (Cassuto et al., 2016). Differential methylation in ANK2, PRDM1 (Camprubi et al., 2016), TP73, GATA3 and VAX2 (Pacheco et al., 2011), and MLPH, SMC1B and KCNJ5 (Laqqan and Hammadeh, 2017) is consistent with previous studies, which makes them strong candidates for inclusion in infertility screening panels. Out of the above, PRDM1 (PR domain containing 1, with ZNF domain), a repressor of beta-interferon gene expression, plays a role in the primordial germ cell induction (Ohinata et al., 2005) and development (Fang et al., 2018). Similarly, MLPH is expressed in testis and plays a role in acrosome biogenesis and the development of spermatid tail (Fukuda and Kuroda, 2002). The importance of ANK2, VAX2, SMC1B and KCNJ5 genes in spermatogenesis remains unknown.
Among imprinted genes, we found DMCs in ASB4, BCAN, CTNNA3, DLGAP2, GATA3, MAGI2 and TP73 genes (Supplementary Table SIV). The ankyrin repeat and SOCS box-containing 4 (ASB4) gene is widely expressed in murine testes from the fourth week after birth extending into adulthood with specifically high expression in the pachytene spermatocytes, suggesting its pivotal role in testes development and spermatogenesis (Kim et al., 2008). Interestingly, under acute heat stress conditions, the CTNNA3 gene is down-regulated in chicken testis (Wang et al., 2015), suggesting its role in the regulation of spermatogenesis. MAGI2 plays an important role in sperm cell maturation by engaging in the regulation of tight junctions (Ihara et al., 2012). DLGAP2 is paternally expressed only in testis (Ranta et al., 2000), but requires further investigation for its role in spermatogenesis.
We found significant hypermethylation in spermatogenesis-associated gene family members, SPATA5, SPATA7, SPATA16 and SPATA22, which play various functions in spermatogenesis or fertility (Dam et al., 2007; Dorosh et al., 2013; Tanaka et al., 2015). We also observed differential methylation in testis-specific genes, TSPY and TTTY4B, which are important for spermatogenesis (Krausz et al., 2010; Lu et al., 2014). SMYD3 transcript shows 2.3-fold higher expression in adult human testes as compared to fetal testes (Zhou et al., 2005) and mutations in this gene have been reported in patients with azoospermia (Venkatesh et al., 2014). Other genes harboring DMCs included ten eleven translocase (TET2), histone demethylases (KDM4C and JPJD1C), histone deacetlyases (HDAC4), histone methyltranferases (EZH2), which play important roles in gene regulation. At least EZH2 has been shown to play a role in the regulation of spermatogonial differentiation (Jin et al., 2017).
Complementation genes constitute another interesting category in which we observed significant differential methylation. We observed that HLA-C gene was hypomethylated in infertile subjects while HLA-DRB6 and HLA-DQA1 were hypermethylated. Interestingly, van der Ven et al. (2000) demonstrated that HLA classes II MHC are strongly associated with gamete quality and embryonic development. HLA-DQA1 and HLA-DRB1 are associated with severe andrological problems in infertile men when compared with normozoospermic men. Mori et al. (1990) elucidated that HLA antigens are expressed in spermatozoa and are beneficial for spermatozoa and oocyte interactions in the mouse. Interestingly, ~50 differentially-methylated genes belonged to the immune response system, which opens up new avenues for investigating their functional roles in male infertility. It is interesting to note that epigenetic alterations in autoimmune processes affect fertility (Schutte et al., 2013).
Among other differentially-methylatyed genes, CRISPLD1 may have a role in cellular adhesion, which is essential for fertilization (Gibbs et al., 2008). In addition, CRISPLD1 was identified as a seminal plasma biomarker, upregulation of which affects sperm motility, acrosome reaction and capacitation (Antoniassi et al., 2016). LPHN3, which encodes a member of the G-protein coupled receptors (GPCR), showed differential expression in asthenozoospermia, normozoospermic infertile patients and controls in our previous study (Bansal et al., 2015). CPEB2, which encodes the cytoplasmic polyadenylation element binding protein, is expressed post-meiotically in mouse spermatogenesis and may have a role in translational regulation of stored mRNAs in the transcriptionally inactive haploid spermatids (Kurihara et al., 2003). KEGG pathway analysis showed that candidate genes participate in key cellular processes associated with spermatogenesis, such as MAPK, which participates in signal transduction cascades and cell adhesion molecules, which participate in the blood testis barrier and the Sertoli cell dependent maturation of germ cells.
Deep sequencing revealed significant hypermethylation in HOXB1, GATA3 and EBF3 genes and hypomethylation in BCAN and TCERG1L genes, making them excellent candidates for further investigation. Interestingly, GATA3 in the Sertoli cells has been shown to coordinate with androgen receptor to regulate gene expression (Bhardwaj et al., 2008). Further research is required to understand the importance of BCAN and EBF3 in spermatogenesis. Another research candidate is TP73 since knockout mice for TP73 are vulnerable to increased DNA damage and cell death in spermatogonia, disorganized apical ectoplasmic specialization, malformed spermatids and marked hyperspermia, suggesting its importance in spermatogenesis (Inoue et al., 2014). We also found that the association between sperm transcript levels and DNA methylation may not be straightforward as sperm are transcriptionally inactive.
In conclusion, the present study highlights the association between methylation changes and loss of fertility. Mapping of the DMCs with respective genes showed HOXB1, EBF3, ANK2, PRDM1, TP73, GATA3, VAX2, MLPH, SMC1B, KCNJ5, ASB4, BCAN, CTNNA3, DLGAP2, MAGI2, SPATA5, SPATA7, SPATA16, SPATA22, TET2, KDM4C, TCERG1L, JMJD1C, HDAC4, SMYD3, HLA-C, HLA-DRB6, HLA-DQA1, CRISPLD1, LPHN3 and CPEB2 genes to be the top candidates showing alterations. The genes consistent with previous methylation studies include ANK2, PRDM1, TP73, GATA3, VAX2, MLPH, SMC1B and KCNJ5, which makes them excellent candidates for inclusion in infertility screening panels. Besides the above, this study has identified other differentially-methylated genes, DLGAP2, HLA-C, HLA-DRB6, HLA-DQA1, CPEB2 and EBF3, which need further investigation for their roles in spermatogenesis. The genes showing hypermethylated (HOXB1, GATA3 and EBF3) and hypomethylated CpGs (BCAN and TCERG1L) in deep sequencing are strong candidates for investigation of their roles in spermatogenesis. In addition to identifying new genes that may play important roles in spermatogenesis, this study has identified methylation markers that can become a part of methylation-based infertility screening panels.
Funding
The study was funded by the Council of Scientific and Industrial Research (CSIR), Govt. of India under a network scheme of projects, with grant number BSC0101.
Conflict of interest
All authors have declared no conflict of interest.
Authors’ contributions
S.R., G.G., S.S., K.M.S. and K.S. conceived the study design. K.M.S., S.S., V.S., R.P. and S.R. conducted experiments. K.M.S., V.S., K.S., N.K.A. and S.T. contributed samples. R.P., G.G., K.S. and S.R. supervised the study. S.S. and K.M.S. analyzed the data. All authors have read and approved the manuscript.
References
Author notes
Kumar Mohanty Sujit and Saumya Sarkar Equal contribution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}