





Transfer RNA is an essential adapter molecule that is found across all three domains of life. The primary role of transfer RNA resides in its critical involvement in the accurate translation of messenger RNA codons during protein synthesis and, therefore, ultimately in the determination of cellular gene expression. This review aims to bring together the results of intensive investigations into the synthesis, maturation, modification, aminoacylation, editing and recycling of bacterial transfer RNAs. Codon recognition at the ribosome as well as the ever-increasing number of alternative roles for transfer RNA outside of translation will be discussed in the specific context of bacterial cells.
INTRODUCTION TO tRNA
Transfer ribonucleic acid (tRNA) is an essential component of the cellular protein synthesis machinery that was found at the origin of life (Altman 1978; Söll and Abelson 1979; Söll and RajBhandary 1995). Primary research on tRNA can be dated back to the early 1950s when Hoagland et al. first determined the presence of an enzyme in the pH 5 insoluble fraction of a rat liver extract that could activate amino acids in an ATP-dependent manner with the concurrent production of aminoacyl-adenylates (Hoagland, Keller and Zamecnik 1956; Hoagland, Zamecnik and Stephenson 1957; Hoagland et al.1958). After this initial finding, further work in the Zamecnik laboratory demonstrated that the amino acid acceptor molecule in the extract was in fact tRNA (reviewed in RajBhandary and Kohrer 2006). The first sequence of a tRNA molecule was made available in 1965 after extensive research in the yeast model system (Holley 1965; Holley et al.1965). Since then, enhancement in column chromatography and gel electrophoresis methodologies has resulted in rapid progress in both purification and sequence determination of individual cellular tRNAs (Cherayil and Bock 1965; Gillam et al.1967; Holladay, Pearson and Kelmers 1971; Pearson, Weiss and Kelmers 1971;
Ikemura and Dahlberg 1973). Functionally, each tRNA molecule can be viewed as a specific adaptor that ultimately results in the productive decoding of messenger RNA to protein. This is achieved by transportation of appropriate amino acids to an elongating polypeptide chain that is associated with the ribosome (Fig. 1) (Hopper and Phizicky 2003).
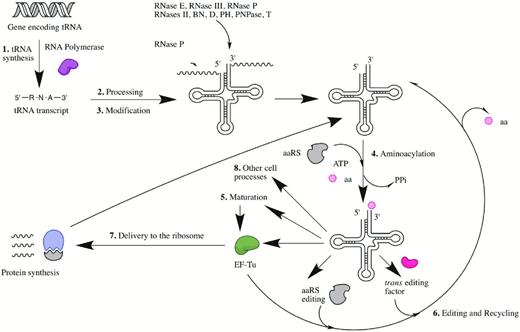
The typical life cycle of a bacterial transfer RNA. The numbering system corresponds to the specific sections of this review. Abbreviations include aminoacyl-tRNA synthetase (aaRS), inorganic pyrophosphate (PPi), amino acid (aa) and elongation factor-TU (EF-Tu).
Before accurate recognition by the aminoacyl-tRNA synthetases (aaRSs) and other components of the protein synthesis machinery can occur, each tRNA must first undergo a specific maturation process. Maturation ensures that the structural integrity and composition of the final tRNA molecule is conductive to its overall function. In all kingdoms of life, maturation of tRNA precursors is comprised of multiple steps. Prior to aminoacylation of the 3′ end by the aaRSs, each synthesized tRNA precursor transcript must be efficiently processed at both the 5′ and the 3′ ends followed, in most cases, by additional modifications at specific residues within the structure (Grosjean 2009). Following these processes, mature aminoacylated tRNA molecules will either proceed directly into protein synthesis or a non-coding pathway such as those concerned with peptidoglycan biosynthesis, antibiotic biosynthesis and antibiotic resistance (Shepherd and Ibba 2013b). In a select number of cases, the aminoacylated tRNA molecule will be required to undergo additional maturation steps prior to participation in these pathways (Blanquet et al.2003). This review will focus specifically on bringing together the key findings that have enhanced our understanding of the processes involved in the production of mature functional bacterial tRNA molecules and their interaction with the ribosome and other biosynthetic pathways.
Synthesis of tRNA
In bacteria, the genetic material encoding tRNA is most frequently found in clusters on the chromosome and the presence of multiple gene copies for a single species is common. To date, three different tRNA-encoding transcription units have been described in the literature encompassing those that just contain tRNA genes, those that contain tRNA genes in the spacer regions of ribosomal RNA operons and tRNA operons that also include specific protein-encoding genes such as elongation factor Tu (EF-Tu) (Toh et al.2001). These genetic structures enable RNA polymerase-mediated transcription of a tRNA from a single promoter into precursors that are typically longer than the final mature molecule (Altman 1975). Whilst they contain the information required for constitution of a functionally unique cellular molecule, in structural terms the genes encoding for transfer RNA are often small ranging from only 75 to 90 base pairs in length. The overall nucleotide sequence varies significantly between each tRNA gene but there are always either four or five inverted repeats that are responsible for formation of the stem-loop elements found within each cloverleaf structure (Altman 1978; Fournier and Ozeki 1985). In bacteria, including Escherichia coli, the tRNA gene often encodes the critical 3′ CCA sequence. This removes the requirement seen in eukaryotic systems whereby this feature must be specifically added to the molecule post-transcriptionally. Aside from all of these tertiary structure-encoding components, tRNA genes also contain specific recognition elements that are necessary for recruitment of enzymes involved in precursor maturation, protein synthesis and, in some cases, other non-translational pathways.
In terms of the necessity of specific promoter elements, it has been demonstrated that the upstream regions of both E. coli and Mycobacterium capricolum tRNA transcription units contain the consensus regions of typical bacterial promoters including the −35 and −10 elements. In addition, the spacing between these elements is consistent with that found for other bacterial promoters, ranging from 15 to 18 nucleotides (Söll and RajBhandary 1995). A conserved domain consisting of a 7 base pair sequence that is GC rich and associated with the stringent control response has been identified in the promoter region of all E. coli tRNA genes (Ikemura and Dahlberg 1973a,b). When focusing on termination of tRNA transcription, DNA sequence analysis has determined that, in both E. coli and M. capricolum, there are rho-independent terminator structures that consist of a dyad symmetrical sequence and T-residues downstream of most of the tRNA genes (Söll and RajBhandary 1995). However, it is thought that transcription of some tRNA genes, such as tRNATyr in E. coli, may require rho-dependent termination. Further research is required to determine the specific details of the transcriptional initiation and termination processes that lead to the successful generation of immature tRNA precursor molecules in different bacteria.
Processing of tRNA precursors
Following their transcription, tRNA gene products often have extensions at both the 5′ and the 3′ ends. In addition, many bacterial transcripts are polycistronic in nature, containing more than one tRNA species (Li and Deutscher 2002; Ow and Kushner 2002). This results in the requirement for processing, which proceeds by a series of endonucleolytic cleavage events (Gegenheimer and Apirion 1981; Grosjean 2009). Substantial amounts of intact tRNA precursor transcripts have not been observed in wild-type bacterial cells grown under normal conditions. This suggests that all of these endonucleolytic tRNA precursor processing events happen either during or immediately after transcription (Altman and Smith 1971).
Across all domains of life, essential 5′ tRNA transcript processing is carried out by a ribonucleoprotein called RNase P (reviewed in detail in Altman and Kirsebom 1999). To date, the only known exception to the requirement for RNase P activity is Nanoarchaeum equitans, which is presumed to be able to produce leaderless tRNA transcripts (Randau and Soll 2008). In bacteria RNase P was initially found to play a part in tRNA processing when it was shown to specifically remove a 41 base pair fragment from the 5′ end of E. coli tRNATyr leaving the 3′ end intact (Altman 1971; Altman and Smith 1971). Subsequent analysis of tRNA precursors accumulated in a temperature-sensitive RNase P mutant strain clarified the essential role of the enzyme in maturation of the 5′ end of all E. coli tRNA species (Altman 1975). With a few exceptions (Gupta 1984; Lee et al.1987), maturation of tRNA by RNase P processing results in the generation of a precursor molecule with a monophosphate at the 5′ end and a terminal 2′-3′-cis glycol.
In structural terms, RNase P is defined as a ribozyme and it has been demonstrated to have surprisingly versatile substrate specificity (reviewed in Kirsebom 2002; Harris and Christian 2003; Kazantsev and Pace 2006; Torres-Larios et al.2006). Unlike many other naturally large ribozymes, RNase P can carry out multiple turnovers in vivo, similar to the ribosome. Whilst RNase P activity is conserved across all domains of life, the protein composition of the enzyme is known to vary depending upon whether it is bacterial, archaeal or eukaryotic in nature. In bacteria, RNase P is comprised of one small basic protein that is approximately 18 000 Daltons in size (Stark et al.1978; Guthrie and Atchison 1980). In archaea there are often 4 to 5 protein subunits and in eukarya the number of protein subunits can be as many as 10 (Hartmann and Hartmann 2003). The catalytic activity of the 375–377 nucleotide RNA component of RNase P was first described in 1983 and, in bacteria, this activity can be achieved even in the absence of the protein component of the enzyme (Stark et al.1978; Guthrie and Atchison 1980; Guerrier-Takada et al.1983). Extensive biochemical characterization has indicated that RNase P catalysis proceeds by an SN2 nucleophilic substitution mechanism (Smith and Pace 1993; Beebe and Fierke 1994). More specifically, magnesium ion-hydrate coordinate complexes surround the target phosphodiester bond of the tRNA precursor molecule resulting in the formation of a trigonal bipyramidal transition state (Persson, Cuzic and Hartmann 2003; Cassano, Anderson and Harris 2004). This subsequently enables the 2′-hydroxyl group of the precursor tRNA to facilitate protonation of the leaving 3′oxygen, thus finalizing removal of the leader sequence and signaling completion of the reaction (reviewed in detail in Kazantsev and Pace 2006). Three specific universal features of the tRNA substrate have been implicated in accurate recognition by the RNA component of bacterial RNase P. These are the TΨC loop, the acceptor stem and the CCA end (Grosjean 2009).
Determination of the X-ray crystal structures of some examples of RNase P has resulted in the conclusion that the bacterial versions of the enzyme can be classified into two groups (Krasilnikov et al.2003, 2004; Kazantsev et al.2005; Torres-Larios et al.2005; Kazantsev and Pace 2006; Baird et al.2007). Type A, or ancestral type RNase P, is widely dispersed throughout many of the phylogenetic branches of the bacterial kingdom. In contrast, type B or Bacillus type RNase P is restricted to Gram-positive bacteria (Pace and Brown 1995; Haas and Brown 1998; Baird et al.2007). Whilst there are differences that focus around certain peripheral elements of the structures of type A and B RNase P proteins, both groups have a common catalytic core and tertiary fold (reviewed in detail in Kazantsev and Pace 2006; Grosjean 2009). Within the catalytic core, there are a series of highly conserved residues. These residues occur within five separate regions of sequence conservation encoding parts of the overall structure that are involved in helix formation or the establishment of critical helical contacts. Structural studies on the protein components of type A and B RNase P have demonstrated that they share both structural and functional similarity. This is further supported by in vitro and in vivo studies that demonstrate efficient functional complementation of the protein component of a type A RNase P with that of a type B RNase P and vice versa (Waugh and Pace 1990; Wegscheid, Condon and Hartmann 2006). The protein components of RNase P are known to complete the typical ribonucleoprotein structure by folding into an α-β sandwich with a highly conserved electrostatic surface (Niranjanakumari et al.1998; Stams et al.1998; Spitzfaden et al.2000; Kazantsev et al.2003). A combination of mutational and biochemical data has indicated that the unusual left-handed β-α- β crossover formed by the protein component produces a positively charged face that binds to the RNA component of the enzyme (Stams et al.1998). Whilst the protein component of bacterial RNase P is not required for catalytic activity in vitro, its overall importance has been linked to acceleration of the tRNA processing reaction. This suggests that it has a role in stabilization of the RNA component and possibly the reaction intermediates in addition to a specific sequence in the precursor RNA in the C5 component of RNase P (Walker and Engelke 2006; Smith, Hsieh and Fierke 2007; Guenther et al.2013).
When the tRNA precursor molecule contains one tRNA sequence, RNase P processing may be the only requirement for efficient maturation of the 5′ end. If the tRNA transcript contains more than one tRNA sequence other endoribonucleases, such as RNase E and RNase III, may be required to generate smaller precursor molecules. These smaller precursors can then be much more efficiently processed by RNase P to generate a tRNA molecule with a mature 5′ end. Subsequent processing by exoribonucleases, including RNases II, BN, D, PH, PNPase and T, then account for generation of the mature 3′ end (Sakano and Shimura 1975, 1978; Schedl, Roberts and Primakoff 1976). Recently, RNase P-mediated separation of some E. coli transcripts including valV valW, leuQ leuP leuV and secG leuU has been demonstrated (Mohanty and Kushner 2007, 2008). One particular study reports RNase P-mediated separation of three precursor transcripts in E. coli that encode for the seven valine tRNA molecules found within the cell (Agrawal, Mohanty and Kushner 2014). Interestingly, specific focus on the ValU and LysT transcripts has highlighted a previously uncharacterized role for RNase P in 3′ end processing. Unusually in this case, RNase P processing proceeds in the 3′ to 5′ direction generating one tRNA precursor at a time with activity being initiated by specific endonucleolytic removal of the Rho-independent transcription terminators (Agrawal, Mohanty and Kushner 2014).
Processing at the 3′ end of the tRNA precursor is typically initiated by an endonucleolytic cleavage event that takes place downstream of the critical CCA terminus (Sekiya et al.1979; Morl and Marchfelder 2001). This cleavage event often happens before RNase P processing of the 5′ end and final exonucleolytic trimming of the 3′ end. Endonucleolytic processing at the 3′ end of the precursor is carried out by a combination of two enzymes termed RNase E and RNase III (Ray and Apirion 1981; Apirion and Miczak 1993). Like RNase P, these enzymes can be found associated with the inner membrane of the cell (Miczak, Srivastava and Apirion 1991). In addition, fundamental aspects of primary and secondary RNA structure have been shown to determine the broad substrate specificities of both RNase E and RNase III. More specifically, RNase E has been demonstrated to cleave sequences that are both single stranded and AU-rich whilst RNase III targets double-stranded RNA sequences (Robertson, Webster and Zinder 1968; McDowall, Lin-Chao and Cohen 1994; Nicholson 1999).
RNase E has a molecular weight of approximately 70 kDa and was first identified as an important enzyme in the separation of precursor ribosomal RNA from larger transcripts (Ghora and Apirion 1978). Subsequent work demonstrated that certain tRNA precursors accumulate in E. coli cells that are deficient in RNase E. One of these rRNA-independently transcribed precursors is known to encode tRNALeu and tRNAHis. Since this precursor cannot be processed in the absence of RNase E, this finding strongly supports a critical role for the enzyme in the initial stages of tRNA processing (Ray and Apirion 1981). Extensive biochemical characterization has concluded that RNase E is primarily involved in the cleavage of multimeric and polycistronic tRNA precursors in the spacer region and in the processing of 3′ trailer regions (Li and Deutscher 2002; Ow and Kushner 2002). However, studies using tRNATyrSu3 precursors that lack a 3′ RNase E cleavage site have indicated that this enzyme can also cleave the 5′ leader region of certain tRNA precursor transcripts (Soderbom, Svard and Kirsebom 2005). RNase E activity is critical to initial tRNA precursor processing and results in the provision of accessible substrates for RNase P, which is inhibited by long trailing regions at the 3′ end of transcripts and, subsequently, the 3′ exonucleases (Li and Deutscher 2002).
In contrast to what has been shown for RNase E, RNase III has been reported to have a less significant role in polycistronic tRNA transcript processing. In terms of overall structure, RNase III is comprised of two identical polypeptide subunits that have a molecular weight of 25 kDa (Dunn 1976). Since RNase III was first described to be critical for cleavage of the large 30S polycistronic ribosomal RNA precursor, it was initially considered to have a role in the processing of tRNA transcripts that are co-transcribed with rRNA (Dunn and Studier 1973; Nikolaev, Silengo and Schlessinger 1973). This was later confirmed by the finding that, in vitro, E. coli 30S rRNA transcripts that include tRNAGlu or the tRNAIle-tRNAAla dimer in the spacer regions are cleaved by RNase III to produce intermediate tRNA precursors that are then suitable for RNase P processing (Lund and Dahlberg 1977). However, studies using mutants that are compromised in RNase III activity demonstrated that this enzyme only has a minor role in processing (Gegenheimer and Apirion 1981). In fact, early tRNA maturation only becomes severely compromised if this mutation is introduced into a strain that is also deficient in RNase P and RNase E activities, suggesting the latter two proteins have the primary roles in this process (Plautz and Apirion 1981).
Whilst the activities of RNase E and RNase III generate precursors that allow RNase P to complete 5′ end tRNA transcript maturation, the endonucleolytic cleavage events that take place at the 3′ end of the transcript usually leave a stretch of extra residues that must be removed by exoribonucleases (Schedl, Roberts and Primakoff 1976). Two types of intermediate tRNA precursors have been identified in bacterial cells and they differ in terms of their requirements for exonucleolytic processing at the 3′ end. In the most common type of precursor, the CCA end of the tRNA is already present and is followed by a series of extra nucleotides. Final maturation of the 3′ end of this precursor requires a nuclease that can trim right up to the critical CCA end without disrupting it. This is supported by mutagenesis studies that directly demonstrate that the CCA repair enzyme, nucleotidyltransferase, is not involved in the maturation of this type of precursor (Deutscher, Lin and Evans 1977). In the second type of precursor, which is rare in bacterial cells but common in eukaryotes, other residues substitute the CCA end. In this case, 3′ processing requires a nuclease that will remove only the substituting residues leaving a product that can be repaired by nucleotidyltransferase such that it has a CCA terminus at the completion of maturation.
Originally, it was thought that RNase II, a well-known 3′ exonuclease, was the sole enzyme involved in 3′ exonucleolytic processing of tRNA transcripts. Whilst this enzyme has been shown in vitro to remove extra nucleotides at the 3′ end of an E. coli tRNATyr precursor, the fact that it is processive in nature and cannot stop at the CCA end has led to searches for another more specific exonuclease (Cudny and Deutscher 1980). A more likely candidate was found with the discovery of a monomeric 38 kDa protein termed RNase D. RNase D is specific for tRNA precursors that have the 3′ CCA end. This enzyme specifically removes all extra residues that come before the CCA end in a non-processive manner and, in doing so, generates a mature precursor tRNA molecule that has amino acid accepting capability (Cudny and Deutscher 1980). Perhaps more importantly, RNase D cannot hydrolyze the CCA end and the rate of its hydrolytic activity has been shown to reduce by 30-fold when it reaches this sequence (Ghosh and Deutscher 1978; Cudny and Deutscher 1980).
In the case of the second type of tRNA precursor, which has an incomplete or absent CCA end, another 3′ processing exonuclease was identified termed RNase BN. RNase BN has a molecular weight of approximately 35 kDa and, unlike RNase D, its specificity for tRNA precursors is not determined by the presence of an intact 3′ CCA end (Seidman et al.1975; Schmidt and McClain 1978; Asha et al.1983; Asha and Deutscher 1983). Other exonucleases that have been shown to play a role in 3′ tRNA processing include RNases PH, T and PNPase (described in some detail in Söll and RajBhandary 1995). Studies attempting to fully characterize all of the separate components of the processing machinery have shown that the role and order of activity of the different enzymes is entirely dependent on the specific tRNA precursor in question (Li and Deutscher 1996). The ultimate conclusion of all of the processes discussed in this section is the generation of a mature tRNA molecule, which can then successfully progress into the nucleotide modification and aminoacylation pathways.
Nucleotide modification of tRNA
Post-transcriptional modification has been shown to expand the chemical diversity and fine-tune the functionality of tRNA molecules across all three domains of life (see Table 1; reviewed extensively in Bjork et al.1987; Söll and RajBhandary 1995; Bjork et al.1999; Gustilo, Vendeix and Agris 2008; Grosjean 2009; Motorin and Helm 2010; El Yacoubi, Bailly and de Crecy-Lagard 2012). To date, more than 100 different chemical modifications have been identified, all of which generate various derivatives of the four common nucleosides adenosine, guanosine, cytidine and uridine. Therefore, it is no surprise that between 1 and 10% of the genes within a specific genome encode for proteins that are involved in the tRNA modification process (Motorin and Helm 2010; El Yacoubi, Bailly and de Crecy-Lagard 2012). Whilst the vast majority of tRNA modifications were discovered in the 1970s, specific adaptation of both mass spectrometry and deep-sequencing techniques are responsible for significant recent advances in our understanding of the critical functional roles these modifications play within the cell (Chan et al.2010; Findeiss et al.2011; Globisch et al.2011; Yi and Pan 2011; Wetzel and Limbach 2012). Modified nucleosides can be found at various locations in the tRNA cloverleaf and can increase the surface area of the molecule by as much as 20% (Bjork et al.1987) (Fig. 2). Notably, the highest frequency and diversity of modifications are concentrated at positions 34 and 37 in the anticodon stem loop (Grosjean 2009).
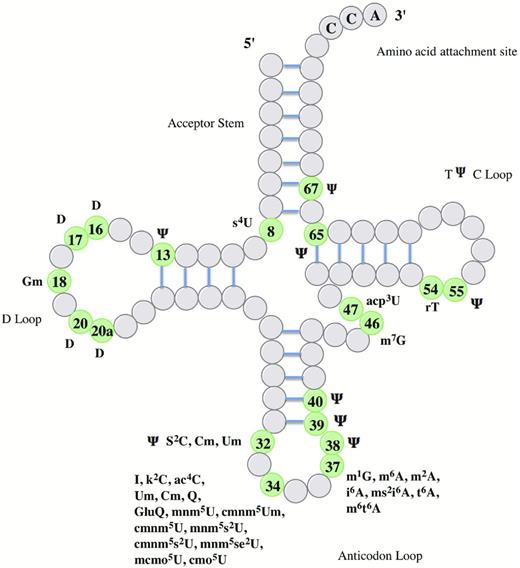
Known modifications and their positions in E. coli tRNA. The information presented in this figure was adapted from El Yacoubi, Bailly and de Crecy-Lagard, (2012). Abbreviations for specific groups include m-, c-, n-, o-, t-, i-, K-, r- and s- for methyl, carbon, amino, oxy, threonine, isopentyl, lysine, ribose and thio groups, respectively. Other abbreviations include acp: 3-amino-3-carboxypropyl, D: dihydrouridine, I: inosine, Ψ: pseudouridine, Q: queuosine. Other capital letters refer to the appropriate bases uracil, cytosine, guanine and adenine. An index and an exponent indicate the number and the position of the substitution, for example 6-dimethyladenosine is written as m62A.
Summary of the potential modifications, their positions and the enzymes required to make them in E. coli tRNA. The information presented in this table was obtained from El Yacoubi, Bailly and de Crecy-Lagard (2012).
| Position | Modification | Enzyme |
|---|---|---|
| 8 | S4U | IscS, Thil |
| 13 | Ψ | TruD |
| 16 | D | Dus A, B, C |
| 17 | D | Dus A, B, C |
| 18 | Gm | TrmH |
| 20 | D | Dus A, B, C |
| 20a | D | Dus A, B, C |
| 32 | S2C Cm, Um Ψ | IscS, TtcA Trml RluA |
| 34 | I k2C ac4C Um, Cm Q, GluQ mnm5U cmnm5Um cmnm5U mnm5s2U cmnm5s2U mnm5se2U mcmo5U cmo5U | TadA TilS TmcA TrmL FolE, QueDCEF, Tgt, QueA and G+YadB MnmC1 and C2+TrmL MnmE, GidA ? ? ? CmoA and B AroBDEKLAC |
| 37 | m1G m6A m2A i6A ms2i6A t6A m6t6A | TrmD TrmN6? MiaA MiaB, IscS, IscU TsaC, D, B and E TsaA |
| 38 | Ψ | TruA |
| 39 | Ψ | TruA |
| 40 | Ψ | TruA |
| 46 | m7G | TrmB |
| 47 | acp3U | ? |
| 54 | rT | TrmA |
| 55 | Ψ | TruB |
| 65 | Ψ | TruC |
| 67 | Ψ | TruC |
| Position | Modification | Enzyme |
|---|---|---|
| 8 | S4U | IscS, Thil |
| 13 | Ψ | TruD |
| 16 | D | Dus A, B, C |
| 17 | D | Dus A, B, C |
| 18 | Gm | TrmH |
| 20 | D | Dus A, B, C |
| 20a | D | Dus A, B, C |
| 32 | S2C Cm, Um Ψ | IscS, TtcA Trml RluA |
| 34 | I k2C ac4C Um, Cm Q, GluQ mnm5U cmnm5Um cmnm5U mnm5s2U cmnm5s2U mnm5se2U mcmo5U cmo5U | TadA TilS TmcA TrmL FolE, QueDCEF, Tgt, QueA and G+YadB MnmC1 and C2+TrmL MnmE, GidA ? ? ? CmoA and B AroBDEKLAC |
| 37 | m1G m6A m2A i6A ms2i6A t6A m6t6A | TrmD TrmN6? MiaA MiaB, IscS, IscU TsaC, D, B and E TsaA |
| 38 | Ψ | TruA |
| 39 | Ψ | TruA |
| 40 | Ψ | TruA |
| 46 | m7G | TrmB |
| 47 | acp3U | ? |
| 54 | rT | TrmA |
| 55 | Ψ | TruB |
| 65 | Ψ | TruC |
| 67 | Ψ | TruC |
Summary of the potential modifications, their positions and the enzymes required to make them in E. coli tRNA. The information presented in this table was obtained from El Yacoubi, Bailly and de Crecy-Lagard (2012).
| Position | Modification | Enzyme |
|---|---|---|
| 8 | S4U | IscS, Thil |
| 13 | Ψ | TruD |
| 16 | D | Dus A, B, C |
| 17 | D | Dus A, B, C |
| 18 | Gm | TrmH |
| 20 | D | Dus A, B, C |
| 20a | D | Dus A, B, C |
| 32 | S2C Cm, Um Ψ | IscS, TtcA Trml RluA |
| 34 | I k2C ac4C Um, Cm Q, GluQ mnm5U cmnm5Um cmnm5U mnm5s2U cmnm5s2U mnm5se2U mcmo5U cmo5U | TadA TilS TmcA TrmL FolE, QueDCEF, Tgt, QueA and G+YadB MnmC1 and C2+TrmL MnmE, GidA ? ? ? CmoA and B AroBDEKLAC |
| 37 | m1G m6A m2A i6A ms2i6A t6A m6t6A | TrmD TrmN6? MiaA MiaB, IscS, IscU TsaC, D, B and E TsaA |
| 38 | Ψ | TruA |
| 39 | Ψ | TruA |
| 40 | Ψ | TruA |
| 46 | m7G | TrmB |
| 47 | acp3U | ? |
| 54 | rT | TrmA |
| 55 | Ψ | TruB |
| 65 | Ψ | TruC |
| 67 | Ψ | TruC |
| Position | Modification | Enzyme |
|---|---|---|
| 8 | S4U | IscS, Thil |
| 13 | Ψ | TruD |
| 16 | D | Dus A, B, C |
| 17 | D | Dus A, B, C |
| 18 | Gm | TrmH |
| 20 | D | Dus A, B, C |
| 20a | D | Dus A, B, C |
| 32 | S2C Cm, Um Ψ | IscS, TtcA Trml RluA |
| 34 | I k2C ac4C Um, Cm Q, GluQ mnm5U cmnm5Um cmnm5U mnm5s2U cmnm5s2U mnm5se2U mcmo5U cmo5U | TadA TilS TmcA TrmL FolE, QueDCEF, Tgt, QueA and G+YadB MnmC1 and C2+TrmL MnmE, GidA ? ? ? CmoA and B AroBDEKLAC |
| 37 | m1G m6A m2A i6A ms2i6A t6A m6t6A | TrmD TrmN6? MiaA MiaB, IscS, IscU TsaC, D, B and E TsaA |
| 38 | Ψ | TruA |
| 39 | Ψ | TruA |
| 40 | Ψ | TruA |
| 46 | m7G | TrmB |
| 47 | acp3U | ? |
| 54 | rT | TrmA |
| 55 | Ψ | TruB |
| 65 | Ψ | TruC |
| 67 | Ψ | TruC |
Modifications at specific locations within the cloverleaf structure are associated with conferring different functions upon the tRNA molecule (Bjork et al.1987). Those found at or around the 3′ side of anticodon loop position 37 are associated with maintaining both the efficiency and fidelity of translation. This is achieved by altering the sensitivity of the tRNA to the reading context of the ribosome-bound messenger RNA, thereby ensuring that the correct reading frame is supported. More specifically, ms2 and ms2i6 tRNA modifications at position 37 are frequent when the base at position 36 is either adenine or uracil. The nature of these modifications has been directly correlated with stabilization of interactions between the codon and the anticodon particularly at the first base pair position (Bjork et al.1987; El Yacoubi, Bailly and de Crecy-Lagard 2012). This is particularly critical for A:U and U:A. codon: anticodon interactions where the modification prevents the formation of intraloop base pairs within the anticodon stem loop structure and reduces the potential for frameshifting (Agris 2008; Grosjean 2009). In Salmonella typhimurium miaA1 (or miaE) mutants, the tRNA pool is missing the ms2i6o6 modification at adenine 37. Subsequently, this strain has been found to have a significantly reduced growth rate, altered regulation of various biosynthetic operons and a reduced rate of polypeptide chain elongation (Ericson and Bjork 1986). Complementary to these findings, tRNAs across all kingdoms of life that are involved in the recognition of triplet codons that begin with adenine typically have a t6A modification at position 37 (Bjork et al.1987). Escherichia coli tRNAIle that does not have this modification has been shown to decode less efficiently in vitro. tRNAs that recognize codons beginning with cytosine or guanine often have a more simple m1G modification at position 37. A mutant of S. typhimurium that can produce tRNAs where m1G is present at 30°C but not at 41°C undergoes a higher rate of frameshifting when it is presented with the challenge of decoding sequences containing runs of cytosines when it is grown at the higher temperature (Bjork et al.1987, 1999). These findings provide further evidence for the role of tRNA modifications at and around this position in the maintenance of translational efficiency (Miller, Hussain and Schweizer 1976).
Modifications at position 34 in the tRNA cloverleaf have been shown to be involved in maintaining the fidelity of translation by influencing codon choice and discrimination. This is particularly important when two amino acids are defined by codons that differ by only one nucleotide in the triplet. For example, methionine is encoded by AUG and isoleucine is encoded by AUA, AUU and AUC. In bacteria, accurate recognition of the AUA codon requires tRNAIle that has a k2C modification at position 34. This particular modification is critical for changing the amino acid specificity and the anticodon identity of this tRNA molecule from methionine to isoleucine (Soma et al.2003). Conversely, E. coli elongator tRNAMet has an ac4C modification at the same position, which compromises ribosome binding but provides a huge advantage to the cell by preventing misreading of the AUA isoleucine codon in vitro (Stern and Schulman 1978). Similarly, modification of position 34 with Cm or Q has been reported to enable tRNATrp or tRNATyr, respectively, to efficiently discriminate tryptophan (UGG) or tyrosine codons (UAU and UAC) from stop codons during translation (Nakamura and Ito 2011; El Yacoubi, Bailly and de Crecy-Lagard 2012). Hydrolytic deamination of the sixth carbon of adenosine 34 to inosine is a more unusual form of modification that enables a single isoacceptor of E. coli tRNAArg (ACG) to alternatively pair with U, C or A nucleotides in the third position of the anticodon and thus successfully decode all three arginine codons (CGU, CGC and CGA) (Crick 1966; Keller et al.1999). Modifications in the anticodon loop other than at positions 34 and 37 have also been shown to influence both the efficiency and fidelity of translation (Bjork et al.1987).
Modified nucleotides outside of the anticodon region are thought to be involved in stabilization of the overall structure of the tRNA molecule (Bjork et al.1987). In addition, the overall level of tRNA modification is proposed to serve a regulatory function. Recently, bacterial tRNA modification has been shown to have a role in the host immune response. Fully modified native tRNATyr isolated from E. coli was found to be non-immunostimulatory towards human peripheral blood mononuclear cells. However, unmodified tRNATyr induced interferon alpha production under the same conditions suggesting that tRNA modification may also be a mechanism employed by pathogenic bacteria in host immune invasion (Gehrig et al.2012). Further studies are required to determine the nature and quantity of modifications involved in this process.
Whilst tRNA modifications have a primary role in maintaining the efficiency and fidelity of translation, their importance is generally underappreciated, and it is thought that they may also take on a much more global regulatory role within the cell by providing connections between protein synthesis, metabolism and stress response pathways. This is reinforced by the finding that many tRNA modifications are directly determined by cellular or environmental factors such as growth rate, oxygen levels and nutrient levels. For example, the synthesis of some proteins might be influenced by the proportion of tRNAs within the cell pool that have a particular modification. This is because some tRNA modifications give a bias for recognition of one codon over another. In the case of the DNA damage response in yeast, tRNA methyltransferase 9 has been found to enhance the translation of DNA damage proteins, whose transcripts are overrepresented by specific glutamic acid and arginine codons, by modifying the uridine wobble bases of tRNAGlu (UUC) and tRNAArg (UCU) (Begley et al.2007). In addition, several thermophilic bacteria show an increase in tRNA modification levels with growth temperature (Noon et al.2003) and the proportion of thiolated tRNAs in E. coli increases with growth rate (Emilsson, Naslund and Kurland 1992). These finding suggest a role for tRNA modifications in regulation of stress responses (El Yacoubi, Bailly and de Crecy-Lagard 2012). Whatever their final function may be, fully modified tRNA species produced at this stage are able to proceed into aminoacylation and subsequently enter translation and other biological pathways in the cell.
Aminoacylation
The generation of aminoacylated tRNA is a critical function that is essential for translation as well as a variety of other cellular processes, which will be discussed in the section ‘Other roles of tRNA in the cell’ of this review. Across all three domains of life, formation of an aminoacyl-tRNA is enzymatically a two-stage process that is catalyzed by one of 23 aaRS. In the first stage of the reaction, an amino acid is specifically activated by an aaRS to form an aminoacyl-adenylate. Aminoacyl-adenylate formation proceeds in an adenosine triphosphate (ATP)-dependent manner and results in the subsequent release of inorganic pyrophosphate (PPi). In the second stage of the reaction, the activated amino acid moiety becomes transferred to either the 2′ or the 3′ hydroxyl group of the terminal adenine (A76) that is located at the critical CCA terminus of the tRNA acceptor molecule. This results in the formation of an aminoacyl-ester bond and the reaction concludes with release of the aminoacylated-tRNA species from the synthetase (summarized in Fig. 3) (Ibba and Soll 2000; Reynolds, Lazazzera and Ibba 2010). Since the aaRS and their roles in biology and medicine have recently been reviewed elsewhere (Pang, Poruri and Martinis 2014), the remainder of this section will focus on providing a brief summary of the specific identity elements within tRNA that are required for efficient aminoacylation.
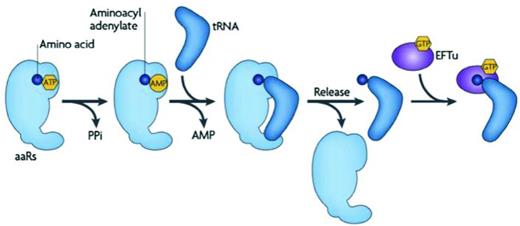
Formation of an aminoacylated-tRNA molecule by an aaRS. An amino acid (blue) is activated by an aaRS to form an aminoacyl-adenylate. This process requires ATP and results in the release of PPi. Following tRNA binding to the aaRS, the activated amino acid is transferred to the 3′ end of the tRNA molecule forming aminoacyl-tRNA with concurrent release of adenosine monophosphate (AMP). The aminoacyl-tRNA product is released from the aaRS and is either subject to binding by EF-Tu for delivery to the ribosome or hijacking by other factors for diversion into biosynthetic pathways outside of translation. Figure reproduced from Reynolds, Lazazzera and Ibba (2010).
Accurate selection of the cognate tRNA from the cellular pool by an aaRS is known to involve a dual discrimination mechanism based upon velocity of the aminoacylation reaction and substrate binding affinity, both of which will only reach optimum levels with the cognate molecule (Yarus and Berg 1967; Ebel et al.1973; Roe, Sirover and Dudock 1973; Lam and Schimmel 1975). It is also known that binding of the cognate tRNA enhances the discriminatory power of some synthetases against non-cognate amino acids (reviewed extensively in Hendrickson and Schimmel 2003). Early research characterizing various aspects of the tRNA cloverleaf structure identified three regions that are important for interaction with and binding to the appropriate aaRS: the acceptor stem, the D loop and the anticodon loop (Schimmel 1977; reviewed extensively in Giege, Sissler and Florentz 1998).
Involvement of the tRNA acceptor stem in recognition by and interaction with the aaRS was first demonstrated using ribonuclease A protection assays and came as no surprise since the activated amino acid is ultimately transferred to the 3′ end of this region (Dickson and Schimmel 1975). More specifically, correct alignment of the 3′ end of the tRNA acceptor stem with the catalytic residues of the enzyme is a requirement for productive aminoacylation (Schimmel 1977). Examples of acceptor stem elements that specifically confer tRNA identity include the G3:U70 wobble in bacterial tRNAAla, which acts as a critical identity element for aminoacylation by AlaRS (Hou and Schimmel 1988), and positions G1, G2, C81 and A82, which have been shown to confer the amino acid accepting specificity of E. coli tRNATyr (Hooper, Russell and Smith 1972; Shimura et al.1972; Smith and Celis 1973). The importance and relevance of specific tRNA acceptor stem elements can also vary between bacterial genera and species. For example, it has recently been shown that Streptococcus pneumoniae AlaRS enzyme is less dependent upon the G3:U70 acceptor stem wobble region for aminoacylation than other bacterial AlaRS proteins (Shepherd and Ibba 2013b, 2014). In addition, the same acceptor stem element can act as an anti-determinant for aminoacylation by other aaRSs. For example, if the G3:U70 element is put into yeast tRNAThr, then it becomes an anti-determinant for aminoacylation by threonyl-tRNA synthetase (Nameki 1995; a full and comprehensive review of tRNA identity elements is given in Giege, Sissler and Florentz 1998).
D-loop residues within tRNA can also act as aminoacylation recognition elements for the aaRSs. This was first demonstrated to be the case for yeast phenylalanyl-tRNA synthetase (PheRS), which can aminoacylate heterologous tRNAPhe species that have a specific sequence of bases in the D-loop that match that of its own tRNA (Dudock et al.1971; Roe, Sirover and Dudock 1973). Strikingly, this region of tRNA structure has also been implicated in editing of misaminoacylated-tRNA species by certain aaRSs (see the Section ‘Editing of aminoacyl-tRNA’ below for more details on the editing process). For example, D-loop residues 16, 20 and 21 in tRNAIle are implicated in the editing of mischarged Val-tRNAIle by isoleucyl-tRNA synthetase (IleRS) (Hale et al.1997; Farrow, Nordin and Schimmel 1999). In addition, D-loop residue 16 in St. pneumoniae tRNAIle was recently shown to determine the aminoacylation capacity of St. pneumoniae IleRS towards both isoleucine and leucine (Shepherd and Ibba 2014).
Whilst the anticodon loop typically comes into close contact with the aaRS in many systems (Schimmel et al.1972; Schoemaker and Schimmel 1974; Schimmel 1977), its importance as a specific identity element is dependent upon the particular tRNA species. For example, early studies in yeast demonstrated that the entire anticodon loop can be removed from tRNAPhe while retaining the capacity for aminoacylation by PheRS (Thiebe, Harbers and Zachau 1972). In contrast, mutation of the third base in the anticodon of E. coli tRNAGly causes a significant decrease in the rate of aminoacylation of this species by glycyl-tRNA synthetase (Carbon and Squires 1971). In some instances, the anticodon loop also confers the amino acid identity of the species. If the second base in E. coli tRNATrp is mutated, then it is readily misaminoacylated with glutamine (Yaniv et al.1974). In addition, modification of either of the first two bases in the anticodon loop of E. coli tRNAfmet inactivates the species entirely (Schulman and Pelka 1977).
After completion of aminoacylation, most charged tRNA species are sequestered by EF-Tu and subsequently proceed into protein synthesis at the ribosome. Alternatively they can enter other specific cellular processes, as discussed below in the Section ‘Other roles of tRNA in the cell’. However, some aminoacylated-tRNA species need to undergo further maturation events prior to participation in these terminal processes as described below.
Maturation of aminoacyl-tRNA
After aminoacylation, most tRNA species are ready to directly participate in protein synthesis at the ribosome or in other cellular pathways. In a relatively small number of cases, aminoacylated-tRNA must first undergo a further maturation step. The most common examples of additional maturation events that take place in bacteria include transamidation, which generates both asparaginylated and glutaminylated tRNA species, production of selenocysteinyl-tRNA and formylation of initiator methionyl-tRNA (summarized in Fig. 4) (Blanquet et al.2003).
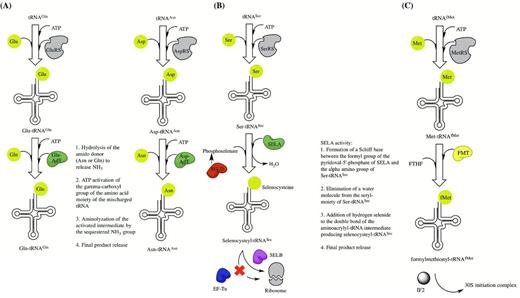
Indirect tRNA aminoacylation pathways. (A) The indirect pathways for generation of Gln-tRNAGln and Asn-tRNAAsn by transamidation. Abbreviations include AdT for amidotransferase. (B) The selenocysteine tRNA modification pathway. (C) The formylation pathway for initiator Met-tRNAfMet. Abbreviations include FMT for methionyl-tRNAfMet transformylase, FTHF for N-10 formyltetrahydrofolate and IF2 for initiation factor 2.
Transamidation is employed by some bacterial species as an indirect mechanism for producing correctly aminoacylated Gln-tRNAGln from Glu-tRNAGln and Asn-tRNAAsn from Asp-tRNAAsn in the absence of an encoded glutaminyl- or asparaginyl-tRNA synthetase, respectively. Transamidation was first identified as a major pathway for generating Gln-tRNAGln in Gram-positive bacteria, cyanobacteria and certain organelles (Wilcox and Nirenberg 1968; Wilcox 1969; Schon, Hottinger and Soll 1988a; Schon et al.1988b). Further studies have since indicated that this pathway, as well as that for Asn-tRNAAsn production, can also be found in some Gram-negative bacteria and archaea (White and Bayley 1972; Strauch, Zalkin and Aronson 1988; Curnow, Ibba and Soll 1996; Gagnon et al.1996; Becker and Kern 1998; Tumbula et al.2000) (reviewed in Blanquet et al.2003). Regardless of bacterial origin, the transamidation reaction can be divided into two steps. Initially, tRNAAsn and tRNAGln are aminoacylated with aspartate and glutamate, respectively. This activity requires either an aspartyl- or a glutamyl-tRNA synthetase that has very poor or no discriminatory power against the non-cognate tRNA but maintains specificity for the cognate amino acid. The second stage of the reaction is then accomplished by an amidotransferase that uses either L-asparagine, in the case of tRNAAsn, or L-glutamate, in the case of tRNAGln, as an amide nitrogen donor to generate the final products (Fig. 4A). The aminoacylated tRNA intermediates produced during the first stage of the reaction typically bind EF-Tu very poorly and so are not efficiently recruited to the ribosome before the next stage of the reaction, which requires the amidotransferase, can proceed. In the case of Asn-tRNAAsn production, it has also been shown that binding of EF-Tu to the mischarged intermediate is prevented by formation of a protective shuttling complex, called the transamidosome, between the non-discriminatory AspRS and the amidotransferase (Bailly et al.2007).
Characterization of the glutamate amidotransferase (GluAdT) from Bacillus subtilis has demonstrated that the protein is produced by expression from three genes, termed gatC, gatA and gatB to produce a final product that is heterotrimeric in nature (Strauch, Zalkin and Aronson 1988; Curnow et al.1997). Since B. subtilis does not have a Gln-tRNA synthetase, transamidation in the presence of a non-discriminatory Glu-tRNA synthetase is the only mechanism for producing correctly acylated Gln-tRNAGln in this bacterium, thus indicating the importance of specific aminoacyl-tRNA maturation events for cell survival (Lapointe, Duplain and Proulx 1986; Schon, Hottinger and Soll 1988a; Chen et al.1990). In many cases, including Deinococcus radidurans, Thermus thermophilus and B. subtilis, GluAdt has been shown to have dual activity in vitro (Curnow et al.1997, 1998; Becker et al.2000b). Therefore, the in vivo specificity of this enzyme against Asp-tRNAAsn or Glu-tRNAGln is entirely dependent on the cellular context, particularly with regard to the presence or absence of the necessary tRNA-synthetase enzymes (Becker et al.1997, 2000a; Curnow et al.1998). Maturation of Glu-tRNAGln and Asp-tRNAAsn to the correctly acylated species is essential for maintaining the fidelity of the genetic code within the bacteria that possess these pathways.
Another form of aminoacyl-tRNA maturation is described by the selenocysteinyl-tRNA pathway, which is found across all three domains of life (reviewed in Commans and Bock 1999). The requirement for this maturation pathway has been found to be most notable in the synthesis of oxidoreductases, which often substitute selenocysteine at cysteine codons because of its lower redox potential (Blanquet et al.2003). The first detection of a protein containing selenocysteine was made in the mid-1970s in a glycine reductase isolated from the anaerobic Gram-positive bacterium, Clostridium sticklandii (Cone et al.1976). Since then the pathway has been well characterized in E. coli, where it was first shown that co-translational insertion of selenocysteine occurs at UGA codons that are present within a specialized mRNA context (Chambers et al.1986; Zinoni et al.1986).
Detailed characterization of the E. coli selenocysteinyl-tRNA pathway has indicated that this bacterium has a specific gene, called selC, which encodes the genetic material required for production of a unique tRNA molecule that is specific for selenocysteine and displays an anticodon that is complimentary to UGA (Leinfelder et al.1988). This tRNASec molecule is a substrate for serylation by seryl-tRNA synthetase. After the production of Ser-tRNASec, a 500 kDa selenocysteine synthase (SelA) modifies the serine into selenocysteine using phosphoselenate (produced by SelD) as a donor (Fig. 4B).
The efficiency of the SerRS:tRNASec mischarging reaction has been demonstrated to be as low as 1% compared to that of the five cognate tRNASer isoacceptors in E. coli (Amberg et al.1996; Commans and Bock 1999). The low rate of serylation of tRNASec by SerRS can be explained by the unusual and unique structure of the tRNA molecule. Bacterial tRNASec is known to be larger than other tRNAs and, in E. coli, reaches a total of 95 nucleotides in length. The increased size is due to a combination of features including a long extra arm that is required for recognition by SerRS, and an extended 13 base pair acceptor helix. In addition, tRNASec has been shown to have a purine: pyrimidine pair between positions 11 and 24, which is rare except in initiator tRNAs, an extended 6 base pair D-stem and a reduced 4 nucleotide D-loop (Baron et al.1993; Sturchler et al.1993; Commans and Bock 1999). These specific and unique tRNA features have been demonstrated to contribute to various stages of the decoding process (Commans and Bock 1999).
Decoding and distinction of selenocysteine codons from normal UGA stop codons also requires the presence of a stem-loop structure, called the SECIS element, to be located approximately 40 nucleotides away from the 3′ end of the UGA selenocysteine codon. In addition, a specific elongation factor, called SelB, is required for recognition of the SECIS element and, thus, efficient delivery of mature selenocysteinyl-tRNASec to the ribosome (Forchhammer, Leinfelder and Bock 1989; Hilgenfeld, Bock and Wilting 1996; Kromayer et al.1996; Bock et al.1997; Haruna et al.2014). Inappropriate sequestration of Ser- or Sec- tRNASec by canonical EF-Tu is prevented by the presence of a specific anti-determinant box in the acceptor stem of tRNASec at the 8th, 9th and 10th base pair positions (Baron and Bock 1991).
A final, and critical, example of maturation prior to use in protein synthesis is given by the highly specific formylation of initiator tRNA that has been pre-charged with methionine (Fig. 4C). This reaction is catalyzed by methionyl-tRNAfMet transformylase (FMT) and requires the formyl-group donor, N-10 formyltetrahydrofolate (FTHF). In E. coli, FMT activity is essential for cell viability at 42°C and its deletion also causes marked growth defects at 37°C (Guillon et al.1992a). Structural studies have indicated that FMT is an elongated protein that is comprised of two separate domains. These domains are joined together by an extended peptide (Schmitt, Blanquet and Mechulam 1996a; Schmitt et al.1996b). The N-terminal region of the protein contains the catalytic center, is comprised of a Rossmann fold and shares high homology with another FTHF-binding protein called glycinamide ribonucleotide transformylase. In contrast, the C-terminal domain adopts a beta-barrel fold. Both domains of the protein and the extended linker region have been shown to make significant contacts with the tRNA substrate. This accounts for the high specificity of FMT for methionylated tRNAs over other aminoacylated species. The activity of FMT has been shown to require specific features of both the acceptor stem and the D-loop for accurate recognition of the aminoacylated initiator tRNA. These features include a mismatched base pair between positions 1 and 72 (Lee, Seong and RajBhandary 1991; Guillon et al.1992b), which along with formylation itself, also prevents misappropriation of the species by EF-Tu (Sundari et al.1976; Petersen et al.1979; Guillon et al.1993, 1996). The maturation of initiator Met-tRNAmet by formylation is essential for initiation of translation in bacteria.
Editing of aminoacyl-tRNA
Under certain circumstances, non-cognate aminoacyl-tRNA needs to be proofread and edited to ensure that the efficiency and fidelity of translation is maintained. In bacteria, this is achieved by a combination of enzymatic activities, which include the pre- and post-transfer editing mechanisms of aaRSs as well as the functions of trans-editing factors, peptidyl-tRNA hydrolase and D-aminoacyl-tRNA deacylase.
The aaRSs represent the first step in quality control of translation (reviewed extensively in Ling, Reynolds and Ibba 2009; Reynolds, Lazazzera and Ibba 2010). Aminoacyl-tRNA synthesis occurs in two stages (described in the Section ‘Aminoacylation’ and Fig. 3) and, if the accuracy of translation is to be adequately maintained, the aaRSs must successfully sequester both cognate tRNA and amino acid from similar non-cognate molecules present in the cellular pool. Generally, the aaRSs have high specificity for their cognate tRNA. This is due to a combination of the large surface area that is available during recognition of this substrate and also kinetic proof reading that occurs during the aminoacylation reaction (Ibba and Soll 2000; Guth and Francklyn 2007). However, near-cognate amino acids can be much harder for the aaRSs to discriminate against since they can differ from the cognate substrate by as little as one methyl group. This particular problem has been investigated in detail for IleRS, which must distinguish between two isosteric amino acids: isoleucine and valine. In this case, IleRS is able to maintain the accuracy of translation by employing both pre- and post-transfer editing mechanisms against the non-cognate amino acid (Baldwin and Berg 1966; Eldred and Schimmel 1972).
Pre-transfer editing refers to specific hydrolysis of a non-cognate aminoacyl-adenylate prior to transfer of the activated amino acid moiety to the tRNA acceptor. In contrast, post-transfer editing occurs after the activated amino acid has been attached to the designated tRNA isoacceptor. This type of editing takes place either prior to (in cis) or after release (in trans) of the aminoacylated tRNA from the synthetase (summarized in Fig. 5) (Ling, Reynolds and Ibba 2009). Whilst many of the aaRSs possess either one or both of these editing mechanisms, inefficient hydrolysis of some incorrectly acylated tRNAs can give rise to a requirement for separate trans-editing factors if the accuracy of translation is to be maintained (Ahel et al.2003).
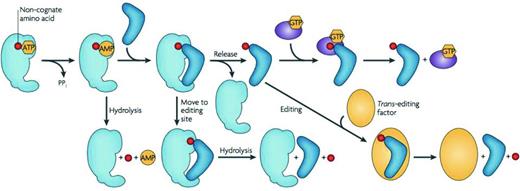
Quality control steps during the formation of a non-cognate aminoacyl-tRNA. After activation of a non-cognate amino acid (red), the aminoacyl adenylate may be hydrolyzed and released. A non-cognate aminoacyl-tRNA may be translocated into the editing site of the aaRS for hydrolysis. If the mischarged aminoacyl-tRNA is released from the aaRS without being edited, it is subjected to editing by trans-editing factors or through resampling by the aaRS. Some non-cognate aminoacyl-tRNAs are discriminated against by EF-Tu, which either binds them too tightly or too weakly for efficient delivery and release in the ribosome. Figure reproduced from Reynolds, Lazazzera and Ibba (2010).
One of the most widespread trans-editing factors is AlaXp, which can be found across all domains of life and shares homology with the editing domain of alanyl-tRNA synthetase (AlaRS) (Fukunaga and Yokoyama 2007; reviewed in Schimmel 2011; Guo and Schimmel 2012). The requirement for AlaXp is associated with the challenge presented by relatively efficient production and release of mischarged Gly-, and more frequently, Ser-tRNAAla from AlaRS (Swairjo and Schimmel 2005; Chong, Yang and Schimmel 2008). Whilst AlaRS does possess its own editing domain against both of these species, serine mischarging is particularly problematic. Furthermore, examples across all three domains of life have indicated that there is a high level of serine sensitivity during protein synthesis, with elevated levels of serine substitution at alanine codons causing toxic effects (Beebe, Ribas De Pouplana and Schimmel 2003; Lee et al.2006; Chong, Yang and Schimmel 2008). Other common examples of bacterial trans-editing factors that are involved in proofreading of misaminoacylated tRNA species included Ybak, which clears ProRS-generated Cys-tRNAPro (An and Musier-Forsyth 2005) and ProXp, which clears ProRS-generated Ala-tRNAPro (Ahel et al.2003; Wong et al.2003; Das et al.2014).
Whilst most trans-editing factors proofread and edit mischarged-tRNAs before they participate in protein synthesis, there is also a requirement for another enzymatic activity that can recycle correctly aminoacylated tRNA species that have already associated with the ribosome and begun to participate in peptide chain elongation. Peptidyl-tRNA hydrolase (PTH) can be found across all three domains of life and the esterase activity of the protein is known to be critical for bacterial cell survival (reviewed in Blanquet et al.2003; Das and Varshney 2006; McFeeters 2013). Requirement for PTH activity is based upon the natural process by which peptidyl-tRNAs dissociate prematurely from the ribosome. This occurs with a translational frequency of approximately 10% and results in ribosomal stalling and sudden abortion of protein chain elongation (Manley 1978; Kurland and Ehrenberg 1985; Jorgensen and Kurland 1990; Heurgue-Hamard et al.1998). If left to accumulate in the cell cytoplasm, these disassociated peptidyl-tRNAs can result in toxicity, imbalance in the aminoacyl-tRNA pool and, ultimately, translational arrest (Atherly and Menninger 1972; Garcia-Villegas et al.1991; Menninger and Coleman 1993; Schmitt et al.1997a, b). These undesirable outcomes are avoided by PTH, which specifically hydrolyzes the peptidyl-tRNAs into uncharged tRNA and N-acyl-L amino acids or peptides. This is achieved by cleavage of the ester bond that is located between the C-terminus of the peptide and the 2′ or 3′ hydroxyl group of the terminal adenine (A76) that is situated at the 3′ end of the tRNA molecule (Cuzin et al.1967; Kossel and RajBhandary 1968).
Structural studies focused on the E. coli enzyme have indicated that PTH is comprised of one alpha-beta globular domain that is folded around a mixed beta-sheet (Schmitt et al.1997). PTH is specific for elongator N-acyl-aminoacyl tRNAs with residues K105 and R133 of the protein responsible for recognition during docking of the 5′ phosphate of the tRNA substrate (Fromant et al.1999). In addition, a cluster of four asparagine residues at positions 10, 21, 68 and 114 are involved in interactions with the TΨC tRNA acceptor stem helix (Fromant et al.1999). In bacteria, fMet-tRNAfmet has a unique wobble region in its acceptor stem between positions 1 and 72 that protects it and consequently the process of translation initiation, from PTH activity (Schulman and Pelka 1975; Dutka et al.1993). This wobble region is missing from bacterial elongator tRNAs thus enhancing the specificity of PTH for them. Residues N10, H20 and D39 have been identified as critical for the catalytic mechanism of the enzyme (Schmitt et al.1997; Goodall, Chen and Page 2004; Das and Varshney 2006).
In bacteria, there is a widespread existence of D-amino acids and they are most commonly found incorporated naturally into some cell wall peptidoglycans or synthesized antibiotics (Shoji, Hinoo and Sakazaki 1976; Peypoux et al.1985; Sivonen et al.1992; Jack and Jung 1998; Friedman 1999). However, accumulation of D-amino acids can be toxic to the cell since not all of the aaRS have adequate discriminatory power against this alternative form of their cognate amino-acid substrate (Friedman 1999; Soutourina, Blanquet and Plateau 2000a). Consequently, there is a requirement for D-aminoacyl-tRNA deacylase activity, which was first identified in bacterial cell extracts after the tyrosyl-tRNA synthetases of both E. coli and B. subtilis were shown to incorrectly aminoacylate their cognate tRNA with D-Tyr in vitro. In E. coli, this enzyme was subsequently named D-Tyr-tRNATyr deacylase (DTD).
Since its identification, structural studies have indicated that bacterial DTD is a homodimer comprised of two 16 kDa subunits. The enzyme has some structural similarity to the editing domains of archaeal threonyl-tRNA synthetases and specifically folds into a beta-barrel structure that is closed on one side by a beta-sheet (Ferri-Fioni et al.2001; Hussain et al.2006). Catalytically, DTD has been shown to have broad substrate specificity against the ester linkage found in D- but not L-aminoacyl tRNAs (Calendar and Berg 1967; Soutourina et al.1999; Soutourina, Blanquet and Plateau 2000a; Soutourina, Plateau and Blanquet 2000b). Although EF-Tu is able to exclude D-aminoacyl-tRNAs from its active site (Yamane, Miller and Hopfield 1981), inactivation of the dtd gene has been shown to exacerbate the toxicity of D-Gln, D-Tyr, D-Ser, D-Asp and D-Trp in E. coli. These findings highlight the importance of DTD activity with regard to its specific ability to proofread and edit D-aminoacyl-tRNAs and thus ensure that an adequately balanced tRNA pool is available for L-aminoacylation by the synthetases and protein synthesis at the ribosome (Soutourina et al.1999; Soutourina, Blanquet and Plateau 2000a).
Codon recognition at the ribosome
Ribosomes are found across all domains of life and represent the primary cellular structure that participates in protein synthesis. Bacterial ribosomes are comprised of a 30S and a 50S subunit. The 30S subunit is made up of 16S ribosomal RNA and approximately 20 protein components. In contrast, the 50S subunit consists of both 23S and 5S ribosomal RNA in complex with approximately 30 proteins. During the process of translation, aminoacylated tRNAs bind to the ribosome in a codon-dependent manner. This ultimately results in translation of the genetic information encoded by a bound mRNA into protein.
Briefly, the process of protein synthesis can be divided into three stages: initiation, elongation and termination. During the first stage, the initiator tRNA, fMet-tRNAfmet, undergoes binding to the 30S ribosomal subunit in the presence of an mRNA bearing the AUG codon. This is promoted by initiation factor two (IF2) whereby appropriate interaction between the initiator tRNA and the mRNA start codon signal rapid recruitment of the 50S ribosomal subunit (Grunberg-Manago et al.1978). During elongation, aminoacyl-tRNA is brought to the ribosome in a complex with GTP-activated EF-Tu (Miller and Weissbach 1977; Johnson, Miller and Cantor 1978). Progression through this stage results in translocation of peptidyl-tRNA from the aminoacyl-site (A) of the ribosome to the peptidyl-site (P). This is then followed by release of deacylated tRNA to the exit site (E) of the ribosome with each of these stages promoted by GTP-activated elongation factor G (Weissbach 1977). In the termination stage, release factors one and two bind the ribosome in a process that is aided by GTP-activated release factor three. This occurs only in response to a stop codon arriving in the aminoacylation site (Weissbach 1977) (reviewed fully in Ogle, Carter and Ramakrishnan 2003; Khade and Joseph 2010; Novoa and Ribas de Pouplana 2012).
During each stage of translation, tRNA undergoes a series of specific interactions with the ribosome that participate directly in the decoding process. In the earliest stages of translation, interactions between the anticodon loop of initiator fMet-tRNAfmet and the P site of the 30S ribosomal subunit are directly responsible for the high accuracy of initiation factor three-mediated discrimination against elongator tRNA. More specifically, residues G1338 and A1339 within the 16S rRNA component of the small subunit interact with conserved GC base pairs found at initiator tRNA anticodon arm positions 29–41 and 30–40, respectively. This stabilizes docking of initiator tRNA to the ribosome and results in full initiation of translation (Abdi and Fredrick 2005; Lancaster and Noller 2005; Berk et al.2006; Korostelev et al.2006; Selmer et al.2006; Qin, Abdi and Fredrick 2007; Voorhees et al.2009).
After completion of early initiation, the acceptor stem of the aminoacylated-tRNA is bound in a complex with EF-Tu and so cannot interact with the A site of the 50S ribosomal subunit. However, the anticodon loop of the tRNA is able to directly interact with the A site of the 30S ribosomal subunit (Moazed and Noller 1989). Conserved residues G530, A1492 and A1493 within the 16S rRNA of the small subunit interact with the resulting codon-anticodon helix (Ogle et al.2001; Cochella, Brunelle and Green 2007). Together, these interactions result in the formation of a decoding intermediate, which is most commonly referred to as the A/T state. This state is not stable during interaction between the ribosome and aminoacyl-tRNA that does not match the codon specified by the bound mRNA sequence (Pape, Wintermeyer and Rodnina 1999; Valle et al.2003).
Interaction of the aminoacylated-tRNA with the A site of the 50S ribosomal subunit is only achieved upon successful codon–anticodon pairing, which triggers EF-Tu-mediated GTP hydrolysis. Accommodation of the aminoacylated-tRNA within the 50S A site subsequently triggers peptide bond formation by the 23S rRNA that comprises the peptidyl-transferase center (Ban et al.2000; Nissen et al.2000; Harms et al.2001). This reaction is known to be aided by the formation of a base pair between G2553 of 23S rRNA and residue C75 of the aminoacyl-tRNA present in the ribosomal A site (Schmeing et al.2005). After this reaction, deacylated tRNA becomes accommodated in the P site of the ribosome, eventually shifting to the E site where the acceptor stem undergoes various interactions with the 50S ribosomal subunit. Dipeptidyl-tRNA in the A site can then progress to the P site making room for accommodation of another aminoacyl-tRNA, thus allowing translation to proceed (fully reviewed in Khade and Joseph 2010). Aminoacylated-tRNA species that are not recruited by the translation machinery typically proceed into other cellular pathways as described below.
Other roles of tRNA in the cell
In addition to their primary role in translation, many bacteria also require aminoacylated-tRNA species to enter the peptidoglycan, antibiotic resistance, antibiotic synthesis and membrane phospholipid modification pathways (reviewed in Shepherd and Ibba 2013b). Due to their critical role in cell survival, some tRNA molecules are also confirmed toxin targets (Lu et al.2005; Jablonowski and Schaffrath 2007; Meineke et al.2011; Nawrot, Sochacka and Duchler 2011; Uthman et al.2011; Lopes et al.2014). In this section, a brief overview of the specific involvement of mature bacterial tRNAs in processes outside of protein synthesis will be given.
Sequestration of aminoacylated-tRNA donors away from the protein synthesis machinery and into the peptidoglycan cross-linking pathway has been demonstrated to occur throughout many genera and species of bacteria. Peptidoglycan is a critical component of the bacterial cell wall where it has an important role in providing protection against osmotic stress and turgor pressure. In Gram-positive bacteria, 30–70% of the total cell wall biomass is defined as peptidoglycan and the structure may be heavily utilized as an anchoring platform for various virulence factors (Schleifer and Kandler 1972; Dramsi et al.2008). In contrast, peptidoglycan makes up 10% or less of the total cell wall biomass in Gram-negative bacteria.
Despite these differences, the basic carbohydrate backbone of peptidoglycan is always comprised of alternating β-1, 4-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues. Furthermore, the lactyl-ether appendage of the MurNAc component is typically modified by the addition of an L-Ala- γ -D-Glu-X-D-Ala-D-Ala-containing pentapeptide side chain. Within this side chain, position X is usually represented by meso-diaminopimelic acid in Gram-negative bacteria and L-lysine in Gram-positive bacteria (Bugg 1999). An additional level of structural rigidity is obtained by the formation of cross-links between the pentapeptide side chains of adjacent peptidoglycan strands. Such cross-links can either be direct, forming between position X on one strand and L-Ala at the fourth position of another, or indirect, which requires the formation of an intersecting amino acid bridge. Overall, cross-linking tends to occur at a higher frequency in Gram-positive bacteria (70–90%) as opposed to Gram-negative bacteria (25–50%). In addition, indirect cross-links are more common in bacteria that have L-lysine at position X in the pentapeptide side chain and formation of the intersecting amino acid bridge usually requires appropriately charged tRNA to serve as the amino acid donor (Garcia-Bustos, Chait and Tomasz 1987; Bugg 1999).
The occurrence of indirect peptidoglycan cross-linking has been reported in several Gram-positive pathogenic bacteria where it is most frequently associated with increased antibiotic resistance levels (reviewed in Shepherd and Ibba 2013b). Briefly, in St. pneumoniae, the peptidoglycan structure is comprised of a combination of both branched and linear muropeptides. Branched muropeptides have an L-Ala-L-Ala or an L-Ser-L-Ala dipeptide bridge attached to the third L-Lysine found at position X in the pentapeptide side chain (Fiser, Filipe and Tomasz 2003). Dipeptide bridge formation is associated with penicillin resistance in this pathogen and requires the activities of the aminoacyl-tRNA-dependent MurM and MurN proteins (Filipe and Tomasz 2000; Weber et al.2000). MurM adds either L-Ala or L-Ser at the first position of the cross-link, which is then subsequently completed by the L-Ala adding activity of MurN (Filipe, Pinho and Tomasz 2000). It has recently been demonstrated that MurM is able to more efficiently use mischarged Ser-tRNAAla as opposed to Ala-tRNAAla and Ser-tRNASer as a substrate for peptidoglycan cross-linking (Shepherd 2011). In addition, MurM can also deacylate Ala- and Ser-tRNAs in the absence of the peptidoglycan substrate, Lipid II, suggesting that this protein has dual functionality in cell-wall biosynthesis and quality control of translation (Shepherd and Ibba 2013b, 2014).
In Staphylococcus aureus, indirect peptidoglycan cross-linking involves the formation of a pentaglycine bridge, which is ultimately appended to the L-Lysine residue occupying position X of the pentapeptide side chain. Formation of the pentaglycine bridge requires the sequential activities of the Gly-tRNAGly-dependent FemX, FemA and FemB proteins (Rohrer et al.1999; Tschierske et al.1999; Schneider et al.2004). Three out of the five tRNAGly isoacceptors encoded in the genome of Sta. aureus have sequence identity elements consistent with weak binding to EF-Tu, thus ensuring their participation in peptidoglycan rather than protein synthesis (Giannouli et al.2009). Participation of aminoacyl-tRNA in Sta. aureus cell-wall biogenesis is critical, and selective inactivation of femA or femB is known to be lethal due to a combination of phenotypic effects that include loss of methicillin resistance (Stranden et al.1997; Ling and Berger-Bachi 1998; Rohrer and Berger-Bachi 2003). Two other Staphylococcal species, Sta. simulans and Sta. capitis, use the aminoacyl-tRNA-dependent Lif and Epr proteins, respectively, to substitute serine at the third and fifth positions of the standard pentaglycine bridge. By doing so, these proteins ultimately confer resistance against the glycyl–glycine endopeptidases that are produced by these bacteria, thus ensuring that peptidoglycan integrity is maintained during competition with other species for occupation of the same niche (Sugai et al.1997; Tschierske et al.1997; Ehlert et al.2000). Other well-characterized examples of the importance of aminoacylated tRNAs in peptidoglycan biosynthesis include Weissella viridescens FemX (Ala addition), Enterococcus faecalis BppA1/BppA2 (Ala addition) and the vancomycin-dependent FemX/VanK system (Gly addition) in Streptomyces coelicolor (reviewed in Shepherd and Ibba 2013a) (Bouhss et al.2002; Hegde and Blanchard 2003; Hong et al.2004, 2005; Maillard et al.2005; Fonvielle et al.2009).
In addition to their involvement in peptidoglycan biosynthesis, aminoacylated-tRNAs also participate in antibiotic resistance and synthesis pathways. Many Gram-positive and Gram-negative bacteria have a specific type of virulence factor, called MprF, encoded in their genome that can catalyze the aminoacylation of inner membrane lipids subsequently altering the permeability of the cell wall to cationic antimicrobial agents (reviewed in Dare and Ibba 2012; Shepherd and Ibba 2013a). This process requires donor aminoacyl-tRNA molecules and is known to adjust the overall negative charge of the cell membrane in a manner that is dependent upon the nature of the amino acid that is being transferred to the lipid (Roy and Ibba 2008; Ernst and Peschel 2011). One of the most well characterized classes of MprF protein is comprised of the aminoacyl-phosphatidylglycerol synthases (aaPGSs). The aaPGSs are integral cytoplasmic membrane proteins that specifically transfer amino acids from appropriately charged tRNA species to the polar head group of phosphatidylglycerol, which is a common bacterial membrane lipid (Roy and Ibba 2008; Ernst et al.2009). Lysylation of phosphatidylglyercol by the aaPGS enzymes of C. perfringens, Sta. aureus and M. tuberculosis is associated with an increase in the net positive charge of the cytoplasmic membranes of these bacteria. This change in membrane polarity has been directly correlated with reduced permeability to defensins, aminoglycosides, glycopeptides and β-lactams (Oku et al.2004; Staubitz et al.2004; Roy and Ibba 2008; Maloney et al.2009). In contrast, recent work focusing on Listeria monocytogenes aaPGS has indicated that lysylation of phosphatidylglycerol in the membrane of this pathogen has a much more global physiological effect on the cell that extends beyond antimicrobial resistance and into regulation of the motility operon (Dare et al.2014). In addition, Pseudomonas aeruginosa has an aaPGS enzyme that aminoacylates phosphatidylglycerol with alanine resulting in neutralization of the overall charge of the membrane. This subsequently causes enhancement in the resistance of the bacterium to a range of negatively charged β-lactams and increased survival upon exposure to acidic or osmotically stressful growth conditions (Roy and Ibba 2008; Klein et al.2009). Whilst the importance of aminoacylated-tRNAs in MprF-mediated antibiotic resistance is clear, these findings suggest that the overall functional relevance of membrane lipid aminoacylation is dependent upon both the species of bacteria and the features of the environment to which it is routinely exposed.
The involvement of aminoacyl-tRNA in antibiotic synthesis has been well characterized in the cases of valanimycin biosynthesis by Str. viridifaciens, and pacidamycin biosynthesis by Str. coeruleorubidus (reviewed with cyclodipeptide synthesis in Shepherd and Ibba 2013a). Essentially, valanimycin biosynthesis requires VlmA. VlmA is a dedicated SerRS synthetase that diverts L-serine from a Ser-tRNASer donor molecule away from protein synthesis and into this antibiotic synthesis pathway (Garg et al.2002, 2008; Garg, Gonzalez and Parry 2006). In contrast, generation of pacidamycin requires PacB activity. PacB transfers L-alanine from donor aminoacyl-tRNA to a critical intermediate in the pathway for synthesis of this antibiotic (Chen et al.1989; Zhang et al.2011a,b). Aminoacyl-tRNA donors are also required for the synthesis of cyclodipeptides by members of the Bacillus and Streptomycete families of bacteria (Lautru et al.2002; Leys et al.2003; Seward et al.2006; Gondry et al.2009; Bonnefond et al.2011).
Given the clear importance of tRNAs to a variety of cellular processes, it is no surprise that they are toxin targets in both eukaryotic and prokaryotic systems. In some cases, possession of a toxin–antitoxin system that targets tRNA can be an advantage to the host bacterium. For example, Shigella flexneri and S. enterica are known to have the VapBC toxin–antitoxin system. The toxin component of this system, VapC, specifically targets the anticodon loop of the initiator tRNAfmet cleaving it between nucleotides A38 and C39 (Winther and Gerdes 2011). In these bacteria, VapC activity has been shown to inhibit translation both in vitro and in vivo in a manner that can be rescued either by VapB, the antitoxin, or addition of extra charged initiator tRNA. In addition, VapC production was specifically activated by an abrupt reduction in growth rate. This suggests that it may be involved in stress response to environmental conditions by affecting the global regulation of cellular translation and causing reprogramming of translational initiation (Winther and Gerdes 2011). This toxin family has also recently been identified in the Gram-negative pathogen Leptospira interrogans where it is believed to have the same regulatory role (Lopes et al.2014).
Other toxins that target tRNAs, including colicin D, colicin E and PrrC, are specifically transported out of the producing cell with the aim of killing competitor cells during niche colonization. Colicins are defined as plasmid-encoded toxins that exert toxicity against other strains of E. coli that do not carry the producing Col plasmid (Konisky 1982; Pugsley 1984a,b). Colicin D is known to cleave the anticodon loops of four isoacceptors of tRNAArg between positions 38 and 39 (Tomita et al.2000). In contrast, Colicin E5 cleaves the anticodon loop of four different tRNAs (His, Tyr, Asn and Asp) between positions 34 and 35 (Ogawa et al.1999). Recently, tRNAs have also been shown to be one of the targets for bacterial contact-dependent growth inhibition (Diner et al.2012).
Targeted-tRNA cleavage is also a documented bacterial antiviral defense mechanism. For example, E. coli can produce an antiviral defense toxin, called PrrC, which specifically cleaves tRNALys at a single site in the anticodon loop just 5′ of the modified wobble uridine base. PrrC activity is induced in response to bacteriophage T4 infection (Amitsur, Morad and Kaufmann 1989; Penner et al.1995; Kaufmann 2000). Subsequent depletion of tRNALys severely impairs synthesis of the late stage viral proteins by the host cell and ultimately arrests rapid spread of the virus throughout the entire bacterial population. Homologues of the PrrC enzyme are found across a diverse range of bacteria, thus indicating the importance of tRNA in antiviral defense strategies. Streptococcal and E. coli PrrC proteins are also known to have fungicidal activity against yeast suggesting that they have the potential to confer a competitive advantage on the host bacterium during niche colonization or pathogenic infection processes (Meineke et al.2011).
OUTLOOK
Recent years have seen new fields opening up in tRNA biology, the success of which is due in no small part to the substantial body of knowledge that has been built up over the last 50 years. Advances in genomics and other technologies that can be applied to the entire cellular population of tRNAs have started to reveal new roles for tRNAs as regulators of numerous cellular processes (Raina and Ibba 2014). For example, the ability to measure the aminoacylation status of all tRNAs and to monitor tRNA modification patterns globally have begun to uncover new roles for tRNA in cellular stress responses (Pan 2013; Gu, Begley and Dedon 2014). Another area of growing interest comes from the increasing awareness that tRNAs directly and indirectly function in numerous roles outside of translation of the genetic code (Banerjee et al.2010; Anderson and Ivanov 2014). Overall, research in the tRNA field is entering a new and exciting era and there are clearly many more surprises still to come from this small but highly versatile RNA.
FUNDING
This work was supported by a grant from the National Institutes of Health (GM065183).
Conflict of interest. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}