




Abstract
De novo truncating mutations in Additional sex combs-like 3 (ASXL3) have been identified in individuals with Bainbridge–Ropers syndrome (BRS), characterized by failure to thrive, global developmental delay, feeding problems, hypotonia, dysmorphic features, profound speech delays and intellectual disability. We identified three novel de novo heterozygous truncating variants distributed across ASXL3, outside the original cluster of ASXL3 mutations previously described for BRS. Primary skin fibroblasts established from a BRS patient were used to investigate the functional impact of pathogenic variants. ASXL3 mRNA transcripts from the mutated allele are prone to nonsense-mediated decay, and expression of ASXL3 is reduced. We found that ASXL3 interacts with BAP1, a hydrolase that removes mono-ubiquitin from histone H2A lysine 119 (H2AK119Ub1) as a component of the Polycomb repressive deubiquitination (PR-DUB) complex. A significant increase in H2AK119Ub1 was observed in ASXL3 patient fibroblasts, highlighting an important functional role for ASXL3 in PR-DUB mediated deubiquitination. Transcriptomes of ASXL3 patient and control fibroblasts were compared to investigate the impact of chromatin changes on transcriptional regulation. Out of 564 significantly differentially expressed genes (DEGs) in ASXL3 patient fibroblasts, 52% were upregulated and 48% downregulated. DEGs were enriched in molecular processes impacting transcriptional regulation, development and proliferation, consistent with the features of BRS. This is the first single gene disorder linked to defects in deubiquitination of H2AK119Ub1 and suggests an important role for dynamic regulation of H2A mono-ubiquitination in transcriptional regulation and the pathophysiology of BRS.
Introduction
Bainbridge–Ropers syndrome (BRS; OMIM 615485) is characterized by failure to thrive, feeding problems, global developmental delay, hypotonia, intellectual disability (ID) and delays in language acquisition (1). De novo dominant truncating mutations in Additional sex combs-like 3 (ASXL3) have emerged as the cause of BRS, while missense mutations in ASXL3 have been identified in individuals with Autism Spectrum Disorder (ASD) (2–4). With massively parallel DNA sequencing, de novo heterozygous mutations have emerged as a prominent cause of ASD and ID. A disproportionate fraction of de novo mutations disrupt chromatin remodeling genes, implicating epigenetic dysregulation of gene transcription as an important molecular mechanism (3,5–7). Here we describe three novel truncating variants in ASXL3 that extend beyond the original cluster of those previously described. These mutations help to define the genotypic spectrum across ASXL3 associated with the clinical features of BRS.
ASXL genes are mammalian homologues of drosophila Additional sex combs (Asx). Members of this gene family enhance transcription regulation by Polycomb-group (PcG) and Trithorax-group (TrxG) complexes (8–10). The ASXL family consists of three members (ASXL1, ASXL2 and ASXL3) that share a common domain architecture: i.e. ASXN, ASXH, ASXM1, ASXM2 domains and a PHD finger (Fig. 1D) (11–13). Germline mutations of ASXL1 and ASXL3 occur in patients with congenital disorders, whereas truncating somatic mutations of all three ASXL family members are observed in human cancers (11,14). Many molecular functions have been predicted for ASXL3 based on its similarity to other ASXL family members, yet few have been confirmed. ASXL1 is the most recognized ASXL family member and functions as an epigenetic scaffold for ubiquitin C-terminal hydrolase BAP1, histone lysine methyltransferase EZH2 and nuclear receptors (15). While ASXL1 exhibits a range of scaffolding interactions, both germline and somatic mutations in ASXL1 have been closely linked to changes in H3K27me3 through its association with EZH2.
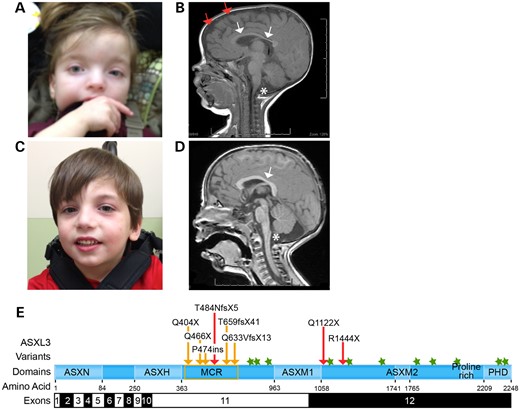
Characterization of pathogenic truncating ASXL3 variants. (A) Facial characteristic of proband I at age 36 months showing low-set ears, broad nasal bridge, sparse arched eyebrows, downslanting palpebral fissures and broad forehead with periorbital fullness. (B) Brain MRI of proband I showing cerebellar vermal hypoplasia (single star), corpus callosum hypoplasia (white arrows), increased subdural space (two red arrows) and decreased white matter volume. (C) Proband II has a small chin, downslanting palpebral fissures and borderline low-set ears. (D) Proband II showing cerebellar vermal hypoplasia (single star) and a shortened corpus callosum (white arrow). (E) Schematic illustration of the main functional units of ASXL3 and pathogenic variants. Novel ASXL3 variants are depicted by red arrows. Previously described truncating ASXL3 variants (orange arrows cluster in the MCR, orange box). Missense ASXL3 variants identified in individuals with Autism spectrum disorder (green stars). ASXL3 functional units are shown with corresponding amino acids. ASXL3 exons 11 and 12 are 1.9 and 3.7 kb in length, respectively, and comprise ∼5.6 kb of the ∼6.8 kb ASXL3 open reading frame.
A series of in vitro assays were important for characterizing the interaction between ASXL1 and BAP1 to form the human Polycomb repressive deubiquitination (PR-DUB) complex. This complex functions to remove the mono-ubiquitin from lysine 119 of histone H2A (H2AK119Ub1), which is primarily ubiquitinated by Polycomb Repressive complex 1 (PRC1) (16). BAP1 is a ubiquitin carboxy-terminal hydrolase that deubiquitinates H2AK119Ub1 as a component of the PR-DUB complex. PR-DUB formation is critical for normal function, as BAP1 alone does not exhibit H2AK119Ub1 deubiquitination activity. However, while BAP1 is a required component of a PR-DUB, ASXL1 may be interchangeable with other ASXL family members to form an enzymatically active complex (17,18). We used primary dermal fibroblasts established from a BRS patient to investigate the molecular defects underlying truncating mutations in ASXL3. De novo truncating transcripts are subject to nonsense-mediated decay, negatively impacting overall ASXL3 expression. We show that ASXL3 interacts with BAP1 forming the key components of the PR-DUB complex. This is an important functional interaction as the level of H2AK119Ub1 is significantly increased in patient fibroblasts, indicating that ASXL3 nonsense mutations disrupt the normal activity of the PR-DUB complex. Transcriptome analysis of proband and control fibroblasts show significant changes in transcriptional regulation that correlate with altered H2AK119Ub1 levels. While germline mutations in PRC1 complex components were previously shown to disrupt H2A mono-ubiquitination in ASD and primary microcephaly (19,20), BRS is the first single gene disorder exhibiting defects in deubiquitination of H2AK119Ub1. These findings highlight a role for dynamic regulation of H2A ubiquitination in development and disease.
Results
Clinical features of novel de novo truncating ASXL3 variants
Clinical exome sequencing (CES) is often performed on probands with a potential genetic disease, but without a clear diagnosis. We describe three cases for which novel de novo heterozygous truncating mutations in ASXL3 were identified by CES. A summary of clinical findings is presented in Table 1. Proband I is a female born by cesarean section at 38 weeks of gestation following a pregnancy complicated by intrauterine growth retardation that was first evident at 18 weeks of gestation. Her birth weight was 2.25 kg (5% for gestational age), and she was 44.4 cm long (<3% for gestational age). Proband I experienced failure to thrive, feeding problems that included a poor suck reflex and delayed gastric emptying that required a G-tube by 8 months of age. When examined at 5 years 6 months of age, her height and weight were 108 cm (24th percentile) and 15.8 kg (4th percentile).
Phenotypic features associated with truncating ASXL3 variants
| ASXL3 variant | c.4330T>C | c.3664C>T | c.1448dupT | c.1210C>T | c.1396C>T | c.1975_1978 delACAG | c.1421insT | c.1897_1898 delCA |
|---|---|---|---|---|---|---|---|---|
| a.a. Position | p.R1444X | p.Q1122X | p.484NfsX5 | p.Q404X | p.Q466X | p.T659fsX41 | p.P474fs | p.Q633VfsX13 |
| Identification | Novel University of Michigan | Bainbridge et al. | Dinwiddie et al. | |||||
| Craniofacial features | ||||||||
| Trigonocephaly | − | − | + | − | − | − | − | + |
| Microcephaly | − | − | − | + | + | − | − | + |
| Prominent forehead | − | − | + | − | − | + | + | NA |
| Arched eyebrows | − | + | + | + | + | − | + | NA |
| Prominent eyes | − | − | − | − | − | − | − | + |
| Astigmatism | − | − | Hyperopia | NA | NA | NA | NA | Myopia |
| Palpebral fissures | Downslanting | − | Downslanting | − | − | − | − | Upslanting |
| Nasal bridge | − | − | Broad | − | − | − | − | Depressed |
| Short/small nose | + | − | + | − | + | + | − | NA |
| Anteverted nares | + | − | − | + | + | + | + | + |
| Posteriorly rotated ears | − | − | − | + | + | − | − | + |
| Low-set ears | − | − | + | − | − | − | + | NA |
| Small chin | + | − | + | NA | NA | NA | NA | NA |
| Neurological features | ||||||||
| Brain MRI | WMa | NA | WMa | NA | NA | NA | WMb | WMc |
| Seizures | − | − | + | − | − | − | − | + |
| ID | + | NA | + | − | − | + | − | + |
| Absence of speech | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + (axial) | − | + | + | − | + (axial) |
| Developmental delay | + | + | + | + | + | + | + | + |
| Other features | ||||||||
| Gastric tube | + | + | + | + | + | − | + | + |
| IUGR | + | − | + | + | + | − | + | + |
| Failure to thrive | + | + | + | + | + | + | + | + |
| Feeding difficulties | + | + | + | + | + | + | + | + |
| Precocious puberty | NA | − | + | NA | NA | NA | NA | NA |
| Recurrent infections | + | − | − | NA | NA | NA | NA | + |
| Ulnar deviation | − | + | − | + | + | − | + | − |
| ASXL3 variant | c.4330T>C | c.3664C>T | c.1448dupT | c.1210C>T | c.1396C>T | c.1975_1978 delACAG | c.1421insT | c.1897_1898 delCA |
|---|---|---|---|---|---|---|---|---|
| a.a. Position | p.R1444X | p.Q1122X | p.484NfsX5 | p.Q404X | p.Q466X | p.T659fsX41 | p.P474fs | p.Q633VfsX13 |
| Identification | Novel University of Michigan | Bainbridge et al. | Dinwiddie et al. | |||||
| Craniofacial features | ||||||||
| Trigonocephaly | − | − | + | − | − | − | − | + |
| Microcephaly | − | − | − | + | + | − | − | + |
| Prominent forehead | − | − | + | − | − | + | + | NA |
| Arched eyebrows | − | + | + | + | + | − | + | NA |
| Prominent eyes | − | − | − | − | − | − | − | + |
| Astigmatism | − | − | Hyperopia | NA | NA | NA | NA | Myopia |
| Palpebral fissures | Downslanting | − | Downslanting | − | − | − | − | Upslanting |
| Nasal bridge | − | − | Broad | − | − | − | − | Depressed |
| Short/small nose | + | − | + | − | + | + | − | NA |
| Anteverted nares | + | − | − | + | + | + | + | + |
| Posteriorly rotated ears | − | − | − | + | + | − | − | + |
| Low-set ears | − | − | + | − | − | − | + | NA |
| Small chin | + | − | + | NA | NA | NA | NA | NA |
| Neurological features | ||||||||
| Brain MRI | WMa | NA | WMa | NA | NA | NA | WMb | WMc |
| Seizures | − | − | + | − | − | − | − | + |
| ID | + | NA | + | − | − | + | − | + |
| Absence of speech | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + (axial) | − | + | + | − | + (axial) |
| Developmental delay | + | + | + | + | + | + | + | + |
| Other features | ||||||||
| Gastric tube | + | + | + | + | + | − | + | + |
| IUGR | + | − | + | + | + | − | + | + |
| Failure to thrive | + | + | + | + | + | + | + | + |
| Feeding difficulties | + | + | + | + | + | + | + | + |
| Precocious puberty | NA | − | + | NA | NA | NA | NA | NA |
| Recurrent infections | + | − | − | NA | NA | NA | NA | + |
| Ulnar deviation | − | + | − | + | + | − | + | − |
NA, not accessed; MRI, magnetic resonance imaging; ID, intellectual disability; IUGR, intrauterine growth restriction; +, present; −, absent/normal.
aGlobal mild white matter volume loss.
bGlobal mild white matter volume loss with normal myelination: secondary brainstem hypoplasia; Hypoplasia/dysplasia of bilateral cerebellar tonsils.
cMild white matter loss with enlarged lateral ventricles and mild prominence of the sulci.
Phenotypic features associated with truncating ASXL3 variants
| ASXL3 variant | c.4330T>C | c.3664C>T | c.1448dupT | c.1210C>T | c.1396C>T | c.1975_1978 delACAG | c.1421insT | c.1897_1898 delCA |
|---|---|---|---|---|---|---|---|---|
| a.a. Position | p.R1444X | p.Q1122X | p.484NfsX5 | p.Q404X | p.Q466X | p.T659fsX41 | p.P474fs | p.Q633VfsX13 |
| Identification | Novel University of Michigan | Bainbridge et al. | Dinwiddie et al. | |||||
| Craniofacial features | ||||||||
| Trigonocephaly | − | − | + | − | − | − | − | + |
| Microcephaly | − | − | − | + | + | − | − | + |
| Prominent forehead | − | − | + | − | − | + | + | NA |
| Arched eyebrows | − | + | + | + | + | − | + | NA |
| Prominent eyes | − | − | − | − | − | − | − | + |
| Astigmatism | − | − | Hyperopia | NA | NA | NA | NA | Myopia |
| Palpebral fissures | Downslanting | − | Downslanting | − | − | − | − | Upslanting |
| Nasal bridge | − | − | Broad | − | − | − | − | Depressed |
| Short/small nose | + | − | + | − | + | + | − | NA |
| Anteverted nares | + | − | − | + | + | + | + | + |
| Posteriorly rotated ears | − | − | − | + | + | − | − | + |
| Low-set ears | − | − | + | − | − | − | + | NA |
| Small chin | + | − | + | NA | NA | NA | NA | NA |
| Neurological features | ||||||||
| Brain MRI | WMa | NA | WMa | NA | NA | NA | WMb | WMc |
| Seizures | − | − | + | − | − | − | − | + |
| ID | + | NA | + | − | − | + | − | + |
| Absence of speech | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + (axial) | − | + | + | − | + (axial) |
| Developmental delay | + | + | + | + | + | + | + | + |
| Other features | ||||||||
| Gastric tube | + | + | + | + | + | − | + | + |
| IUGR | + | − | + | + | + | − | + | + |
| Failure to thrive | + | + | + | + | + | + | + | + |
| Feeding difficulties | + | + | + | + | + | + | + | + |
| Precocious puberty | NA | − | + | NA | NA | NA | NA | NA |
| Recurrent infections | + | − | − | NA | NA | NA | NA | + |
| Ulnar deviation | − | + | − | + | + | − | + | − |
| ASXL3 variant | c.4330T>C | c.3664C>T | c.1448dupT | c.1210C>T | c.1396C>T | c.1975_1978 delACAG | c.1421insT | c.1897_1898 delCA |
|---|---|---|---|---|---|---|---|---|
| a.a. Position | p.R1444X | p.Q1122X | p.484NfsX5 | p.Q404X | p.Q466X | p.T659fsX41 | p.P474fs | p.Q633VfsX13 |
| Identification | Novel University of Michigan | Bainbridge et al. | Dinwiddie et al. | |||||
| Craniofacial features | ||||||||
| Trigonocephaly | − | − | + | − | − | − | − | + |
| Microcephaly | − | − | − | + | + | − | − | + |
| Prominent forehead | − | − | + | − | − | + | + | NA |
| Arched eyebrows | − | + | + | + | + | − | + | NA |
| Prominent eyes | − | − | − | − | − | − | − | + |
| Astigmatism | − | − | Hyperopia | NA | NA | NA | NA | Myopia |
| Palpebral fissures | Downslanting | − | Downslanting | − | − | − | − | Upslanting |
| Nasal bridge | − | − | Broad | − | − | − | − | Depressed |
| Short/small nose | + | − | + | − | + | + | − | NA |
| Anteverted nares | + | − | − | + | + | + | + | + |
| Posteriorly rotated ears | − | − | − | + | + | − | − | + |
| Low-set ears | − | − | + | − | − | − | + | NA |
| Small chin | + | − | + | NA | NA | NA | NA | NA |
| Neurological features | ||||||||
| Brain MRI | WMa | NA | WMa | NA | NA | NA | WMb | WMc |
| Seizures | − | − | + | − | − | − | − | + |
| ID | + | NA | + | − | − | + | − | + |
| Absence of speech | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + (axial) | − | + | + | − | + (axial) |
| Developmental delay | + | + | + | + | + | + | + | + |
| Other features | ||||||||
| Gastric tube | + | + | + | + | + | − | + | + |
| IUGR | + | − | + | + | + | − | + | + |
| Failure to thrive | + | + | + | + | + | + | + | + |
| Feeding difficulties | + | + | + | + | + | + | + | + |
| Precocious puberty | NA | − | + | NA | NA | NA | NA | NA |
| Recurrent infections | + | − | − | NA | NA | NA | NA | + |
| Ulnar deviation | − | + | − | + | + | − | + | − |
NA, not accessed; MRI, magnetic resonance imaging; ID, intellectual disability; IUGR, intrauterine growth restriction; +, present; −, absent/normal.
aGlobal mild white matter volume loss.
bGlobal mild white matter volume loss with normal myelination: secondary brainstem hypoplasia; Hypoplasia/dysplasia of bilateral cerebellar tonsils.
cMild white matter loss with enlarged lateral ventricles and mild prominence of the sulci.
Proband I presented with dysmorphic facial features that included square facies, downslanting palpebral fissures, low-set ears, broad nasal bridge, small short nose and micrognathia (Fig. 1A). Proband I experienced hyperopia and left-sided esotropia that was successfully repaired. Hypotonia was noted at the neonatal neurological examination. She has global developmental delay and at 6 years of age was unable to sit, crawl or stand independently. Proband I does not communicate verbally and presents with ID. A brain MRI performed at 7 months of age showed a reduction in white matter and cerebral volume together with an increased subdural space most predominantly surrounding the frontal lobes (Fig. 1B). Structurally, thinning of the corpus callosum and hypoplasia of the inferior cerebellar vermis were also observed. Unlike other cases of BRS, Proband I experienced staring spells starting at 8 months that resolved by Age 2 and were not accompanied by any electroencephalogram (EEG) changes. Her blood lactate, plasma amino acids and urine organic acids were normal.
Proband II is a male born at 39 weeks by vaginal delivery following a pregnancy complicated by reduced fetal movement. At birth, he weighed 3.4 kg (25th percentile) and was 50.8 cm long (50th percentile). Reduced tone was noted immediately following vaginal delivery. Proband II has a history of failure to thrive and feeding difficulties. Prominent gastrointestinal symptoms included reflux, chronic constipation and oral aversion that required G-tube feedings. Metabolically, his blood lactate, plasma amino acids and urine organic acids were also normal. Proband II presented with minor dysmorphic features, which included a small nose with anteverted nares, a small chin and downslanting palpebral fissures (Fig. 1C).
Neurological exams throughout noted hypotonia and severe delays in meeting developmental milestones. Proband II began pulling to standing at 16 months, but lacks sufficient coordination to walk independently at 4 years of age. He has ID and often acts aggressively. He can mimic a few words but does not use them for communication. The cerebral cortex was structurally normal by MRI, while vermal hypoplasia of the cerebellum and a shortened corpus callosum were noted (Fig. 1D). Proband II experienced recurrent ear infections and had a tonsillectomy. He also experiences joint swelling accompanied by episodes of hand and foot discoloration that are attributed to acrocyanosis.
Proband III is a male who was born at 40 weeks via cesarean section. His birth weight was 3.1 kg (50th percentile) and length 49.53 cm (25th percentile). Proband III has feeding difficulties that have not resolved by 13 months. He was found to have ankyloglossia and had a frenulotomy. Initial concerns about his development started at 7 months of age when he failed to meet gross-motor milestones. At 13 months, he was unable to roll over and sit upright. His neurological exam was remarkable for hypotonia. No dysmorphic features were observed.
All three probands received a detailed clinical evaluation and had a normal oligonucleotide chromosomal microarray before CES was ordered at GeneDx or Ambry Genetics. Trio-CES (proband, mother and father) was performed at GeneDx for Proband I and identified a de novo ASXL3 (NM_030632) c.1448dupT frameshift insertion that creates a premature stop codon 5 amino acids (p.T484NfsX5) downstream of the insertion, in exon 11 of ASXL3 (Fig. 1E). This truncating variant creates a premature stop in the same region of ASXL3 as the five previously described de novo truncating and nonsense ASXL3 variants, supporting a genetic diagnosis of BRS for this proband (2,4).
CES of Proband II and his mother identified a heterozygous ASXL3 c.4330T>C, p.R1444X nonsense mutation in Proband II that was not present in his mother (Fig. 1E). The father was not available for testing. Trio-CES, performed at Ambry Genetics for Proband III, discovered a de novo heterozygous ASXL3 c.3364C>T, p.Q1122X nonsense variant (Fig. 1E). Both of these ASXL3 variants fall outside the original cluster of mutations previously described in exon 11 and instead localize to the 5′ end of exon 12, expanding the clinical significance of nonsense variants throughout ASXL3 in BRS. Mosaicism was not investigated to determine the timing of these de novo events.
Truncating ASXL3 variants result in loss-of-function alleles
The N-terminal ASXN and ASXH domains of ASXL3 are predicted to act as a scaffold to mediate interactions with protein complexes (11,12,17,21,22). In contrast, the C-terminal ASXM2 domain and PHD finger recognize chromatin histone post-translational modifications (PTMs) (12,23,24). The first pathogenic variants identified in ASXL3 clustered between these two C- and N-terminal functional components, raising the possibility that stably expressed truncated proteins may uncouple the ASXL3 scaffolding interactions and chromatin reader activities, allowing the truncated protein to act as a dominant negative (2). To investigate this scenario, a primary dermal fibroblast line was established from Proband I, who is heterozygous for a frameshift variant within the mutational cluster region of ASXL3 (ASXL3+/fs fibroblasts; Fig. 1E). A C-terminally V5/His tagged full-length ASXL3 (fl-ASXL3-V5/His) and truncated ASXL3 (tr-ASXL3-V5/His) fragment potentially expressed from the ASXL3 c.1448dupT allele were also generated (Fig. 2A). A total of 293 T overexpressed tr-ASXL3-V5/His was analyzed concurrently with whole cell lysate from ASXL3+/+ and ASXL3+/fs fibroblasts. The α-ASXL3 antibody recognizes tr-ASXL3-V5/His. However, a similarly sized band was not detected in either ASXL3+/+ or ASXL3+/fs fibroblasts, indicating the ASXL3 c.1448dupT variant does not express a stable protein (Fig. 2D). To evaluate the impact of this frameshift mutation on expression of full-length ASXL3, the 280 KDa band, corresponding to the fl-ASXL3-V5/His band, was quantified from ASXL3+/+ and ASXL3+/fs fibroblast whole cell lysates (Fig. 2B). Expression of endogenous ASXL3 in ASXL3+/fs fibroblasts was 50% of that quantified in control fibroblasts after normalization to β-actin (Fig. 2C), reflecting the sensitivity of BRS pathology to reduced dosage of ASXL3.
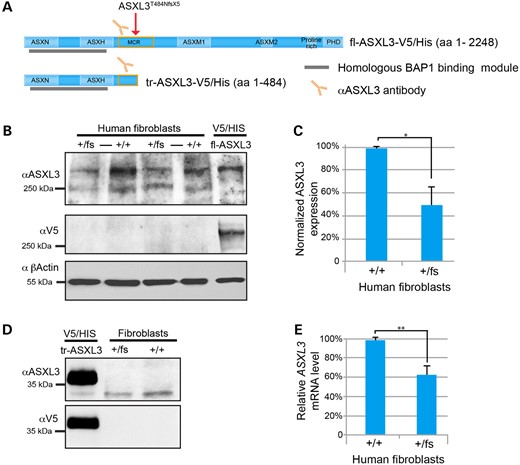
Characterization of truncating ASXL3 variants in patient fibroblasts. (A) Schematic of ASXL3 C-terminally V5/His tagged constructs and length in amino acids (aa), the region of ASXL3 recognized by the α-ASXL3 antibody (orange antibody), ASXL3 N-terminus that shares 89% homology with the BAP1-binding region of ASXL1 and 2 (gray bar), proband I truncating variant (red arrow). (B) Western blot of two biological replicates comparing endogenous full-length ASXL3 expression in patient ASXL3+/fs and control ASXL3+/+ fibroblasts (+/fs to +/+). Overexpressed fl-ASXL3-V5/His runs at ∼280 kDa, corresponding to the highest molecular weight band from fibroblast cell lysates recognized by the α-ASXL3 antibody. fl-ASXL3-V5/His is recognized by α-ASXL3 and α-V5 antibodies. β-Actin was used as a loading control. (C) Quantification of endogenous full-length ASXL3 protein in ASXL3+/fs and control ASXL3+/+ fibroblasts relative to expression of β-actin, n = 3 (Error bars denote SD; *P = 0.04). (D) Overexpressed tr-ASXL3-V5/His is recognized by α-ASXL3 and α-V5 antibodies. A band of comparable molecular weight is not detected in ASXL3+/fs and control ASXL3+/+ cell lysate. (E) qRT–PCR analysis of ASXL3 transcripts from ASXL3+/fs and control ASXL3+/+ fibroblasts cDNA relative to GAPDH, n = 3 (Error bars denote SD; **P = 0.005).
De novo truncating ASXL3 variants are predicted to promote nonsense-mediated decay (NMD). To investigate the dynamics of mRNA stability, mutant ASXL3 mRNA transcripts were quantified from control and ASXL3+/fs fibroblast cDNA. Quantitative real-time polymerase chain reaction (qRT–PCR) of ASXL3 and GAPDH were performed in triplicate. Normalized ASXL3 transcripts from patient cDNA were 61% of the transcripts detected in controls (Fig. 2E), suggesting the c.1448dupT transcripts undergo NMD. To directly quantify the ratio of ASXL3 reference to c.1448dupT transcripts, an amplicon spanning the pathogenic variant was sequenced. The ASXL3 c.1448dupT genotype was detected in 12.8% of a total 112 000 reads covering the amplicon generated from ASXL3+/fs fibroblast cDNA. The ASXL3 c.1448dupT transcript was not detected in 127 000 reads covering the amplicon generated from ASXL3+/+ fibroblast cDNA. These results suggest that the ASXL3 c.1448dupT transcript is sensitive to NMD and contributes to the reduced ASXL3 transcript level and protein expression in Proband I fibroblasts (Fig. 2D).
Truncating ASXL3 mutations alter H2A ubiquitination
ASXL1 and ASXL2 possess a conserved N-terminal BAP1-binding site, with ASXL3 exhibiting 89% homology across these same amino acids (11). The BAP1-binding site is 5′ of the ASXL3 p.T484NfsX5 variant and covered by the tr-ASXL3-V5/His ASXL3 fragment. To determine whether ASXL3 also interacts with BAP1, flag-tagged BAP1 (BAP1-Flag) and tr-ASXL3-V5/His were overexpressed and co-immunoprepitpated (co-IP) from 293 T cells (Fig. 3A). When co-overexpressed with BAP1-Flag, a fraction of tr-ASXL3-V5/His is post-translationally modified, generating a larger band by western blot not observed when tr-ASXL3-V5/His is overexpressed alone. The PTM shifts the tr-ASXL3-V5/His band size in excess of what would be predicted for phosphorylation alone and was confirmed following incubation of co-overexpressed cell lysates with calf intestinal phosphatase (CIP) (Supplementary Material, Fig. S1). Reciprocal co-IP was performed with both α-Flag and α-V5 antibodies. As shown in Figure 3A, the two proteins interact when co-immunoprecipitated with either antibody; however, BAP1-Flag preferentially interacts with the post-translationally modified tr-ASXL3-V5/His. This indicates that ASXL3 interacts with BAP1, making up the components of the PR-DUB complex. In addition to phosphorylation, ASXL3 can be poly-ubiquitinated at K350, which may account for the additional tr-ASXL3-V5/His band shifted ∼20 kDa larger when co-expressed with BAP1-flag (PhosphoSite.org). The specific PTM that contributes to this interaction will be investigated in future experiments.
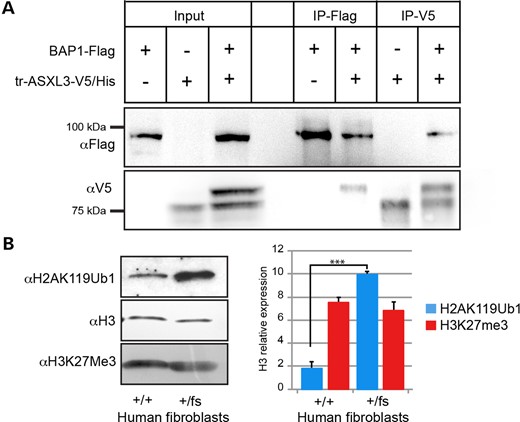
ASXL3 interacts with BAP1 and truncating mutations alter histone H2A ubiquitination. (A) BAP1-Flag and tr-ASXL3-V5/His were individually and co-overexpressed in 293 T. The N-terminal fragment of ASXL3 is post-transcriptionally modified when co-overexpressed together with BAP1-Flag and is seen as a doublet in the corresponding lane. Reciprocal co-immunoprecipitations were performed with α-V5 and α-flag antibodies. BAP1-Flag preferentially binds the post-transcriptionally modified tr-ASXL3-V5/His fragment. (B) H2AK119Ub1 and H3K27Me3 acid extracted histones levels compared between ASXL3+/+ and ASXL3+/fs fibroblasts. H3 used as loading control. A 5-fold increase in H2AK119Ub1 was quantified in ASXL3+/fs fibroblasts compared with H3 levels. No change detected in normalized H3K27Me3 acid extracted histone levels between ASXL3+/+ and ASXL3+/fs fibroblasts, n = 3 (Error bars denote SD; ***P = 0.001).
PcG proteins form multi-subunit, chromatin-modifying complexes that maintain transcriptional repression in multicellular organisms. While the molecular functions of Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) have been subject to intense scrutiny, the role of PR-DUB is less well described (16). PRC2 is well known for its role in histone H3 trimethlyation of lysine 27 (H3K27me3). PRC1 is responsible for the majority of H2AK119Ub1 across the genome, while PR-DUB removes this histone PTM (16). Loss of Ring1b, an E3 ubiquitin ligase associated with PRC1, leads to reduced H2AK119Ub1, release of the paused polymerase and gene de-repression (25–27). To investigate the impact of ASXL3 mutations on the global level of H2A mono-ubiquitination, we measured the H2AK119Ub1 level of acid extracted histones from control and ASXL3+/fs fibroblasts. Genome-wide H2AK119Ub1 level in ASXL3+/fs fibroblasts was elevated 5-fold above control cells when normalized to histone H3 (Fig. 3B). ASXL1 loss-of-function mutations have been shown to also alter the levels of H3K27me3 (28,29); however, the level of H3K27me3 was unchanged in ASXL3+/fs fibroblasts. These studies suggest the ASXL3-specific PR-DUB complex may confer distinct molecular functions independent of other ASXL family members.
Transcriptome altered in ASXL3+/fs skin fibroblasts
To analyze the impact of elevated H2AK119Ub1 on transcriptional regulation in ASXL3+/fs fibroblasts, we conducted a genome-wide analysis of mRNA levels for three biological replicates of ASXL3+/fs and ASXL3+/+ fibroblast lines. Highly reproducible changes in the transcriptome were observed in ASXL3+/fs cells with 564 genes showing significant changes in transcription (false discovery rate adjusted P-values defined by DEseq2, q < 0.05 and log2 fold change in expression of ≥1). Despite the genome-wide increase in H2AK119Ub1 and its potential to repress transcription overall, the transcriptional levels observed in fibroblasts from both genotypes were highly correlated (Pearson correlation coefficient r2—0.96), indicating that global transcriptional activity is not altered in ASXL3+/fs fibroblasts. Of the 564 differentially expressed genes (DEGs) in ASXL3+/fs fibroblasts, 48% (254) were significantly downregulated and 52% (310) significantly up-regulated (Fig. 4A). Five percent of the transcripts were non-coding RNA that were distributed between up- and downregulated transcripts. Fifteen of the significant DEGs were confirmed by qRT–PCR from both proband and control fibroblasts and normalized to GAPDH (Supplementary Material, Fig. S2). The comparable number of up- and downregulated genes suggests that the function of ASXL3 is required to both silence and activate genes in a cellular-dependent context, a function of this gene family that has been conserved across evolution (8–10,30).
![Transcriptome analysis of ASXL3+/fsfibroblasts. (A) Percentage of differentially up- and down- regulated transcripts in ASXL3+/fs fibroblasts. Five percent of differentially expressed transcripts were identified as non-coding RNA. (B) Volcano plot: all 564 significant DEGs (green dots) are represented in terms of their measured expression change (log2 fold change) and the significance of their change [−log10 (P-value)]. The significance is represented in terms of the negative log of the P-value. (C) Red circles are KEGG pathway with significant normalized enrichment scores according to the false discovery rate P-value (FDR P-value) corrected for multiple hypothesis testing. The impact of each pathway is plotted relative to the number of DEGs enriched in each pathway (p-Acc) and the perturbation (p-ORA) of the pathway based on the measured expression changes across the pathway topology. Black dots represent KEGG pathways that do not reach significance according to FDR P-value calculated. (D) Significantly enriched KEGG pathways and their FDR P-value.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/25/3/10.1093_hmg_ddv499/3/m_ddv49904.jpeg?Expires=1750441215&Signature=kmp0yyRFlA5jWGa6Sg3QnkV11ssb3QKII4oSplSCDVpOyjHSyiI6kMzDsOWJW3I~myYIs9M8DG0BA45BdKMD1g4jizDbw5AiCWa9jfG40kMRQE~NDbI42LLTNMlj0khMVBzVAvk3r6LNuFi5TEwV-FoLujJyO2JXVnjrDH71Quo7z5HPwwI8s3efIgUZl0bcYfs2qLNZlsfgVb9ue98owmU4OpyIA1MytggmP5EJ2i13M7McrWye-DtSbGS635TfNFNtMcDfdDYwXgwIWITow7wG3YFJZnew8QkBX-pbrrEnuEsYlHyw2M89YMn4xWpOFV-CiItPToFpUh48Ac7FGg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transcriptome analysis of ASXL3+/fsfibroblasts. (A) Percentage of differentially up- and down- regulated transcripts in ASXL3+/fs fibroblasts. Five percent of differentially expressed transcripts were identified as non-coding RNA. (B) Volcano plot: all 564 significant DEGs (green dots) are represented in terms of their measured expression change (log2 fold change) and the significance of their change [−log10 (P-value)]. The significance is represented in terms of the negative log of the P-value. (C) Red circles are KEGG pathway with significant normalized enrichment scores according to the false discovery rate P-value (FDR P-value) corrected for multiple hypothesis testing. The impact of each pathway is plotted relative to the number of DEGs enriched in each pathway (p-Acc) and the perturbation (p-ORA) of the pathway based on the measured expression changes across the pathway topology. Black dots represent KEGG pathways that do not reach significance according to FDR P-value calculated. (D) Significantly enriched KEGG pathways and their FDR P-value.
In ASXL3+/fs fibroblasts, HOX transcripts were highly repressed. Differentially expressed HOX transcripts were specific to contiguous HOX genes, the HOXA and HOXC clusters, and included the intervening long non-coding RNA (lncRNA) at both loci. On Chromosome 7, HOXA7, HOXA10, HOXA11 and HOXA13 are repressed, as well as the HOXA10-AS and HOXA11-AS lncRNA that reside in the intervening sequence at that chromosomal locus. A similar configuration is observed on chromosome 12 where HOXC10, HOXC11, HOTAIR, HOXC-AS1, HOXC-AS2 and HOXC-AS3 are all significantly repressed. The top up-regulated genes depicted on the volcano plot were components of the extracellular matrix (Fig. 4B).
To better understand the functional impact of transcriptional changes occurring, we performed enrichment analysis of Gene Ontology (GO) categories on pooled proband I and control fibroblast transcriptomes (31–33). DEGs from our dataset were enriched in GO terms that underscore the role of ASXL3 in transcriptional regulation, development and proliferation, i.e. positive regulation of transcription from RNA polymerase II promoter, endoderm differentiation, proximal/distal limb development, early digestive track development, negative regulation of astrocyte differentiation, growth factor activity and negative regulation of cell proliferation. The top GO terms from this analysis are specified in Table 2. These findings are consistent with the features of failure to thrive, global developmental delay, reduced brain volume, ID, dysmorphic features and feeding problems observed in BRS. It will be important to determine whether DEGs enriched in the GO terms ‘extracellular matrix organization’ and ‘integral components of plasma membrane’ contribute to the clinical features of BRS.
Gene ontology categories enriched with BRS fibroblast DEGs
| GO accession | Name | P-value (elim pruninga) |
|---|---|---|
| Biological process | ||
| GO:0030198 | extracellular matrix organization | 2.400e−7 |
| GO:0045944 | positive regulation of transcription from RNA polymerase II promoter | 2.00e−5 |
| GO:0009954 | proximal/distal pattern formation | 3.500e−5 |
| GO:0048566 | embryonic digestive tract development | 6.300e−5 |
| GO:0008285 | negative regulation of cell proliferation | 7.600e−5 |
| GO:0070836 | caveola assembly | 7.900e−5 |
| GO:0001706 | endoderm formation | 1.000e−4 |
| GO:0048712 | negative regulation of astrocyte differentiation | 1.100e−4 |
| Cellular component | ||
| GO:0005887 | integral component of plasma membrane | 6.000e−13 |
| GO:0005615 | extracellular space | 2.100e−9 |
| GO:0005578 | proteinaceous extracellular matrix | 1.600e−8 |
| GO:0001725 | stress fiber | 7.700e−6 |
| Molecular function | ||
| GO:0008083 | growth factor activity | 1.300e−6 |
| GO:0008201 | heparin binding | 2.800e−5 |
| GO:0043565 | sequence-specific DNA binding | 4.400e−5 |
| GO:0001077 | transcriptional activator activity, RNA polymerase II core | 1.900e−4 |
| promoter proximal region sequence-specific binding | ||
| GO:0008330 | protein tyrosine/threonine phosphatase activity | 2.800e−4 |
| GO:0042803 | protein homodimerization activity | 4.800e−4 |
| GO accession | Name | P-value (elim pruninga) |
|---|---|---|
| Biological process | ||
| GO:0030198 | extracellular matrix organization | 2.400e−7 |
| GO:0045944 | positive regulation of transcription from RNA polymerase II promoter | 2.00e−5 |
| GO:0009954 | proximal/distal pattern formation | 3.500e−5 |
| GO:0048566 | embryonic digestive tract development | 6.300e−5 |
| GO:0008285 | negative regulation of cell proliferation | 7.600e−5 |
| GO:0070836 | caveola assembly | 7.900e−5 |
| GO:0001706 | endoderm formation | 1.000e−4 |
| GO:0048712 | negative regulation of astrocyte differentiation | 1.100e−4 |
| Cellular component | ||
| GO:0005887 | integral component of plasma membrane | 6.000e−13 |
| GO:0005615 | extracellular space | 2.100e−9 |
| GO:0005578 | proteinaceous extracellular matrix | 1.600e−8 |
| GO:0001725 | stress fiber | 7.700e−6 |
| Molecular function | ||
| GO:0008083 | growth factor activity | 1.300e−6 |
| GO:0008201 | heparin binding | 2.800e−5 |
| GO:0043565 | sequence-specific DNA binding | 4.400e−5 |
| GO:0001077 | transcriptional activator activity, RNA polymerase II core | 1.900e−4 |
| promoter proximal region sequence-specific binding | ||
| GO:0008330 | protein tyrosine/threonine phosphatase activity | 2.800e−4 |
| GO:0042803 | protein homodimerization activity | 4.800e−4 |
aFor each Gene Ontology (GO) term (31,32), the number of DEG annotated to the term is compared with the number of genes expected just by chance. Given the nature of the gene ontology, this introduces errors by considering genes multiple times. To overcome this limitation, the elim method was used to iteratively remove genes that map to significantly enriched GO term (33).
Gene ontology categories enriched with BRS fibroblast DEGs
| GO accession | Name | P-value (elim pruninga) |
|---|---|---|
| Biological process | ||
| GO:0030198 | extracellular matrix organization | 2.400e−7 |
| GO:0045944 | positive regulation of transcription from RNA polymerase II promoter | 2.00e−5 |
| GO:0009954 | proximal/distal pattern formation | 3.500e−5 |
| GO:0048566 | embryonic digestive tract development | 6.300e−5 |
| GO:0008285 | negative regulation of cell proliferation | 7.600e−5 |
| GO:0070836 | caveola assembly | 7.900e−5 |
| GO:0001706 | endoderm formation | 1.000e−4 |
| GO:0048712 | negative regulation of astrocyte differentiation | 1.100e−4 |
| Cellular component | ||
| GO:0005887 | integral component of plasma membrane | 6.000e−13 |
| GO:0005615 | extracellular space | 2.100e−9 |
| GO:0005578 | proteinaceous extracellular matrix | 1.600e−8 |
| GO:0001725 | stress fiber | 7.700e−6 |
| Molecular function | ||
| GO:0008083 | growth factor activity | 1.300e−6 |
| GO:0008201 | heparin binding | 2.800e−5 |
| GO:0043565 | sequence-specific DNA binding | 4.400e−5 |
| GO:0001077 | transcriptional activator activity, RNA polymerase II core | 1.900e−4 |
| promoter proximal region sequence-specific binding | ||
| GO:0008330 | protein tyrosine/threonine phosphatase activity | 2.800e−4 |
| GO:0042803 | protein homodimerization activity | 4.800e−4 |
| GO accession | Name | P-value (elim pruninga) |
|---|---|---|
| Biological process | ||
| GO:0030198 | extracellular matrix organization | 2.400e−7 |
| GO:0045944 | positive regulation of transcription from RNA polymerase II promoter | 2.00e−5 |
| GO:0009954 | proximal/distal pattern formation | 3.500e−5 |
| GO:0048566 | embryonic digestive tract development | 6.300e−5 |
| GO:0008285 | negative regulation of cell proliferation | 7.600e−5 |
| GO:0070836 | caveola assembly | 7.900e−5 |
| GO:0001706 | endoderm formation | 1.000e−4 |
| GO:0048712 | negative regulation of astrocyte differentiation | 1.100e−4 |
| Cellular component | ||
| GO:0005887 | integral component of plasma membrane | 6.000e−13 |
| GO:0005615 | extracellular space | 2.100e−9 |
| GO:0005578 | proteinaceous extracellular matrix | 1.600e−8 |
| GO:0001725 | stress fiber | 7.700e−6 |
| Molecular function | ||
| GO:0008083 | growth factor activity | 1.300e−6 |
| GO:0008201 | heparin binding | 2.800e−5 |
| GO:0043565 | sequence-specific DNA binding | 4.400e−5 |
| GO:0001077 | transcriptional activator activity, RNA polymerase II core | 1.900e−4 |
| promoter proximal region sequence-specific binding | ||
| GO:0008330 | protein tyrosine/threonine phosphatase activity | 2.800e−4 |
| GO:0042803 | protein homodimerization activity | 4.800e−4 |
aFor each Gene Ontology (GO) term (31,32), the number of DEG annotated to the term is compared with the number of genes expected just by chance. Given the nature of the gene ontology, this introduces errors by considering genes multiple times. To overcome this limitation, the elim method was used to iteratively remove genes that map to significantly enriched GO term (33).
The molecular pathways impacted by DEGs in BRS are poorly understood. To explore the disrupted molecular pathways that can be inferred from our data, we performed impact analysis with iPathway tools to identify interactions among DEGs that predict significantly affected Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (34–39). This analysis quantifies two independent probabilities: (i) over-representation of DEGs in a given pathway (P-Acc) and (ii) perturbation of a pathway computed by propagating the measured expression changes across the pathway topology (P-ORA). Five KEGG pathways were found to be significantly impacted: (i) Cell adhesion molecules, (ii) Neuroactive ligand-receptor interaction, (iii) Axon guidance, (iv) Rheumatoid arthritis and (v) Transcription misregulation in cancer (Fig. 4D). DEGs were both enriched and highly dysregulated in the ‘Rheumatoid arthritis’, ‘Axon guidance’ and ‘Neuroactive ligand-receptor interaction’ pathways, compared with the ‘Transcriptional misregulation in cancer’, and ‘Cell adhesion molecules’ pathways were specified by the enrichment of DEGs alone. The DEGs in each KEGG pathway are outlined in Supplementary Material, Figure S3. Understanding the gene expression signatures and their impact on molecular pathways will provide a better understanding of key gene targets responsible for features of BRS.
Discussion
Here we describe three novel variants in ASXL3 that contribute to our understanding of the clinical spectrum and molecular impact of truncating mutations in BRS. The three probands we describe fit within the phenotypic spectrum previously described for BRS. These data suggest that the phenotypic variability is not directly correlated with the placement of the truncating mutations in ASXL3, as two of the variants we describe do not fall within the original cluster of mutations. The distinct phenotypic features observed in individual BRS probands are outlined in Table 1; however, the most pervasive characteristics are non-specific features. Most prominent among them are feeding difficulties that are present from birth and often require intervention. Psychomotor delays are observed, starting with the earliest milestones. Several probands were small at birth and failed to thrive. Likewise, the degree of ID is variable; however, all are non-verbal or experience significant communication difficulties.
The genetic variation originally published for ASXL3 suggested that pathogenic truncating variants in ASLX3 might function in a dominant-negative manner. First, the original mutations clustered in the penultimate exon of ASXL3 (2,4), bisecting the N-terminal protein scaffolding functions of the gene from the C-terminal chromatin/DNA-targeting activities (12). Second, nonsense mutations both 5′ and 3′ of the mutational cluster region (MCR) are observed in databases composed of sequence variants from phenotypically normal individuals (2). Our data show that the ASXL3 c.1448dupT variant is subject to NMD (Fig 2D) and not expressed in ASXL3+/fs fibroblasts consistent with loss-of-function mutations (Fig 2B). While these data argue against a dominant-negative genetic mechanism, these findings do not address the ASXL3 nonsense mutations that populate sequence databases composed of phenotypically normal individuals. In these cases, ASXL3 nonsense mutations may represent mutations arising post-zygotically or during later embryogenesis and thus may be explained by the timing of these de novo mutational events and mosaicism (40,41). This explanation is supported by the high incidence of somatic mutations in ASXL genes observed in various types of cancers (14,42–52). The timing of de novo mutational events during development may also account for some of the phenotypic variability exhibited between BRS patients with similar ASXL3 nonsense variants.
While ASXL3 and ASXL1 share homology that allows both proteins to interact with similar binding partners, the divergent genetics and molecular pathology corresponding to their respective germline and somatic mutations suggest distinct molecular mechanisms. In ASXL3 genetics, somatic mutations are observed primarily in melanoma, and only ASXL3 missense have has been observed in individuals with ASD. These genetic findings also link ASXL3 to other encoded chromatin remodelers, namely KDM5B, KMT2C, KMT2H and NSD2 (3). Molecularly, our data revealed elevated H2AK119Ub1 in ASXL3+/fs primary fibroblasts, while changes in the H3K27Me3 were unchanged (Fig 3B). In contrast, ASXL1, ASXL2, DNMT3A, EZH2, IDH1, IDH2, KMT2A and TET2 genes are altered in myelodysplastic syndrome and acute myeloid leukemia (14,42,43,53–56), while ASXL3 mutations are conspicuously lacking (57). Likewise, changes in H3K27Me3 are the molecular signature attributed to germline and somatic mutations in ASXL1. Consequently, despite some overlapping clinical features, BRS may be molecularly distinct from Bohring-Opitz syndrome (OMIM 605039) caused by de novo mutations in ASXL1.
Few genes that impact the dynamic regulation of H2A mono-ubiquitination have been identified in human genetic studies. Human variants have been observed in autism susceptibility candidate 2 (AUTS2) and Polyhomeotic-like 1 (PHC1), which encode non-canonical and canonical PRC1 components, respectively, and disrupt mono-ubiquitination of H2A (19). Copy number variants of AUTS2 have been observed in individuals with ID (OMIM 615834) (58,59), while homozygous missense mutations in PHC1 were identified in a consanguineous family with primary microcephaly (OMIM 615414) (20). Likewise, reduced H2AK119ub1 levels have also been described in Down syndrome (OMIM 190685) due to increased dosage of Ubiquitin-specific protease 16 (USP16), which encodes ubiquitin carboxyl-terminal hydrolase 16. Reduced H2AK119ub1 was correlated to decreased proliferation of normal fibroblasts and postnatal neural progenitors and accelerated senescence (60). Conversely, Usp16 null mouse embryonic stem cells exhibit delayed lineage commitment and persistent self-renewing proliferation (61). These findings accentuate the sensitivity of growth and developmental mechanisms to the dosage of genes that impact H2A mono-ubiquitination regulatory mechanisms. Truncating mutations in ASXL3 are the first to link the pathology of a single gene disorder to elevated levels of H2AK119Ub1. This finding also illuminates an important role for dynamic regulation of H2A mono-ubiquitination in development, that when disrupted can result in features of Autism and ID.
Consistent with the growth deficits observed in BRS, many DEGs identified from transcriptome analysis of ASXL3+/fs and ASXL3+/+ fibroblasts were enriched in GO terms defined by proliferation and growth. Cdkn2a is a well-characterized PRC1 target locus in fibroblasts, which encodes p16Ink4a and p19Arf, tumor suppressors involved in senescence-induced loss of proliferation (62–65). The Cdkn2a locus is also sensitive to changes in H2AK119KUb1 (60,66); however, it is not differentially expressed in ASXL3+/fs fibroblasts. Instead, ASXL3+/fs DEGs were enriched in the GO terms ‘negative regulation of cell proliferation’ (GO:0008285; Table 2) and ‘growth factor activity’ (GO:0008083), which may represent alternative mechanisms that contribute to the reduced growth phenotypes in BRS. Likewise, seven DEGs were significantly enriched in the GO term for embryonic digestive tract development (GO:0048566; PCSK5, RARRES2, ALDH1A2, PKDCC, FOXF2, ADA and GLI2), which is pertinent given the feeding difficulties observed in BRS patients.
A number of repressed HOX genes were enriched in the proximal/distal pattern formation GO term (GO:0009954), and differential regulation of HOX genes may contribute to the ulnar deviation phenotype that is observed in 4 of the 8 described BRS patients. However, the role of ASXL3 in transcriptional regulation of HOX genes during development has yet to be determined. Human genetics has revealed the clinical manifestations of germline pathogenic variants in HOXA11 and HOXA13, but few phenotypic features overlap with BRS. Dominant truncating variants in HOXA11 lead to radioulnar synostosis with amegakaryocytic thrombocytopenia (OMIM 605432), while dominant truncating and missense variants in HOXA13 are associated with hand-foot-genital syndrome (OMIM 140000) or Guttmacher syndrome (OMIM 176305), respectively (67). These disorders highlight the digit and urogenital malformations that are often associated with HOXA mutations and suggest that heterozygous truncating variants in ASXL3 may not be critical for spatial and temporal developmental repression of HOXA genes required to generate these phenotypes.
KEGG analysis highlights the combinations of DEGs that may contribute to BRS pathology. Our pathway analysis identified several KEGG pathways consistent with BRS features. Additional experimental work will need to be performed to determine whether DEGs enriched in the ‘Axon guidance’ pathway contribute to the thin corpus callosum, observed in Proband I. This approach could provide insights concerning the best treatment options to pursue (Supplementary Material, Fig. S3). Given the role of ASXL family members in cancer, enrichment of DEGs in the ‘Transcriptional misregulation in cancer’ pathway is well supported by the published literature. The added cancer risk conferred by germline mutations in ASXL3 has not been determined and will need to be assessed for children and adults carrying mutations in this gene.
In conclusion, BRS patient primary fibroblasts were used to investigate the molecular defects associated with truncating variants in ASXL3. Our data highlight an important role for ASXL3 in specifying H2AK119Ub1 levels, a molecular feature sensitive to the dosage of genes that impact H2A mono-ubiquitination. These findings will aid in the future development of appropriate models to investigate the role of this chromatin modification in BRS.
Materials and Methods
Establishment primary skin fibroblast line
Punch skin biopsies were taken from the upper back for proband I and controls at the University of Michigan. All procedures followed were in accordance with the ethical standards of the University of Michigan Medical School IRB, and an informed consent was obtained. A primary skin fibroblast line was established according to a previous published protocol (68).
Western blot analyses and antibodies
Primary fibroblast cells were lysed with 50 mm Tris–HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm Na-vanadate and 1 mm phenylmethylsulfonyl fluoride (PMSF), supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail 3 obtained from Sigma-Aldrich (P8340 and P0044; St Louis, MO, USA) and were normalized for protein content using Bradford assay and β-actin. Cell lysates were separated using electrophoresis on 6 and 10% SDS-polyacrylamide gels and transferred to PVDF membrane (Millipore, Billerica, MA, USA). For western blot, after the transfer, the PVDF membrane was blocked with 4% milk and incubated with following antibodies overnight. Primary antibodies used were: α-ASXL3 (NAP2-14791; Novus Biologicals, Littleton, CO, USA), α-ubiquityl-Histone H2A, α-V5 (05-678 and 46-0705; Millipore, Billerica, MA, USA), α-Histone H3 tri methyl K27, α-Histone H3 antibody (ab6002, and ab1791; Abcam Cambridge, MA, USA), α-FLAG (F3165; Sigma-Aldrich). HRP-conjugated secondary antibodies sc-2005, sc-2030 from Santa Cruz Biotechnology (Dallas, TX, USA) were used for 1 h incubation at room temperature. Antibody incubation and chemiluminescence detection were performed according to manufacturer's instruction [ThermoFisher Scientific (Waltham, MA, USA) cat no. 34095].
Co-immunoprecipitation
In total, 293 T cells were transiently co-transfected with plasmids expressing FLAG-BAP1 and tr-ASXL3-V5/His (aa1-aa484) gene constructs using lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA, USA). Two days following transfection, total cell lysates were prepared with modified RIPA buffer. α-V5 and α-Flag antibodies were incubated with the protein G-coupled (protein G dynabeads, Life Technologies, 1004D) to form a beads-Ab complex. Target antigen was precipitated from the total cell lysate using dynabead-Ab complex. Bound beads were washed 3 times with PBS. Substrate was eluted by boiling in 50 mM glycine buffer pH 2.8 and analyzed by western blot.
Acid extraction of histones from primary fibroblast cell lines
Confluent cells (80–100%) were harvested with TEB [0.5% Triton X 100 (v/v), 2 mm PMSF, 0.02% (w/v) NaN3], supplemented with 5 mm sodium butyrate at a density of 107 cells per ml and lysed on ice for 10 min. Nuclei were pelleted, resuspended in 1/4th volume of 0.2 m HCl overnight. Insoluble material was pelleted, and supernatant containing histones was incubated with sample loading buffer for 10 min. Protein concentration were quantified using Bradford assay.
Generation of fl-ASXL3-V5/His and tr-ASXL3-V5/His constructs
Total RNA was extracted from human neural progenitor cells using RNeasy mini kit (74104, Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. cDNA synthesis on the isolated RNA was then carried out with SuperScript III Reverse Transcriptase using oligo dT primer according to the instructions provided (18080, Invitrogen, Grand Island, NY, USA). Full-length ASXL3 (fl-ASXL3 forward primer 5′-GCCACCATGAAAGACAAGAGGAAGAAG-3′, fl-ASXL3 reverse primer 5′-TGCTCGTACAACCAGGCATGCTACAC-3′) and truncated ASXL3 fragment (tr-ASXL3 forward primer 5′-GCCACCATGAAAGACAAGAGGAAGAAG-3′ and tr-ASXL3 reverse primer 5′-CTGGCATTCACAGATACTAGTCTCTACCTC-3′) were amplified using Phusion Hot Start II High-Fidelity DNA Polymerase (ThermoFisher Scientific, Waltham, MA, USA). Fragments were gel purified using QIAquick Gel Extraction kit (28704, Qiagen) and incubated at 72°C with dNTP and Taq polymerase (201205, Qiagen) for 10 min to create 3′ A overhang. Product was ligated into pCDNA3.1-V5-His-Topo (45-0005, Invitrogen).
RNA isolation, library construction and sequencing
Transcriptome libraries were prepared using 200–1000 ng of total RNA. PolyA + RNA isolation, cDNA synthesis, end-repair, A-base addition and ligation of the Illumina indexed adapters were performed according to the TruSeq RNA protocol (Illumina). Libraries were size selected for 250–300 bp cDNA fragments on a 3% Nusieve 3:1 (Lonza) gel, recovered using QIAEX II reagents (QIAGEN) and PCR amplified using Phusion DNA polymerase (New England Biolabs). Total transcriptome libraries were prepared as above, omitting the poly A selection step and captured using Agilent SureSelect Human All Exon V4 reagents and protocols. Library quality was measured on an Agilent 2100 Bioanalyzer for product size and concentration. Paired-end libraries were sequenced with the Illumina HiSeq 2500, (2 × 121 nucleotide read length) with sequence coverage to 50 m paired reads and 100 m total reads.
RNA-sequencing data analysis
Alignment: Paired-end reads were aligned to the human reference genome (GRCh37/hg19) and transcriptome using a RNA-seq spliced read mapper Tophat2 (Tophat 2.0.4) with default settings (69,70). Fragment quantification was computed using feature Counts and annotated according to RefSeq genes. Fragments are only assigned to a gene if it overlaps features of that gene. DESeq2 was used to calculate estimates of dispersion and logarithmic fold changes to perform the expression normalization and differential expression analysis (71). Significantly DEGs were declared to have FDR ≤ 0.1 and log2 fold change of ≥1. We annotated genes and isoforms with NCBI Entrez GeneIDs and text descriptions. We used ipathway guide (http://www.advaitabio.com/ipathwayguide.html) for enrichment analysis of the set of DEGs to identify significantly enriched functional categories.
Preparation and analysis of miRNA-seq libraries
Total RNA was isolated using RNeasy Kit (Invitrogen) with DNase I digestion according to the manufacturer's instructions from the patient fibroblast cells. cDNA was synthesized from total RNA using Superscript III (Invitrogen) and random primers (Invitrogen). The region harboring the patient mutation c.1448dupT was amplified using the PCR with the product size of 454 bp. Barcode-indexed shotgun sequencing libraries were prepared from ASXL3 RT-PCR products using Tn5 transposase as previously described (72,73). Shotgun reads were mapped to the human reference assembly build 37 with BWA-MEM (biorxiv, http://arxiv.org/abs/1303.3997) (74). Freebayes (http://arxiv.org/abs/1207.3907) was used to identify variants including the ASXL3 frameshift and using option ‘--pooled-continuous’ to count the allelic distribution in each sample.
qRT–PCR verification
RNA sequencing findings of several top of DEGs were confirmed by qRT–PCR using the SYBR PCR master mix (Roche Applied Science, Indianapolis, USA). Total RNA was isolated using RNeasy Kit (Invitrogen) with DNase I digestion according to the manufacturer's instructions. RNA integrity was verified on an Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA). cDNA was synthesized from total RNA using Superscript III (Invitrogen) and random primers (Invitrogen). Quantitative RT–PCR was performed using Eppendorf Mastercycler® ep realplex according to manufacturer's instructions. Each target was run in triplicate, and expression levels relative to the housekeeping gene GAPDH were determined on the basis of the comparative threshold cycle CT method (2−ΔΔCT). The primer sequences used in these experiments are given in Supplementary Material, Figure S3C.
Funding
This work was supported by the National Institutes of Health (R00HD069624 to S.L.B., R01 DC009410 to D.M.M.) and by the Donita B. Sullivan Professorship funds to D.M.M.
Acknowledgements
We thank Amanda Moccia for assistance with manuscript preparation, Dr Jacob Kitzman and Dr Dan Robertson for technical advice, and genetic counselors Bridget O'Connor and Kailey Owens, who continue to counsel families with BRS patients seen at University of Michigan Pediatrics Medical Genetics.
Conflict of Interest statement. None declared.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}