





The increasing numbers of characterized base-pairing small RNAs (sRNAs) and the identification of these regulators in a broad range of bacteria are allowing comparisons between species and explorations of sRNA evolution. In this review, we describe some examples of trans-encoded base-pairing sRNAs that are species-specific and others that are more broadly distributed. We also describe examples of sRNA orthologs where different features are conserved. These examples provide the background for a discussion of mechanisms of sRNA evolution and selective pressures on the sRNAs and their mRNA target(s).
INTRODUCTION
Bacteria have evolved a variety of sophisticated regulatory mechanisms to adjust gene expression in response to a wide range of growth conditions. Key modulators of protein synthesis at the post-transcriptional level are small RNAs (sRNAs) that range in size from 50 to 500 nucleotides and base pair with mRNAs encoded at a genomic location distinct from the sRNA gene (reviewed in Storz et al.2011). Generally, these trans-encoded sRNAs have limited complementarity with their mRNA targets. Thus, single sRNAs can target multiple mRNAs, and single mRNAs can be the target of multiple sRNAs. However, a consequence of the limited base pairing is that the functions of many of these sRNAs, particularly in enteric bacteria, are dependent on the Hfq chaperone protein, which facilitates the intermolecular contacts between the sRNA and mRNA (reviewed in Vogel and Luisi 2011; Sobrero and Valverde 2012).
The majority of the base-pairing sRNAs are induced in response to specific environmental cues such as iron limitation, availability of a particular carbon source, cell envelope stress or cell density (reviewed in Storz et al.2011; Hoe et al.2013). The fact that many of these responses also have associated protein transcription factors has raised questions about the advantages of sRNA regulators. A number of benefits have been suggested (reviewed in Beisel and Storz 2010). The ability of sRNAs to directly target mRNAs for RNase degradation, suppress the degradation of other mRNAs and repress or enhance translation, permits rapid decreases or increases in the synthesis of the encoded protein. The codegradation of some sRNAs together with their targets allows the inactivation of the regulator in a way that is not possible for transcription factors. In addition, the higher overall levels of sRNAs compared to transcription factors provides a better buffer against stochastic noise. The relatively low energy expenditure required to synthesize an sRNA is also considered an advantage. Finally, given that the extent of base pairing can influence regulatory output, gene expression can easily be fine-tuned with the addition or subtraction of sRNA–mRNA base-pairing sites as well as with single nucleotide changes in these interaction sites.
The action of sRNAs in concert with transcription factors to simultaneously or reciprocally repress or enhance target RNA expression at both transcriptional and post-transcriptional levels places sRNAs in large regulatory networks (reviewed in Beisel and Storz 2010; Storz et al.2011). The incorporation of sRNAs in such networks allows more nuanced regulation with respect to the strength and speed of a response or can change the regulation for a subset of genes; for example, allowing a transcription activator to also downregulate genes.
While many of the initial studies of base-pairing sRNAs were conducted in Escherichia coli and Salmonella enterica, the ever-improving ability to carry out deep sequencing has led to the identification of sRNAs in increasing numbers of bacterial species (reviewed in Sharma and Vogel 2009, 2014). Although in most organisms the full repertoire of sRNAs has not been characterized as extensively as in E. coli and S. enterica, the sequencing data allow prediction of sRNA candidates in different species and facilitate cross-species comparisons.
With better understanding of the physiological roles and mechanisms of action of base-pairing sRNAs and the large sets of detected or predicted sRNAs, it is becoming more feasible to address questions about the evolution of sRNAs. How have sRNA genes evolved and become so pervasive and integral to bacteria physiology? This review will consider the requirements for base-pairing sRNAs and their targets. We will then discuss sRNAs that show extremely limited distribution as well as sRNAs found more broadly. Examples of sRNA orthologs where different features are conserved will also be considered. Finally, we will explore mechanisms of DNA change that lead to the formation and decay of sRNA genes as well as the physiological and structural constraints that impact the evolution of sRNA–mRNA pairs.
sRNA AND mRNA FEATURES REQUIRED FOR PRODUCTIVE BASE PAIRING
Several features need to be present for an sRNA to be expressed, stable and able to base pair with mRNAs (illustrated for E. coli Spot 42 in Fig. 1a). mRNAs similarly require specific features to be a target of an sRNA (illustrated for Spot 42 targets in Fig. 1c).
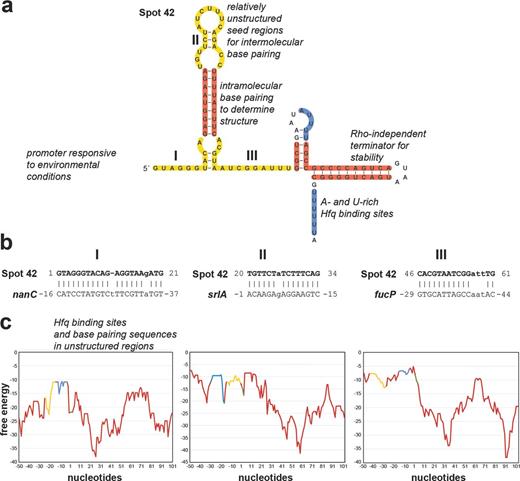
Features of sRNAs and mRNAs required for effective regulation illustrated for E. coli Spot 42 (a) and three of its target mRNAs, nanC, srlA and fucP (b and c), that base pair with the three different single-stranded regions of Spot 42 (I, II and III, respectively). The structure and regions of base pairing and Hfq binding in (a) are based on (Møller et al.2002a, 2002b; Beisel and Storz 2011; Beisel et al.2012). The graphs in (c) show the free-energy profiles (kcal/mol, y-axis) for RNA local secondary structure along the sequences of the Spot 42 targets (x-axis, numbering is relative to the start codon). These graphs reveal that sequences involved in base pairing and Hfq binding have less secondary structure. Yellow denotes complementary sequences, blue denotes Hfq-binding sequences, red denotes sequences involved in forming secondary structures and green denotes start codons.
Required sRNA features
A recurring theme is that the transcription of base-pairing sRNAs is strongly regulated. In fact, in many instances, the sRNA genes are among the most highly regulated genes in a particular regulon. For example, aside from rpoE itself (encoding σE), the genes encoding the MicA, RybB and MicL RNAs are the most strongly σE-induced genes in E. coli (Guo et al.2014). In addition to depending on other alternative sigma factors for expression, sRNA genes are regulated by transcription activators such as hydrogen peroxide-responsive OxyR or repressors such as iron-responsive Fur, as well as by two-component systems that are activated by a range of signals such as low magnesium in the case of PhoQ and PhoP (reviewed in Göpel and Görke 2012; Hoe et al.2013). In a number of cases, the gene encoding the corresponding transcription regulator is divergent from the sRNA gene. Overall, a regulated promoter that responds to environment signals is a feature of most base-pairing sRNA genes.
A key parameter in one early computational search for sRNAs genes in intergenic regions was the presence of a Rho-independent terminator comprised of a stable stem loop followed by a stretch of U residues (Chen et al.2002). As more and more sRNAs have been characterized, it has become clear that the presence of a Rho-independent terminator is associated with almost every base-pairing sRNA. The strong terminator, which confers resistance to degradation by ribonucleases (Ishikawa et al.2012) and, as will be discussed below, provides an Hfq-binding site, can be considered a second critical feature of a sRNA gene.
Another essential sRNA feature is a region where base pairing with the mRNA is initiated, frequently called the seed region. Some sRNAs such as RybB are only known to have one region that base pairs with the mRNA targets (Papenfort et al.2010) while others such as Spot 42 or FnrS have multiple seed regions that each base pair with different mRNA targets (Durand and Storz 2010; Beisel and Storz 2011). Several lines of evidence indicate that the seed region needs to be unstructured. First, structural analysis of individual sRNAs has repeatedly shown that the sequences required for base pairing are largely single stranded (Sharma et al.2007; Papenfort et al.2010; Fröhlich et al.2013; Shao et al.2013). Second, global structural predictions show a clear correlation between the regions of base pairing and the regions of the sRNA predicted to have the least secondary structure (Peer and Margalit 2011; Richter and Backofen 2012). Similarly, both individual and global analyses point to the seed region being the most conserved part of the sRNA (Sharma et al.2007; Peer and Margalit 2011). Some sRNAs such as ArcZ are processed with the result that the base-pairing sequence becomes the 5′ end of the sRNA, possibly increasing accessibility of the seed region (Papenfort et al.2009). Additionally, there are sRNA examples such as DsrA for which the initial sRNA–mRNA contact via the seed region leads to the unwinding of flanking structural elements ultimately leading to more extended base pairing with the target mRNA and strengthening the interaction (Lease, Cusick and Belfort 1998).
A requirement for sRNA function in many bacteria is association with the Hfq RNA chaperone protein. Numerous studies have shown that Hfq protects sRNAs from ribonuclease degradation and facilitates intermolecular contacts between sRNAs and cognate mRNA targets (reviewed in Vogel and Luisi 2011; Sauer 2013). The Hfq protein, which is a homolog of Sm and Lsm proteins that are components of RNA splicing and degradation complexes in eukaryotes and archaea, forms a homohexameric ring (reviewed in Wilusz and Wilusz 2013). The two faces of the Hfq ring, designated proximal and distal with respect to the positions of the N-and C-termini, are important for binding RNAs in E. coli. The proximal side shows strong specific binding to the single-stranded U-rich sequences associated with Rho-independent transcription terminators (Otaka et al.2011; Sauer and Weichenrieder 2011), while the distal surface preferentially binds polyA and ARN (A, A/G, any nucleotide) repeats (Link, Valentin-Hansen and Brennan 2009; Robinson et al.2014). Recently, the rim or lateral surface of the Hfq ring has been shown to interact with RNA as well, with two U residues contacted by rim residues in a co-crystal structure of Hfq and the RydC RNA (Sauer, Schmidt and Weichenrieder 2012; Panja, Schu and Woodson 2013; Dimastrogiovanni et al.2014). While sRNAs in many bacteria require Hfq binding for function, mutational studies in E. coli suggest that there is significant variation in the way different sRNAs bind to Hfq (Zhang et al.2013). These data are consistent with in vivo and in vitro studies showing that sRNAs have differing capacities to compete with each other for Hfq binding (Moon and Gottesman 2011; Olejniczak 2011; Małecka et al.2015). The differences in binding, though not fully understood, probably have functional consequences. Binding sites for Hfq and for other proteins, particularly in organisms that do not possess Hfq family members, are another critical feature of most base-pairing sRNAs.
Finally, base-pairing sRNAs all possess double-stranded regions that help to stabilize the sRNAs as well as allow for the appropriate orientation of the seed region and binding sites for Hfq and other proteins in relation to the mRNA targets. While specific sequences in the stable duplexes may not be conserved, they are enriched for nucleotides that co-vary to retain intramolecular base pairing and the secondary structure elements, allowing the single-stranded sequences to be accessible for interactions with other RNAs or proteins (Chen et al.2002; Ishikawa et al.2012).
Required target mRNA features
Several of the features required by sRNAs must also be present on the mRNA target. First and foremost, the mRNA must have a sequence that can base pair with the sRNA. For mRNAs that are subject to repression, this region needs to be unstructured (Fig. 1c). Despite strong base-pairing potential, Spot 42-dependent regulation was not detected for some predicted targets because the base-pairing sequence in the mRNA was sequestered in a secondary structure (Beisel et al.2012). Nucleotide changes that weakened the secondary structure without compromising the extent of base-pairing potential converted these mRNAs into Spot 42-regulated transcripts. The position of the seed sequence in the mRNA also impacts how the sRNA can affect expression of the target. In cases where the sRNA modulates mRNA stability, base pairing must be in a position to block access to the ribonuclease or recruit the ribonuclease to a specific site (Pfeiffer et al.2009; Prévost et al.2011; Bandyra et al.2012; Fröhlich et al.2013; Papenfort et al.2013). In cases where the sRNA blocks translation, the site of base pairing is generally near the ribosome-binding site (Bouvier, Sharma and Mika 2008), while for translational activation, base pairing must result in the opening of the secondary structure which otherwise occludes the ribosome-binding site (reviewed in Papenfort and Vanderpool 2015).
Studies of individual targets have suggested that, at least in enteric bacteria, mRNAs subject to regulation by base-pairing sRNAs all have an Hfq-binding site (reviewed in Vogel and Luisi 2011). For the mRNAs where this has been examined, the Hfq-binding site is in the form of an ARN repeat near, either in linear sequence or in the context of the tertiary structure, the unstructured sRNA-binding site (Soper and Woodson 2008; Salim and Feig 2010; Salim et al.2012). mRNAs that show extensive complementarity to the Spot 42 sequence but do not show regulation can be converted into targets by the addition of an ARN repeat near the site of base pairing, or by replacing an ARN repeat that overlaps the target-binding site (Beisel et al.2012). Furthermore, the distance between the Hfq-binding site and region of base pairing is important. If the ARN repeat in the rpoS mRNA, for which translation is activated by the ArcZ, DsrA and RprA RNAs, is engineered to be closer or further from the sRNA target site, the mRNA becomes inert to sRNA regulation despite being functional for Hfq binding (Peng, Soper and Woodson 2014).
Thus, both sRNAs and target mRNAs require sites of interaction that are unobstructed by secondary structures or where sRNA–mRNA base pairing can easily lead to opening of the secondary structures, along with Hfq-binding sites positioned at an optimal distance from the base-pairing sequences. Additional studies are required to understand the distance requirements and how protein binding correlates with the type and extent of regulation observed. It is intriguing that many mRNAs found to be the target of one sRNA, subsequently are shown to be regulated by other sRNAs. Once features that allowed an mRNA to become a target are in place, it may be easier to evolve base pairing with additional sRNAs regulators.
sRNAs WITH LIMITED OR WITH BROAD DISTRIBUTION
It is clear from phylogenetic comparisons that some base-pairing sRNAs show very limited distribution (Lindgreen et al.2014). However, a few sRNAs are more broadly conserved (Peer and Margalit 2014). An examination of sRNAs from both ends of this spectrum can provide insights into differences between these sRNAs.
Strain- and species-specific sRNAs
The acquisition or loss of sRNAs can facilitate rapid diversification of regulatory networks between bacterial species and even contribute to strain-to-strain variability. A dRNA deep sequencing study to map and compare transcription start sites between four different clinical isolates of the food-borne pathogen Campylobacter jejuni revealed a number of strain-specific sRNAs whose expression was documented by Northern analysis (Dugar et al.2013). For two of these chromosomally encoded variable sRNAs (CJnc30 and CJnc80), there is no sequence similarity to other known sRNAs. A computational study that examined the distribution of sRNAs across 27 E. coli and Shigella genomes found that 23 out of the 83 known or predicted sRNAs were variable in the genomes analyzed (Skippington and Ragan 2012). Given that the variation is sporadic, this study concluded that it is primarily due to gene loss, not lateral transfer. The observation that the variable sRNAs were less well integrated into regulatory circuits with fewer connections to other regulators compared to more conserved sRNAs led to the prediction that loss of variable sRNAs is less disruptive to regulatory networks than loss of conserved sRNAs. A similar phylogenetic comparison of predicted sRNAs within the Vibrio genus found that close to 50% of the sRNAs were not conserved in all Vibrio cholerae strains (Toffano-Nioche et al.2012). The large numbers of unique sRNAs in different Campylobacter, Escherichia, Shigella and Vibrio species and strains point to more rapid evolution of sRNAs genes than neighboring protein-coding genes.
More broadly distributed sRNAs
Examples of more widely conserved sRNAs can also be found. An effort to trace the evolution of known E. coli sRNA families using sequence and structure information revealed that most of the base-pairing sRNAs only came into existence after the split of the Enterobacteriales order from the rest of ϒ-proteobacteria (Peer and Margalit 2014). However, five well-characterized E. coli base-pairing sRNAs (Spot 42, GcvB, RyhB, SgrS, SdsR) can be traced outside of Enterobacteriales (Spot 42 is shown in Fig. 2a). All of these sRNAs are induced by specific environmental signals, have multiple mRNA targets and function as hubs in regulatory circuits.
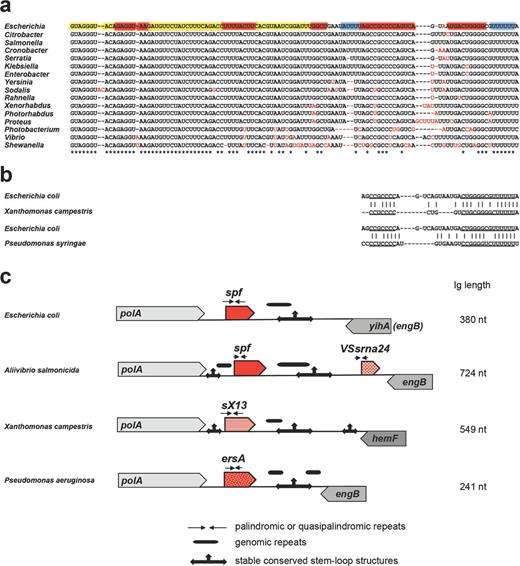
Mechanisms of sRNA evolution illustrated for Spot 42 (encoded by the spf gene), VSsrna24, sX13 and ErsA encoded downstream of polA. Alignment of Spot 42 in Gammaproteobacteria showing the contributions of single nucleotide substitutions and deletions to Spot 42 evolution (a) and similarity of terminators among the sRNAs found downstream of polA in E. coli, Xanthomonas campestris and P. syringae (b). The coloring of the E. coli sequence in (a) is the same as in Fig. 1a. (c) Architecture of the intergenic region (Ig) downstream of polA showing the location of variable repeats and conserved stable hairpins and suggesting the contribution of palindrome misalignment to sRNA evolution. The different sRNA families are indicated by the different patterns of the red arrows. The corresponding trees for the sRNAs found downstream of polA in γ-proteobacteria, Xanthomonadaceae and Pseudomonadaceae are given in Fig. S1 (Supporting Information).
Another sRNA family, the AbcR family, also has been found to be broadly distributed in the α-proteobacterial phylum including the plant pathogen Agrobacterium tumefaciens, the plant symbiont Sinorhizobium meliloti and the human pathogen Brucella abortus (reviewed in Becker et al.2014). The first members of this family were characterized in A. tumefaciens where AbcR1 and AbcR2 were found to be encoded in the same intergenic region, and AbcR1 was shown to act as a quintessential Hfq-binding sRNA repressing the translation of three mRNAs encoding components of ABC transport systems (Wilms et al.2011). Subsequently, other AbcR orthologs were characterized in S. meliloti and B. abortus. The paralogs within some strains are encoded at distinct genetic locations and in some cases show differential regulation; however, all appear to repress the synthesis of ABC transport proteins, many of which likely transport amino acids. The property of repressing the synthesis of amino acid importers is shared with the broadly distributed GcvB family, raising the question whether these two families evolved convergently (Sharma et al.2011).
PERMUTATIONS IN sRNA EVOLUTION
A cross-species comparison of sRNAs and the genomic positions of their genes illustrate how different features of specific sRNAs as well as their targets are conserved in various permutations across species. As we will discuss, the high variability and continuum of conserved features of sRNAs and their targets allows for the fine-tuning of regulation under the range of environmental conditions associated with the varied life styles of bacteria.
Same sequence but different regulation
sRNAs paralogs or orthologs can have similar sequences but show different regulation. For example, the CJnc180 and CJnc190 RNAs of C. jejuni showed conservation in four strains examined but had different expression patterns throughout growth (Dugar et al.2013). The levels of GcvB RNA in S. enterica are high in exponential phase and decrease as the cells enter stationary phase (Sharma et al.2007). In contrast, in V. cholerae, the levels of GcvB are higher in stationary phase than in exponential phase (Papenfort et al.2015). A third informative example is provided by the homologous GlmY and GlmZ RNAs, which show distinct regulation in different Enterobacteriales species (Göpel et al.2011). σ54 and the response regulator GlrR control the expression of these sRNAs in Yersinia pseudotuberculosis and S. enterica. However, while σ54 and GlrR are absolutely required for GlmY and GlmZ expression in Y. pseudotuberculosis (and Dickeya, Erwinia and Serratia), both genes are still expressed in S. enterica mutants lacking σ54 due to overlapping σ70 promoters. Overlapping σ54 and σ70 promoters were also observed for GlmY in E. coli (and Citrobacter, Cronobacter and Enterobacter species). In contrast, a single σ70 promoter gives rise to constitutive GlmZ expression in E. coli (and probably also Klebsiella and Shigella). These phylogenetic comparisons suggest that glmY and glmZ originated, via gene duplication, from a single sRNA gene controlled exclusively by σ54 in an ancestral Enterobacteriales strain. Gradually promoter mutations caused divergent expression of these two sRNAs in different species.
Same regulation and sequence but different mRNA target preference
Despite possessing similar regulation and sequences, sRNA orthologs can display different mRNA target preferences. An example is provided by the RyhB orthologs in E. coli and V. cholerae (Davis et al.2005; Mey, Craig and Payne 2005). Expression of both orthologs is repressed by Fur in the presence of high iron, and the central portion of these two sRNAs is well conserved, though the Vibrio ortholog has ∼60 nt extensions on either side of the conserved region. Despite the conserved region, the two sRNAs differ in the extent and function of their regulons. A number of mRNAs encoding iron-containing proteins known to be repressed by RyhB in E. coli (Massé, Vanderpool and Gottesman 2005) are similarly regulated in V. cholerae. Other transcripts regulated by RyhB in E. coli, however, showed no RyhB-dependent regulation in V. cholerae, and conversely several genes that showed RyhB dependence in V. cholerae are not RyhB targets in E. coli. The novel V. cholerae targets encode proteins involved in aerobic and anerobic respiration, energy metabolism, motility, chemotaxis and biofilm formation. Thus, RyhB has a core function with respect to iron homeostasis in both species, but the V. cholerae sRNA has acquired new physiological roles. Additionally, different regions of some of the common targets are subject to base pairing with the E. coli and V. cholerae RyhB RNAs. For instance, while RyhB from both species regulates sodB, the sequences in the 5′ untranslated regions (UTR) showing the greatest complementarity to RyhB differ.
Same mRNA target preference but different regulation and sequence
sRNAs can also show unique regulation and expression and be encoded in very different genomic locations, but have similar unstructured seed regions that allow for regulation of overlapping targets. This is exemplified by the GcvB (Sharma et al.2007) and DapZ RNAs of S. enterica (Chao et al.2012). Unlike GcvB, which is highly expressed in log phase, DapZ is exclusively expressed later in stationary phase and is regulated by the horizontally acquired HilD virulence transcription factor. Additionally, while GcvB is encoded in an intergenic region, DapZ is encoded at the 3′ end of the dihydrodipicolinate reductase gene dapB, whereby DapZ and dapB utilize the same 3′ UTR but are transcribed from separate promoters. Transcriptomic analysis upon pulse expression of DapZ showed that the sRNA regulates the same ABC transporters as GcvB via a G/U-rich seed domain that is very reminiscent of the sequence found in GcvB. Thus, although the two sRNAs are part of different regulatory networks, they base pair with overlapping targets. Less striking examples of this overlapping regulation are mRNAs such as the E. coli rpoS and csgD mRNAs (reviewed in Boehm and Vogel 2012; Mika and Hengge 2014), which are regulated by multiple sRNAs at the same general region but where each of the sRNAs also controls a distinct regulon.
Same regulation and function but different sequence
In addition to sRNAs having similar targets as a consequence of some sequence similarity, there are functional analogs with no sequence conservation. Examples are RyhB of E. coli and PrrF1 and PrrF2 of Pseudomonas aeruginosa (Wilderman et al.2004). RyhB, PrrF1 and PrrF2 are all induced by low iron upon Fur depression and target some of the same mRNAs. The RyhB sequence, its –10 and –35 promoter and Fur operator sequence are well conserved in Enterobacteriaceae, but the RyhB gene is absent in the Pseudomonas. Yet Fur is an essential repressor in this organism and was observed to upregulate genes known to be repressed by E. coli RyhB. PrrF1 and PrrF2 were identified using a bioinformatics approach of looking for Fur consensus sequences preceding predicted Rho-independent transcription terminators within intergenic regions of the P. aeruginosa genome. While PrrF1 and PrrF2 are encoded in tandem and are >95% identical, they share very little sequence similarity with RyhB. However, all regulate the common RyhB targets sodB and sdhC by targeting the 5′ end of the messages and blocking ribosome binding. Other examples of sRNAs sharing the same regulation and function but little sequence similarity are E. coli MicA and V. cholerae VrrA (Udekwu et al.2005; Song et al.2008). Like MicA, VrrA is induced by membrane stress via σE and represses translation of the outer membrane porin OmpA by base pairing with a region overlapping the ribosome-binding site of the ompA mRNA.
Same genetic location but different sequence and regulation
For some sRNAs, little is conserved aside from the genetic location. An example is the intergenic region downstream of the broadly conserved polA gene, which encodes DNA polymerase I. In E. coli, this intergenic region encodes the Spot 42 RNA whose expression is repressed by CRP in low glucose and which represses the synthesis of transporters of alternative carbon sources (Beisel and Storz 2011). In Aliivibrio salmonicida, the same intergenic region encodes two sRNAs: a Spot 42 ortholog and the unrelated VSsrna24 RNA (Hansen et al.2012). Like in E. coli, expression of A. salmonicida Spot 42 is activated by glucose and repressed by cAMP, whereas VSsrna24 is repressed by glucose independently of cAMP. Interestingly though, there is very little overlap in the genes regulated by Spot 42 in these two organisms; many of the genes encoding mRNA targets in E. coli are not present in A. salmonicida and homologs of the remaining E. coli targets show no Spot 42 dependence. sRNA genes have also been found downstream of polA in Xanthomonas (Schmidtke et al.2013) and Pseudomonas species (Ferrara et al.2014; Park et al.2014), but the nucleotide sequences of these two families of sRNAs are distinct from Spot 42 with the exception of some similarity in the sequences comprising a stable hairpin structure at the 3′end (Fig. 2b and c). Expression of these sRNAs is induced by various stress conditions, and the sRNAs have been suggested to play roles in virulence. Thus, while multiple sRNA genes are found downstream of polA in Enterobacteriaceae, Vibrionaceae, Xanthomonadaceae and Pseudomonadaceae, they encode three distinct sRNA families (see Fig. S1, Supporting Information), indicating that this region may be a hotspot for the evolution of new sRNAs.
MECHANISMS OF sRNA EVOLUTION
The preceding examples illustrate how different sRNA features can vary between bacteria and lead to the question of how the changes came about. As we will describe next, single nucleotide polymorphisms, gene duplication, palindrome misalignment, chromosomal rearrangements, horizontal gene transfer or combinations of these genomic alterations are all possible mechanisms of change that can lead to both the evolution and the erosion of sRNA genes (illustrated for Spot 42 and sRNAs encoded downstream of polA in Fig. 2 and summarized in Fig. 3). Before embarking on a discussion of these mechanisms, it should be noted that not all detected small transcripts are likely to be functional. A subset of small transcripts could be intermediates in evolution, while others might be transcriptional noise that could serve as substrates for future evolution.
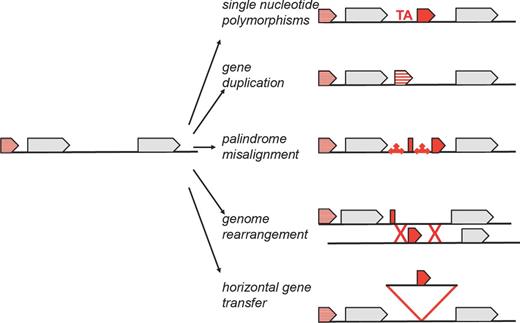
Schematic summary of possible mechanisms of sRNA gene evolution, which include single nucleotide polymorphisms, gene duplication, palindrome misalignment, genome rearrangement and horizontal gene transfer.
Additionally, it is worth noting that increasing numbers of functional sRNAs derived from or overlapping mRNAs are being discovered (reviewed in Miyakoshi, Chao and Vogel 2015b). 3′ UTRs are particularly good candidates for base-pairing sRNA evolution given the sequences often already contain one essential sRNA feature, a Rho-independent terminator stem loop, and there are few other constraints in the region downstream of the stop codon (Chao et al.2012; Guo et al.2014; Kim et al.2014). Some sRNAs have been found to share a terminator with a convergently transcribed protein-coding gene (Argaman et al.2001), again possibly taking advantage of an existing feature. Two examples of functional sRNAs derived from the 5′ UTR encompassing an S-adenosylmethionine riboswitch in Listeria monocytogenes have been described (Loh et al.2009). Given that many riboswitches, particularly those regulating transcription termination, give rise to stable RNA fragments, it is conceivable that other 5′ UTRs function as base-pairing sRNAs as well. Several sRNAs are encoded in the same orientation as the downstream gene. Possibly, some of these RNAs were once the 5′ UTR of a longer transcript for the downstream gene, but over time mutations that generated a stable terminator and new promoter led to independent functions. A recent study also showed that a stable fragment from the S. enteria gltIJKL mRNA acts as a regulator by base pairing with GcvB to inhibit the activity of the sRNA (Miyakoshi, Chao and Vogel 2015a, in press). Finally, there are examples of RNAs that have both protein-coding and base-pairing regulatory functions. Interestingly, there is variation in which of these two functions is more broadly conserved. The SgrT protein encoded by the E. coli SgrS RNA is only detected for a subset of SgrS orthologs (Horler and Vanderpool 2009), while the base-pairing function of the RNAIII of Staphylococcus aureus is less conserved than the encoded Hld protein (Queck et al.2008).
Single nucleotide polymorphisms
Some of the differences in the expression of two orthologs or targeting of different mRNAs in the examples described above are a result of single nucleotide changes. Examination of the variable regions of the Spot42 RNA (Fig. 2a) shows that these sequences are rich in A and U nucleotides suggesting that some of the single nucleotide polymorphisms are introduced by slippage during replication, though other mechanisms such as nucleotide misincorporation or DNA damage are also likely to contribute to spontaneous changes in sRNA genes. The introduction of these single nucleotide polymorphisms into one of the many regions found to be transcribed by deep sequencing (reviewed in Sharma and Vogel 2009, 2014) could lead to the evolution of an RNA with the ability to base pair with an mRNA in a manner that provides a selective advantage. This conclusion is supported by a systematic study of the effects of limited nucleotide changes in DsrA and RyhB (Peterman, Lavi-Itzkovitz and Levine 2014). One or two nucleotide changes in the seed region of the sRNA were found to fine-tune the strength of regulation in a target-specific manner. Mutations in the neighboring structural elements also impacted regulatory activity by affecting sRNA abundance and, in some cases, resulting in base pairing with non-specific targets, thus rewiring the sRNA into new regulons.
Gene duplication
There are multiple examples of sRNA genes derived from gene duplication (reviewed in Caswell, Oglesby-Sherrouse and Murphy 2014). The most obvious are those where the sequence similarity between two genes is still very high. As already mentioned above, there are two copies of both the AbcR and PrrF RNAs in multiple organisms. For other base-pairing sRNAs, such as the Qrr RNAs that modulate quorum sensing in Vibro species (reviewed in Bardill and Hammer 2012) and the csRNAs that have pleiotropic effects in Streptococcus species (Schnorpfeil et al.2013), there are even more copies of the sRNA gene. Some duplicated sRNAs have extremely similar regulation and target specificities while for others these features have diverged, allowing the regulation of additional mRNAs, particularly if there are limitations on the evolution of the targets. It is not yet clear what constrains the total number of sRNAs genes, and why some are commonly found as two copies while others vary in number from one to five in different species (reviewed in Caswell, Oglesby-Sherrouse and Murphy 2014).
Some sRNAs might have resulted from the duplication of regions to which similarity is less obvious. For instance, S. enterica DapZ, which has a seed region that is very similar to GcvB, could have been derived from a duplication of the gcvB gene (Chao et al.2012). An sRNA gene also could be derived from a duplication of a portion of the ultimate target gene. The E. coli MicL RNA encoded in the 3′ end of the cutC protein-coding gene is a possible example of such evolution (see graphical abstract). This sRNA has only been found to have a single target, lpp, but shares extended complementarity with this mRNA (Guo et al.2014) (see also Fig. S2, Supporting Information). We propose that duplications of portions of protein-coding genes should be considered potential sources of sRNA genes.
Palindrome misalignment
Another probable mechanism for the generation of sRNA genes is the misalignment of palindromic or quasi-palindromic sequences, consisting of near-perfect or imperfect inverted repeats separated by a short spacer, which have the potential to form hairpin structures with stable stems and short loops. During DNA synthesis, the close proximity of the repeats makes it possible for newly synthesized DNA to mispair with incorrect complementary sequences through intramolecular and intermolecular interactions giving rise to point mutations and duplications (reviewed in Lovett 2004). For example, the nascent and lagging template strands may undergo intramolecular pairing forming stable hairpin structures. During this process, the 3′ end of the nascent strand becomes open to exonucleolytic degradation that may lead to mutations. This mechanism might have led to sRNA evolution in the sequences downstream of the polA where there are signatures of various repeat elements (Fig. 2c). The 3′ ends of some mRNAs, especially the regions between stop codons and terminators, could be hotspots for sRNA generation by palindrome misalignment. Since the nucleotide sequences comprising and surrounding terminator stem-loop structures frequently have C- or CU-tracks, they could interact with the AG-rich sequences in different parts of the gene.
Genome rearrangement
A recent comparison of intergenic regions, which differ between E. coli and S. typhimurium but show clear transcription in one or the other species, revealed that larger genomic rearrangements, most likely caused by homologous recombination between repeat and bacteriophage sequences, can also lead to both the genesis and decay of sRNA genes (Raghavan et al.2015). As one example, the E. coli sRNA EcsR1 was likely lost in S. enterica due to a genome rearrangement that split the intergenic region into two fragments located ∼200 kb apart. In another example, the SesR2 gene arose in an intergenic region formed via phage-mediated genome rearrangement in a subset of Salmonella species, probably via point mutations that created a σ70-like promoter.
Horizontal gene transfer
There is clear evidence that horizontal gene transfer is also a mechanism for sRNA dissemination. Horizontally acquired genes are frequently transferred between bacteria via bacteriophage and plasmids, and a number of sRNAs have been dispersed in this way. In a recent study of enterohemorrhagic E. coli, it was estimated that 55 non-coding sRNAs are encoded within the extra 1.4 Mb of horizontally acquired DNA elements (Tree et al.2014). In fact, pathogenicity islands of horizontally acquired DNA consisting of active and cryptic prophages are enriched ∼1.8-fold for predicted sRNA genes relative to the core genome. Predicted sRNA genes were especially prevalent in specific locations within lambdoid phages; numerous sRNA genes were found to be encoded downstream of the bacteriophage Q antiterminated promoter (PR). Two of these sRNAs were characterized and found to function as anti-sRNA regulators that act by base pairing with FnrS and GcvB, thereby repressing the sRNA function and indirectly activating the targets of these sRNAs.
Other examples of cryptic bacteriophage-derived base-pairing sRNAs in non-pathogenic E. coli include the DicF RNA, which inhibits cell division by base pairing with ftsZ, and the IpeX RNA, which inhibits synthesis of the OmpC porin (Faubladier and Bouché 1994; Castillo-Keller et al.2006). Neither of these sRNAs has been characterized extensively, but it is striking that dicF and its flanking sequences, which are encoded in the immunity region of lambdoid prophage, are detected in a widespread family of prophage-like elements that are present in distantly related species (Faubladier and Bouché 1994). Thus, DicF-like sRNAs might be present in many different bacteria.
There are also multiple examples of sRNAs encoded on horizontally acquired pathogenicity islands. Targeted searches of these sequences lead to the identification of 19 island-encoded sRNAs in S. enterica (Padalon-Brauch et al.2008) and 7 in S. aureus (Pichon and Felden 2005), some of which show significant variation between pathogenic strains. The island-encoded sRNAs can regulate core host genes. For example, the InvR RNA encoded by the Salmonella pathogenicity island I repressed the synthesis of the OmpD outer membrane porin encoded by the core genome (Pfeiffer et al.2007). Conversely, core genome-encoded sRNAs can regulate mRNA targets encoded in the pathogenicity islands. For example, the broadly conserved SgrS RNA, which evolved before the acquisition of the virulence factors and plays an important role in combating phosphate sugar stress in E. coli and S. enterica, has been repurposed to repress the synthesis of the secreted effector protein, which is encoded by the horizontally acquired sopD mRNA and helps to establish Salmonella virulence (Papenfort et al.2012). Base pairing between SgrS and sopD occurs within the same conserved seed region of SgrS that maintains post-transcriptional regulation of a number of sugar transporters. Interestingly, this conserved region allows for the regulation of sopD but not its paralog sopD2. The discrimination is achieved by a single G-C pair with sopD which is a G-U wobble pair with sopD2, highlighting the effectiveness of base pairing to regulate a whole swath of targets while being deaf to thousands of other mRNAs in the cell.
CONSTRAINTS ON sRNA-mRNA TARGET EVOLUTION
In considering how sRNAs as well as their mRNA targets evolve, constraints on their co-evolution should also be evaluated. There is an apparent overrepresentation of base-pairing sRNAs that are induced by certain physiological conditions such as iron starvation, changes in carbon source availability, outer membrane stress and stationary-phase growth. This skew in distribution may solely be a reflection of the sRNAs studied. For example, the characterization of an sRNA in one organism might spur studies of similar sRNAs in other organisms. Alternatively, the skew may be a consequence the specific cellular needs, the mRNA features required for sRNA-mediated regulation or, most likely, all of these factors. It is worth noting that, in general, only limited growth defects have been associated with the lack of sRNAs consistent with modulatory, but not essential, roles in the cell.
Physiological constraints
Strikingly, certain mRNAs reoccur as the target of sRNAs in a range of bacteria. One example is the sdh transcript encoding succinate dehydrogenase. This polycistronic mRNA is regulated by sRNAs in several different species. In E. coli, the sdhCDAB mRNA is repressed by RyhB in response to limited iron availability, as well as by Spot 42 and RybB in response to high glucose and cell envelope stress, respectively (Desnoyers and Massé 2012). It is also repressed by the Fur-regulated NrrF RNA in Neisseria meningitides (Mellin et al.2010), and by the FsrA RNA in response to low iron in B. subtilis (Gaballa et al.2008). There may be high selective pressure for the precise regulation of succinate dehydrogenase synthesis given that this enzyme connects the TCA cycle and electron transport chain and requires multiple iron atoms.
Likewise, the synthesis of outer membrane porins and inner membrane transporters is regulated by a multitude of sRNAs in γ- and α-proteobacteria. The σE-induced MicA, RybB and MicL RNAs together repress the synthesis of most major outer membrane proteins in order to alleviate cell envelope stress (Guo et al.2014). GcvB, DapZ and AbcR all control the synthesis of ABC transporters of amino acids in response to changes in nutrient availability, while SgrS regulates the expression of both importers and exporters of phosphorylated sugars to combat sugar-phosphate stress (Sharma et al.2007; Rice and Vanderpool 2011; Sun and Vanderpool 2011; Wilms et al.2011). Synthesis of the outer membrane porin OmpD is even repressed by four different sRNAs, RybB, MicC, SdsR and InvR in S. enterica (Fröhlich et al.2012). Possibly, sRNAs are needed to regulate the levels of these abundant membrane proteins whose activities may be difficult to modulate once the proteins are in the membrane and for which some mRNAs have long half-lives. As we will discuss below, the prevalence of mRNA targets encoding membrane proteins may also be a consequence of the features required to export these proteins.
mRNAs encoding transcription factors additionally are frequently the targets of sRNAs. In Vibrio species, the redundant Qrr sRNAs target the mRNAs encoding LuxO, LuxR and AphA, key transcription regulators of the quorum-sensing response, and thus contribute to the virulence of V. cholerae (reviewed in Bardill and Hammer 2012). In E. coli, synthesis of the master regulators of flagellar synthesis encoded by the flhDC mRNA is repressed by ArcZ, OmrA, OmrB and OxyS, and activated by McaS, and the master transcription regulator of curli biogenesis encoded by csgD is repressed by OmrA, OmrB, McaS, RprA, GcvB and RydC, making biofilm formation a physiological response that is multiply regulated by sRNAs (reviewed in Boehm and Vogel 2012; Mika and Hengge 2014). In E. coli, three sRNAs, DsrA, RprA and ArcZ, increase the translation of σS, and the levels of the two-component proteins EnvZ/OmpR, DpiA/DpiB and PhoP/PhoQ are regulated by the sRNAs OmrA/OmrB, ChiX and GcvB, respectively (reviewed in Mandin and Guillier 2013). The multiply-regulated mRNAs all encode transcription factors that are hubs in regulatory networks and are also multiply regulated at the level of transcription. Thus, another force driving the evolution of mRNAs as targets of sRNAs may be the need for nuanced modulation of key regulators in response to a wide range of environmental signals.
Mechanistic constraints
Evolution of sRNA–mRNA pairs necessarily is also impacted by mechanistic constraints. On the mRNA side, while secondary structures typically build up in coding sequences, many bacterial mRNAs retain a relaxed structure that facilitates ribosome binding within the first 40–50 bases of the coding sequences (Kudla et al.2009). This relative lack of secondary structure simultaneously would allow sRNA base pairing with the mRNA. Targeting of an mRNA by an sRNA would further reinforce the constraints to maintain an unstructured region. It is also worth noting that the GA-rich sequences associated with ribosome-binding sites can conform to the ARN motif bound by Hfq and also base pair with C- and U-rich motifs found in many sRNAs, particularly in St. aureus (Geissmann et al.2009). In addition to these general features, mRNAs that have sequences promoting translational pausing, for example, allowing the mRNA to be transferred to the signal recognition particle during the translation of a secreted or membrane protein, might be particularly good targets of sRNA regulation. The observation that introduction of a site that reduces translation was required for optimal repression by SgrS (Kawamoto et al.2005) provides a plausible explanation for why so many membrane proteins are the targets of sRNA regulation.
On the sRNA side, there is a mosaic pattern of constraints (Shabalina, Ogurtsov and Spiridonov 2006; Shabalina, Spiridonov and Kashina 2013), where short stretches of intramolecular (sRNA–sRNA) and intermolecular (sRNA–mRNA and sRNA–protein) interactions can alternate. This feature permits simple contacts with many different mRNAs (Fig. 1). The flexibility also allows for further elaborations such as different regulatory outcomes for individual targets (Figueroa-Bossi et al.2009; Bossi et al.2012; Salvail et al.2013; Feng et al.2015), base pairing with an anti-sRNA mentioned above (Tree et al.2014; Miyakoshi, Chao and Vogel 2015a, in press) or binding by other proteins in addition to or instead of Hfq (Jørgensen et al.2013).
Overall, the structural flexibility in sRNAs facilitates the emergence of complex regulatory networks as well as rapid evolution of connections within the networks. Regions of base pairing can readily evolve depending on the physiological constraints. The MicL RNA, which has only a single known target and shares extensive complementarity with this one target (Guo et al.2014) (Fig. S2, Supporting Information), likely is on the young side of the evolutionary spectrum, while Spot 42, which has a broad distribution and targets many mRNAs with multiple seed regions (Beisel and Storz 2011) (Fig. 1), can be considered an older regulator from an evolutionary standpoint.
Interesting questions that arise are which mRNAs are the ‘founder’ targets for a particular sRNA in an organism and is there a correlation between target age and the strength of regulation. In addition, for mRNAs regulated by multiple sRNAs, which sRNA came first? The recent analysis of the evolutionary ages of sRNAs and their mRNA targets showed that for several of the more broadly distributed sRNAs such as Spot 42, the sRNAs were evolutionarily older than their binding sites on mRNA targets (Peer and Margalit 2014). The earliest interactions of Spot42 involved the gltA, sthA and xylF mRNAs, while base pairing with galK and nanC mRNAs evolved later. For a few mRNAs, an sRNA-binding site may have co-appeared with or evolutionarily preceded sRNAs. However, it is likely that for most mRNAs, the establishment of the founder interactions forced selective pressure on the sRNAs and additional targets were acquired by fitting a binding site to the active accessible region of the sRNA. Differences between the evolutionary ages of target mRNAs and the sRNA-binding sites embedded in them highlight the importance of evolutionary analysis at nucleotide resolution.
BROADER PERSPECTIVE
Eukaryotic miRNAs are similar to trans-encoded base-pairing sRNAs in bacteria in their ability to modulate the translation and stability of multiple mRNAs via limited complementarity to their targets (reviewed in Gottesman 2005). The evolutionary forces shaping these two classes of regulatory RNAs can similarly be compared. The highly conserved secondary structures of both sRNAs and miRNAs dictate the mechanisms for generating the transcripts. The RNA regulators also all form stable paired hairpins alternated with single-stranded nucleotides that are under selective pressure owing to their involvement in intermolecular interactions (Shabalina, Spiridonov and Kashina 2013). miRNAs, like sRNAs, can act on multiple target mRNAs with variable levels of complementarity, allowing fine-tuned regulation based on the extent of complementarity (Shabalina, Spiridonov and Kashina 2013). Another shared feature between sRNAs and miRNAs is the predominantly relaxed secondary structure on their mRNA target sites and, accordingly, high accessibility of these regions for base pairing. One difference between bacterial sRNAs and eukaryotic miRNAs is the proteins required for the RNAs to carry out their regulatory functions. However, it is intriguing that the protein machinery of eukaryotic RNA interference seems to have been pieced together from ancestral archaeal, bacterial and phage proteins involved in DNA repair and RNA processing (reviewed in Shabalina and Koonin 2008), while Hfq is an ortholog of Sm and Lsm proteins found in eukaryotes (reviewed in Wilusz and Wilusz 2013). It is likely that further comparisons of sRNAs and miRNAs as well as their associated proteins will advance our understanding of the evolution of these regulatory RNAs.
We anticipate that a deeper understanding of sRNA–mRNA target evolution will also have practical consequences. Programs that incorporate parameters based on features discussed here are already improving the prediction of mRNA target sites (Wright et al.2013). A better appreciation of the constraints on both the sRNAs and the mRNAs undoubtedly will allow for the design of more effective synthetic base-pairing sRNAs, which, until now, frequently have had smaller effects than expected or desired (reviewed in Kang et al.2014). Finally, knowledge of sRNA evolution, combined with the advantages of short replication times and easy manipulation, should make bacteria an ideal system in which to carry out experimental evolution of regulatory sRNAs to further study the processes and forces shaping these molecules.
We thank S. Gottesman, A. Kouse, K. Papenfort and the reviewers for comments.
FUNDING
This work was supported by the Intramural Research Programs of the National Library of Medicine (S.A.S.) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (G.S.).
Conflict of interest. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}