





Abstract
Post-streptococcal glomerulonephritis (PSGN) is primarily associated with preceding group A streptococcal skin or throat infections, now mainly observed in economically disadvantaged communities. This condition significantly predisposes individuals to later-life chronic kidney disease and concurrent renal complications, with the elderly experiencing increased severity and less favourable outcomes. Streptococcal pyrogenic exotoxin B and nephritis-associated plasmin receptor are identified nephritogenic antigens (nephritogens). Pathogenesis of PSGN is multifactorial. It can involve the formation of antigen-antibody immune complexes, causing inflammatory damage to renal glomeruli. Deposition of circulating immune complexes or in situ formation of immune complexes in glomeruli, or both, results in glomerulonephritis. Additionally, molecular mimicry is hypothesized as a mechanism, wherein cross-reactivity between anti-streptococcal antibodies and glomerular intrinsic matrix proteins leads to glomerulonephritis. Besides, as observed in clinical studies, streptococcal inhibitor of complement, a streptococcal-secreted protein, can also be associated with PSGN. However, the interplay between these streptococcal antigens in the pathogenesis of PSGN necessitates further investigation. Despite the clinical significance of PSGN, the lack of credible animal models poses challenges in understanding the association between streptococcal antigens and the disease process. This review outlines the postulated mechanisms implicated in the development of PSGN with possible therapeutic approaches.
Introduction
Streptococci are ubiquitous Gram-positive bacteria found as normal microbial flora in humans (Wong and Stevens 2013). It causes a wide range of non-invasive and invasive diseases (Martin and Green 2006, Avire et al. 2021). Streptococci are also associated with immune-mediated complications, including post-streptococcal glomerulonephritis (PSGN), acute rheumatic fever, rheumatic heart disease, paediatric autoimmune neuropsychiatric disorders, and reactive arthritis (Carapetis et al. 2005, Prato et al. 2021, Brouwer et al. 2023). In most cases of post-streptococcal immune complications, the primary infections may not be detected. The global incidence of streptococcal infections, including pyoderma and pharyngitis, exceeds 700 million cases annually, with PSGN cases estimated to exceed 4.7 million (Carapetis et al. 2005, Marshall et al. 2011, Walker et al. 2014, Alhamoud et al. 2021).
PSGN is associated with prior skin or throat infections, mainly caused by Streptococcus pyogenes, a group A streptococcus (GAS), and less frequently by Streptococcus dysgalactiae subspecies equisimilis (SDSE; group C or G streptococcus) (Rodriguez-Iturbe and Haas 2016). Approximately 1–2 weeks after untreated streptococcal throat infections, and 3–6 weeks following unresolved streptococcal skin infections, a portion of individuals develop PSGN (Walker et al. 2014). PSGN is prevalent among children (5–12 yr), although it can affect the elderly (≥60 yr) who are immunocompromised with concurrent co-morbidities (Nasr et al. 2011, Nasr et al. 2013). The elderly experience more severity and a poor prognosis compared to children (Nasr et al. 2011). Approximately 25%–50% of PSGN cases are asymptomatic and are diagnosed incidentally during routine urinalysis (Yoshizawa et al. 1996, Rawla et al. 2022). The affected individuals present with macroscopic haematuria, oliguria, facial oedema, and hypertension, but may also display other symptoms (Table 1). Subsequent haematobiochemical assays and urinalysis reveal hypoalbuminemia, proteinuria, increased serum creatinine and urea, reduced estimated glomerular filtration rate, elevated C-reactive protein, decreased complement C3, along with increased white blood cell count, increased neutrophil/lymphocyte ratio, and elevated anti-streptolysin O titres (Satoskar et al. 2014, Demircioglu et al. 2018, Satoskar et al. 2020, Alhamoud et al. 2021, Rawla et al. 2022).
Common clinical presentations in PSGN.
| Clinical manifestations | Case frequency | References |
|---|---|---|
| Gross haematuria | 30%–50% | Rawla et al. (2022) |
| Oliguria | <50% | Xu and Somers (2020) |
| Hypertension | 60%–80% | Rawla et al. (2022) |
| Periorbital oedema | 65%–90% | Elzouki (2012) |
| Proteinuria (nephrotic range) | 2%–5%a, >20%b | Tasic (2008), Luo et al. (2010) |
| Azotaemia | 83%b | Rodriguez-Iturbe and Haas (2016) |
| Congestive heart failure | 43%b | Rodriguez-Iturbe and Haas (2016) |
| Clinical manifestations | Case frequency | References |
|---|---|---|
| Gross haematuria | 30%–50% | Rawla et al. (2022) |
| Oliguria | <50% | Xu and Somers (2020) |
| Hypertension | 60%–80% | Rawla et al. (2022) |
| Periorbital oedema | 65%–90% | Elzouki (2012) |
| Proteinuria (nephrotic range) | 2%–5%a, >20%b | Tasic (2008), Luo et al. (2010) |
| Azotaemia | 83%b | Rodriguez-Iturbe and Haas (2016) |
| Congestive heart failure | 43%b | Rodriguez-Iturbe and Haas (2016) |
Paediatric case.
Geriatric case.
Abbreviation: PSGN: post-streptococcal glomerulonephritis.
Common clinical presentations in PSGN.
| Clinical manifestations | Case frequency | References |
|---|---|---|
| Gross haematuria | 30%–50% | Rawla et al. (2022) |
| Oliguria | <50% | Xu and Somers (2020) |
| Hypertension | 60%–80% | Rawla et al. (2022) |
| Periorbital oedema | 65%–90% | Elzouki (2012) |
| Proteinuria (nephrotic range) | 2%–5%a, >20%b | Tasic (2008), Luo et al. (2010) |
| Azotaemia | 83%b | Rodriguez-Iturbe and Haas (2016) |
| Congestive heart failure | 43%b | Rodriguez-Iturbe and Haas (2016) |
| Clinical manifestations | Case frequency | References |
|---|---|---|
| Gross haematuria | 30%–50% | Rawla et al. (2022) |
| Oliguria | <50% | Xu and Somers (2020) |
| Hypertension | 60%–80% | Rawla et al. (2022) |
| Periorbital oedema | 65%–90% | Elzouki (2012) |
| Proteinuria (nephrotic range) | 2%–5%a, >20%b | Tasic (2008), Luo et al. (2010) |
| Azotaemia | 83%b | Rodriguez-Iturbe and Haas (2016) |
| Congestive heart failure | 43%b | Rodriguez-Iturbe and Haas (2016) |
Paediatric case.
Geriatric case.
Abbreviation: PSGN: post-streptococcal glomerulonephritis.
The incidence of PSGN is high in middle- to low-income countries (Barnett et al. 2018, Demircioglu et al. 2018, Satoskar et al. 2020, Thorgrimson et al. 2022). It is also observed among the First Nations Peoples of high-income countries (May et al. 2016, Ramanathan et al. 2017, Demircioglu et al. 2018, Thorgrimson et al. 2022). Ethnic background, socioeconomic status, and access to healthcare, including antibiotic usage are considered factors for differences in the disease incidence. Although an equal sex difference in GAS throat and skin infections is observed, a 2:1 male-to-female ratio in PSGN has been reported (Wannamaker 1970, Baldwin et al. 1974, Tewodros et al. 1992). Genetic predispositions have also been suggested (Layrisse et al. 1983, Mori et al. 1996, Bakr et al. 2007). Currently, the worldwide incidence of PSGN has declined remarkably, particularly in middle- to high-income countries, due to improved living standards, hygiene practices, better access to antibiotic therapy, and healthcare (WHO 2005, Rodriguez-Iturbe and Musser 2008, Rodriguez-Iturbe and Haas 2016). Despite these advances, the diagnosis of PSGN may be delayed in patients who do not show overt clinical signs on presentation (Pais et al. 2008). Besides, genetic variations within prevalent streptococcal strains and host susceptibility have led to the re-emergence of the disease (Ralph and Carapetis 2013, You et al. 2018).
PSGN is a significant risk factor for later-life chronic kidney disease (CKD) (Hoy et al. 2012, Balasubramanian and Marks 2017). Approximately 30%–40% of individuals with a prior history of immune-mediated nephropathy and acute kidney complications progress to CKD and kidney failure within 20 yr (Julian et al. 1988, Mehta et al. 2015). Clarkson et al. (2019) reported that nearly one in eight, who suffer from CKD, progress to kidney failure with high mortality. No specific treatment and prevention strategies for PSGN have been developed yet, and early diagnosis and symptom-based medical intervention are essential in mitigating further risks (Satoskar et al. 2020, Brant et al. 2022, Khou et al. 2022). In this narrative review, multifactorial mechanisms that may contribute to kidney damage in PSGN have been outlined. The need for preclinical models to test novel therapeutic approaches based on these mechanisms is also highlighted.
Streptococcal virulence and nephritogenicity
The major virulence factors (Table 2) expressed by streptococci include M protein, streptolysins O and S, C5a peptidase, streptococcal pyrogenic exotoxins (Spe), streptococcal protective antigens (Spa), streptococcal inhibitor of complement (SIC), streptokinase, and superantigens (chromosome-encoded: SpeG, SpeJ, SpeQ, SpeR, and SmeZ; prophage-encoded: SpeA, SpeC, SpeH, SpeI, SpeK–M, and SSA) (Sagar et al. 2008, Brouwer et al. 2023). Some of these factors, such as Spa or SIC, are found to be restricted in their distribution to specific emm types (Hartas and Sriprakash 1999). Streptococcal isolates were traditionally characterized based on serotype-specific antiserum that was developed against the M protein. Streptococci are now categorized according to the sequence of the 5′ end (variable region) of the emm gene that encodes the M protein (this is referred to as emm typing) (Walker et al. 2014).
Streptococcal virulence factors associated with PSGN.
| Virulence factors | Ex vivo and in vitro (clinical samples) | In vivo (animal models) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serology | Histology | BioChem | References | Serology | Histology | BioChem | References | |||||
| HM | IHC | IF | HM | IHC | IF | |||||||
| SpeBa | √ | − | − | √ | − | Cu et al. (1998), Batsford et al. (2005) | √ | √ | − | √ | √ | Kuo et al. (1998), Kuo et al. (2004), Luo et al. (2007) |
| NAPlr/GAPDHa | √ | − | √ | √ | √ | Yamakami et al. (2000), Yoshizawa et al. (2004), Oda et al. (2010), Oda et al. (2012) | − | − | − | − | − | − |
| SICb | √ | − | − | − | √ | Sriprakash et al. (2002), Karmarkar et al. (2013) | − | − | − | − | − | − |
| M proteinc | √ | √ | √ | √ | √ | Markowitz and Lange (1964), Michael et al. (1966), Treser et al. (1969), Goroncy-Bermes et al. (1987), Goodfellow et al. (1999) | √ | √ | √ | √ | − | Kaplan (1958), Kantor (1965), Lindberg and Vosti (1969), Burova et al. (2013) |
| Streptokinasec | √ | − | − | √ | √ | Villarreal et al. (1979), Ohkuni et al. (1983), Johnston and Zabriskie (1986) | √ | √ | √ | √ | √ | Nordstrand et al. (1998), Nordstrand et al. (2000) |
| Virulence factors | Ex vivo and in vitro (clinical samples) | In vivo (animal models) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serology | Histology | BioChem | References | Serology | Histology | BioChem | References | |||||
| HM | IHC | IF | HM | IHC | IF | |||||||
| SpeBa | √ | − | − | √ | − | Cu et al. (1998), Batsford et al. (2005) | √ | √ | − | √ | √ | Kuo et al. (1998), Kuo et al. (2004), Luo et al. (2007) |
| NAPlr/GAPDHa | √ | − | √ | √ | √ | Yamakami et al. (2000), Yoshizawa et al. (2004), Oda et al. (2010), Oda et al. (2012) | − | − | − | − | − | − |
| SICb | √ | − | − | − | √ | Sriprakash et al. (2002), Karmarkar et al. (2013) | − | − | − | − | − | − |
| M proteinc | √ | √ | √ | √ | √ | Markowitz and Lange (1964), Michael et al. (1966), Treser et al. (1969), Goroncy-Bermes et al. (1987), Goodfellow et al. (1999) | √ | √ | √ | √ | − | Kaplan (1958), Kantor (1965), Lindberg and Vosti (1969), Burova et al. (2013) |
| Streptokinasec | √ | − | − | √ | √ | Villarreal et al. (1979), Ohkuni et al. (1983), Johnston and Zabriskie (1986) | √ | √ | √ | √ | √ | Nordstrand et al. (1998), Nordstrand et al. (2000) |
These virulence factors are well established as nephritogenic antigens.
This virulence factor has yet to be investigated to substantiate its potential nephritogenicity.
Despite associations with PSGN, these virulence factors are not considered reliable candidates for nephritogenic antigens, as reviewed by Rodríguez-Iturbe and Batsford 2007, Mosquera and Pedreañez 2021.
Abbreviations: BioChem: Biochemical analysis of urine/blood, GAPDH: glyceraldehyde-3-phosphate dehydrogenase, HM: histomorphology with light microscopy (staining: mainly- haematoxylin-eosin and periodic acid-Schiff, in some cases- periodic acid silver methenamine and Wright-Giemsa) and/or electron microscopy, IF: immunofluorescence, IHC: immunohistochemistry, NAPlr: nephritis-associated plasmin receptor, PSGN: post-streptococcal glomerulonephritis, Ref: references, SIC: streptococcal inhibitor of complement, SpeB: streptococcal pyrogenic exotoxin B.
Streptococcal virulence factors associated with PSGN.
| Virulence factors | Ex vivo and in vitro (clinical samples) | In vivo (animal models) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serology | Histology | BioChem | References | Serology | Histology | BioChem | References | |||||
| HM | IHC | IF | HM | IHC | IF | |||||||
| SpeBa | √ | − | − | √ | − | Cu et al. (1998), Batsford et al. (2005) | √ | √ | − | √ | √ | Kuo et al. (1998), Kuo et al. (2004), Luo et al. (2007) |
| NAPlr/GAPDHa | √ | − | √ | √ | √ | Yamakami et al. (2000), Yoshizawa et al. (2004), Oda et al. (2010), Oda et al. (2012) | − | − | − | − | − | − |
| SICb | √ | − | − | − | √ | Sriprakash et al. (2002), Karmarkar et al. (2013) | − | − | − | − | − | − |
| M proteinc | √ | √ | √ | √ | √ | Markowitz and Lange (1964), Michael et al. (1966), Treser et al. (1969), Goroncy-Bermes et al. (1987), Goodfellow et al. (1999) | √ | √ | √ | √ | − | Kaplan (1958), Kantor (1965), Lindberg and Vosti (1969), Burova et al. (2013) |
| Streptokinasec | √ | − | − | √ | √ | Villarreal et al. (1979), Ohkuni et al. (1983), Johnston and Zabriskie (1986) | √ | √ | √ | √ | √ | Nordstrand et al. (1998), Nordstrand et al. (2000) |
| Virulence factors | Ex vivo and in vitro (clinical samples) | In vivo (animal models) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serology | Histology | BioChem | References | Serology | Histology | BioChem | References | |||||
| HM | IHC | IF | HM | IHC | IF | |||||||
| SpeBa | √ | − | − | √ | − | Cu et al. (1998), Batsford et al. (2005) | √ | √ | − | √ | √ | Kuo et al. (1998), Kuo et al. (2004), Luo et al. (2007) |
| NAPlr/GAPDHa | √ | − | √ | √ | √ | Yamakami et al. (2000), Yoshizawa et al. (2004), Oda et al. (2010), Oda et al. (2012) | − | − | − | − | − | − |
| SICb | √ | − | − | − | √ | Sriprakash et al. (2002), Karmarkar et al. (2013) | − | − | − | − | − | − |
| M proteinc | √ | √ | √ | √ | √ | Markowitz and Lange (1964), Michael et al. (1966), Treser et al. (1969), Goroncy-Bermes et al. (1987), Goodfellow et al. (1999) | √ | √ | √ | √ | − | Kaplan (1958), Kantor (1965), Lindberg and Vosti (1969), Burova et al. (2013) |
| Streptokinasec | √ | − | − | √ | √ | Villarreal et al. (1979), Ohkuni et al. (1983), Johnston and Zabriskie (1986) | √ | √ | √ | √ | √ | Nordstrand et al. (1998), Nordstrand et al. (2000) |
These virulence factors are well established as nephritogenic antigens.
This virulence factor has yet to be investigated to substantiate its potential nephritogenicity.
Despite associations with PSGN, these virulence factors are not considered reliable candidates for nephritogenic antigens, as reviewed by Rodríguez-Iturbe and Batsford 2007, Mosquera and Pedreañez 2021.
Abbreviations: BioChem: Biochemical analysis of urine/blood, GAPDH: glyceraldehyde-3-phosphate dehydrogenase, HM: histomorphology with light microscopy (staining: mainly- haematoxylin-eosin and periodic acid-Schiff, in some cases- periodic acid silver methenamine and Wright-Giemsa) and/or electron microscopy, IF: immunofluorescence, IHC: immunohistochemistry, NAPlr: nephritis-associated plasmin receptor, PSGN: post-streptococcal glomerulonephritis, Ref: references, SIC: streptococcal inhibitor of complement, SpeB: streptococcal pyrogenic exotoxin B.
The streptococcal strains associated with renal dysfunctions are confined to specific M protein serotypes (i.e. M1, M2, M4, M12, M25, M42, M49, M55, M57, M60, and M61) (Cunningham 2000, Balasubramanian and Marks 2017, Satoskar et al. 2020). The nephritogenic fractions, including streptococcal plasma membrane and other secretory proteins are considered to be implicated in PSGN. Recent investigations suggest that two ‘nephritogenic’ antigens (nephritogens), namely streptococcal pyrogenic exotoxin B (SpeB) and nephritis-associated plasmin receptor (NAPlr) are primarily associated with PSGN, rather than the M proteins (Batsford et al. 2005, Balasubramanian and Marks 2017, Satoskar et al. 2020). In addition, SIC, found within some of these M types has been documented to be associated with renal complications (Sriprakash et al. 2002, Karmarkar et al. 2013). Further investigations are required to identify the role of SIC in renal pathology. In addition, the combined effects of streptococcal antigens in causing or exacerbating renal impairment remain unexplored. Investigating these interactions is crucial for the development of more effective diagnosis, treatment, management, and prevention strategies.
Pathogenic mechanisms implicated in PSGN
Immune complex formation is the main precondition for PSGN, although several other host- and pathogen-related factors (Fig. 1, Table 3) may contribute to the pathogenesis (Yoshizawa et al. 1992, Nordstrand et al. 1999, Satoskar et al. 2020, Mosquera and Pedreañez 2021). We hypothesize that the pathogenesis of PSGN may involve three distinct pathways or a combination of these (Fig. 1). First, streptococcal nephritogenic antigens (e.g. NAPlr/SpeB) themselves can convert plasmin from the plasminogen, facilitating the activation of glomerular matrix metalloproteinases (MMPs). These antigens can also activate the complement system through the alternative pathway (Satoskar et al. 2020, Mosquera and Pedreañez 2021). All these factors can lead to a defective extracellular matrix composition, along with the chemotaxis of inflammatory cells that release cytokines/chemokines (Mosquera and Pedreañez 2021), resulting in the degradation of the glomerular basement membrane (GBM) (Fig. 1a; plasmin-mediated injury).

Postulated mechanisms implicated in the development of PSGN. GAS initially causes pharyngitis and pyoderma. In a proportion of individuals, untreated or repeated streptococcal infections lead to post-streptococcal complications of the kidney. Three distinct pathways, or a combination of these, may be associated with the pathogenesis. (a) Plasmin-mediated injury: Streptococcal nephritogenic antigens (e.g. SpeB/NAPlr) can convert plasminogen to plasmin that can activate glomerular MMPs and modify extracellular matrix composition, including chemotaxis of inflammatory cells, leading to degradation of GBM. (b) Immune complex-mediated injury: This involves glomerular subepithelial immune complex deposition via three different mechanisms; (b1) streptococcal antigens (e.g. SpeB/NAPlr/SIC) could interact with antibodies in circulation to form complexes that deposit in the GBM, (b2) streptococcal antigens (e.g. SpeB/NAPlr/SIC) could deposit in the GBM, and circulatory antibodies specific to these antigens can cross the glomerular capillary endothelial lining and bind these exogenous antigens already embedded in GBM, (b3) anti-streptococcal antibodies might transverse the capillary endothelium and cross-react with glomerular intrinsic matrix proteins (i.e. endogenous/native antigens). Immune complexes formed through any of these mechanisms can activate complement cascade (C5b-9), monocyte/macrophage and neutrophil recruitment, activation, and cytokine release resulting in inflammation and injury to glomerular capillary tufts and podocytes. (c) SIC-mediated injury: Circulatory SIC may penetrate the podocyte, bind, and co-localize with Ezrin intracellularly. This may cause cytoskeletal defects leading to podocytopathy. GBM: glomerular basement membrane, ICs: immune complexes, MMPs: matrix metalloproteinases, NAPlr: nephritis-associated plasmin receptor, PSGN: post-streptococcal glomerulonephritis, SIC: streptococcal inhibitor of complement, SpeB: streptococcal pyrogenic exotoxin B.
Pathological features of immune-mediated renal diseases.
| Renal diseases | Antibodies | Histopathological features | References |
|---|---|---|---|
| PSGN | Anti-SpeB, anti-NAPlr, anti-M, anti-SIC(?)a | Deposition of IgG and C3 (occasionally C4, IgM) along glomerular capillary loops and mesangium, glomerular endocapillary hypercellularity, tubulointerstitial oedema | Kraus and Beachey (1988), Balasubramanian and Marks (2017), Satoskar et al. (2020) |
| IgA nephropathy | Anti-Gd IgA1 | Mesangial deposition of IgA and C3, mesangial hypercellularity, tubular atrophy, interstitial fibrosis | Kim et al. (2012), Habas et al. (2022) |
| LN | Anti-dsDNA, anti-nucleosome, anti-histone, anti-C1q | Full-house glomerular deposits (IgA, IgM, IgG, C3, and C1q), endocapillary hypercellularity, hyaline deposition and fibrinoid necrosis in glomeruli, glomerular crescent formation, tubular atrophy, interstitial fibrosis | Yang et al. (2015), Choi et al. (2023) |
| Goodpasture's syndrome (Anti-GBM disease) | Anti-type IV collagen α3 chain | Linear deposition of IgG and/or C3 along the GBM, crescent formation in glomeruli, glomerulosclerosis, interstitial fibrosis, tubular atrophy | Kalluri et al. (1995) |
| ANCA-associated vasculitis and GN | Anti-PR3, anti-MPO | Excessive activation of neutrophil, subsequent degranulation, and NETosis leading to endothelial injury and complement activation, pauci-immune necrotizing, or crescentic GN | Hilhorst et al. (2015), Jennette and Nachman (2017) |
| C3GN | Anti-C3, anti-C4, anti-C5, anti-factor B | Deposition of granular C3 in the mesangium and glomerular capillary walls, mesangial and endocapillary hypercellularity, tubular atrophy, interstitial fibrosis | Sethi et al. (2012) |
| NS (steroid-sensitive) | Anti-nephrin | Subtle dusting pattern of IgG at podocyte slit, focal segmental glomerulosclerosis, MCD | Doublier et al. (2001) |
| PMN | Anti-semaphorin 3B, anti-PLA2R, anti-PCDH7, anti-contactin 1, anti-TSHD7A, anti-netrin G1, anti-HTRA1, anti-NELL1 | Deposition of granular IgG1, IgG3, IgG4, and/or C3 along the GBM, thickened glomerular capillary wall, focal segmental glomerulosclerosis, tubular atrophy, interstitial fibrosis | Stangou et al. (2019), Tesar and Hruskova (2021) |
| MPGN | Anti-C1q, anti-C3 convertase, anti-factor B and H | Deposition of monoclonal IgG and C3 in glomerular capillary walls and mesangium, global glomerulosclerosis, tubular atrophy, interstitial fibrosis | Nasr et al. (2004) |
| HUS (atypical) | Anti-factor H | Over-activation of alternative complement pathway leading to complement deposition on endothelial cells resulting in endothelial damage and thrombosis in blood vessels | Raina et al. (2022) |
| CV | RF activity involving the binding of IgM to Fc region of IgG | Luminal deposition of cryoglobulin and IgG leading to glomerular vasculitis | Silva et al. (2019) |
| Renal diseases | Antibodies | Histopathological features | References |
|---|---|---|---|
| PSGN | Anti-SpeB, anti-NAPlr, anti-M, anti-SIC(?)a | Deposition of IgG and C3 (occasionally C4, IgM) along glomerular capillary loops and mesangium, glomerular endocapillary hypercellularity, tubulointerstitial oedema | Kraus and Beachey (1988), Balasubramanian and Marks (2017), Satoskar et al. (2020) |
| IgA nephropathy | Anti-Gd IgA1 | Mesangial deposition of IgA and C3, mesangial hypercellularity, tubular atrophy, interstitial fibrosis | Kim et al. (2012), Habas et al. (2022) |
| LN | Anti-dsDNA, anti-nucleosome, anti-histone, anti-C1q | Full-house glomerular deposits (IgA, IgM, IgG, C3, and C1q), endocapillary hypercellularity, hyaline deposition and fibrinoid necrosis in glomeruli, glomerular crescent formation, tubular atrophy, interstitial fibrosis | Yang et al. (2015), Choi et al. (2023) |
| Goodpasture's syndrome (Anti-GBM disease) | Anti-type IV collagen α3 chain | Linear deposition of IgG and/or C3 along the GBM, crescent formation in glomeruli, glomerulosclerosis, interstitial fibrosis, tubular atrophy | Kalluri et al. (1995) |
| ANCA-associated vasculitis and GN | Anti-PR3, anti-MPO | Excessive activation of neutrophil, subsequent degranulation, and NETosis leading to endothelial injury and complement activation, pauci-immune necrotizing, or crescentic GN | Hilhorst et al. (2015), Jennette and Nachman (2017) |
| C3GN | Anti-C3, anti-C4, anti-C5, anti-factor B | Deposition of granular C3 in the mesangium and glomerular capillary walls, mesangial and endocapillary hypercellularity, tubular atrophy, interstitial fibrosis | Sethi et al. (2012) |
| NS (steroid-sensitive) | Anti-nephrin | Subtle dusting pattern of IgG at podocyte slit, focal segmental glomerulosclerosis, MCD | Doublier et al. (2001) |
| PMN | Anti-semaphorin 3B, anti-PLA2R, anti-PCDH7, anti-contactin 1, anti-TSHD7A, anti-netrin G1, anti-HTRA1, anti-NELL1 | Deposition of granular IgG1, IgG3, IgG4, and/or C3 along the GBM, thickened glomerular capillary wall, focal segmental glomerulosclerosis, tubular atrophy, interstitial fibrosis | Stangou et al. (2019), Tesar and Hruskova (2021) |
| MPGN | Anti-C1q, anti-C3 convertase, anti-factor B and H | Deposition of monoclonal IgG and C3 in glomerular capillary walls and mesangium, global glomerulosclerosis, tubular atrophy, interstitial fibrosis | Nasr et al. (2004) |
| HUS (atypical) | Anti-factor H | Over-activation of alternative complement pathway leading to complement deposition on endothelial cells resulting in endothelial damage and thrombosis in blood vessels | Raina et al. (2022) |
| CV | RF activity involving the binding of IgM to Fc region of IgG | Luminal deposition of cryoglobulin and IgG leading to glomerular vasculitis | Silva et al. (2019) |
This mechanism has been proposed based on evidence of kidney patients with serum antibodies against SIC, but has not been established yet.
Abbreviations: ANCA: antineutrophilic cytoplasmic antibody, SpeB: streptococcal pyrogenic exotoxin B, C1q: complement component C1q, C3: complement component 3, C3GN: complement 3 glomerulonephritis, C4: complement component 4, C5: complement component 5, CV: cryoglobulinemic vasculitis, dsDNA: double-stranded deoxyribonucleic acid, Fc region: fragment crystallizable region, GBM: glomerular basement membrane, Gd IgA1: galactose deficient IgA1, GN: glomerulonephritis, HTRA1: human high temperature requirement A1, HUS: haemolytic uremic syndrome, IC: immune complex, IgA: immunoglobulin A, IgG: immunoglobulin G, IgM: immunoglobulin M, LN: lupus nephritis, M: M protein, MCD: minimal change disease, MPGN: membranoproliferative glomerulonephritis, MPO: myeloperoxidase, NAPlr: nephritis-associated plasmin receptor, NELL1: NEL-like protein 1, NET: neutrophil extracellular traps, NS: nephrotic syndrome, PCDH7: protocadherin 7, PLA2R: phospholipase A2 receptor, PMN: primary membranous nephropathy, PR3: proteinase 3, PSGN: post-streptococcal glomerulonephritis, RF: rheumatoid factor, SIC: streptococcal inhibitor of complement, TSHD7A: thrombospondin type 1 domain-containing protein 7A.
Pathological features of immune-mediated renal diseases.
| Renal diseases | Antibodies | Histopathological features | References |
|---|---|---|---|
| PSGN | Anti-SpeB, anti-NAPlr, anti-M, anti-SIC(?)a | Deposition of IgG and C3 (occasionally C4, IgM) along glomerular capillary loops and mesangium, glomerular endocapillary hypercellularity, tubulointerstitial oedema | Kraus and Beachey (1988), Balasubramanian and Marks (2017), Satoskar et al. (2020) |
| IgA nephropathy | Anti-Gd IgA1 | Mesangial deposition of IgA and C3, mesangial hypercellularity, tubular atrophy, interstitial fibrosis | Kim et al. (2012), Habas et al. (2022) |
| LN | Anti-dsDNA, anti-nucleosome, anti-histone, anti-C1q | Full-house glomerular deposits (IgA, IgM, IgG, C3, and C1q), endocapillary hypercellularity, hyaline deposition and fibrinoid necrosis in glomeruli, glomerular crescent formation, tubular atrophy, interstitial fibrosis | Yang et al. (2015), Choi et al. (2023) |
| Goodpasture's syndrome (Anti-GBM disease) | Anti-type IV collagen α3 chain | Linear deposition of IgG and/or C3 along the GBM, crescent formation in glomeruli, glomerulosclerosis, interstitial fibrosis, tubular atrophy | Kalluri et al. (1995) |
| ANCA-associated vasculitis and GN | Anti-PR3, anti-MPO | Excessive activation of neutrophil, subsequent degranulation, and NETosis leading to endothelial injury and complement activation, pauci-immune necrotizing, or crescentic GN | Hilhorst et al. (2015), Jennette and Nachman (2017) |
| C3GN | Anti-C3, anti-C4, anti-C5, anti-factor B | Deposition of granular C3 in the mesangium and glomerular capillary walls, mesangial and endocapillary hypercellularity, tubular atrophy, interstitial fibrosis | Sethi et al. (2012) |
| NS (steroid-sensitive) | Anti-nephrin | Subtle dusting pattern of IgG at podocyte slit, focal segmental glomerulosclerosis, MCD | Doublier et al. (2001) |
| PMN | Anti-semaphorin 3B, anti-PLA2R, anti-PCDH7, anti-contactin 1, anti-TSHD7A, anti-netrin G1, anti-HTRA1, anti-NELL1 | Deposition of granular IgG1, IgG3, IgG4, and/or C3 along the GBM, thickened glomerular capillary wall, focal segmental glomerulosclerosis, tubular atrophy, interstitial fibrosis | Stangou et al. (2019), Tesar and Hruskova (2021) |
| MPGN | Anti-C1q, anti-C3 convertase, anti-factor B and H | Deposition of monoclonal IgG and C3 in glomerular capillary walls and mesangium, global glomerulosclerosis, tubular atrophy, interstitial fibrosis | Nasr et al. (2004) |
| HUS (atypical) | Anti-factor H | Over-activation of alternative complement pathway leading to complement deposition on endothelial cells resulting in endothelial damage and thrombosis in blood vessels | Raina et al. (2022) |
| CV | RF activity involving the binding of IgM to Fc region of IgG | Luminal deposition of cryoglobulin and IgG leading to glomerular vasculitis | Silva et al. (2019) |
| Renal diseases | Antibodies | Histopathological features | References |
|---|---|---|---|
| PSGN | Anti-SpeB, anti-NAPlr, anti-M, anti-SIC(?)a | Deposition of IgG and C3 (occasionally C4, IgM) along glomerular capillary loops and mesangium, glomerular endocapillary hypercellularity, tubulointerstitial oedema | Kraus and Beachey (1988), Balasubramanian and Marks (2017), Satoskar et al. (2020) |
| IgA nephropathy | Anti-Gd IgA1 | Mesangial deposition of IgA and C3, mesangial hypercellularity, tubular atrophy, interstitial fibrosis | Kim et al. (2012), Habas et al. (2022) |
| LN | Anti-dsDNA, anti-nucleosome, anti-histone, anti-C1q | Full-house glomerular deposits (IgA, IgM, IgG, C3, and C1q), endocapillary hypercellularity, hyaline deposition and fibrinoid necrosis in glomeruli, glomerular crescent formation, tubular atrophy, interstitial fibrosis | Yang et al. (2015), Choi et al. (2023) |
| Goodpasture's syndrome (Anti-GBM disease) | Anti-type IV collagen α3 chain | Linear deposition of IgG and/or C3 along the GBM, crescent formation in glomeruli, glomerulosclerosis, interstitial fibrosis, tubular atrophy | Kalluri et al. (1995) |
| ANCA-associated vasculitis and GN | Anti-PR3, anti-MPO | Excessive activation of neutrophil, subsequent degranulation, and NETosis leading to endothelial injury and complement activation, pauci-immune necrotizing, or crescentic GN | Hilhorst et al. (2015), Jennette and Nachman (2017) |
| C3GN | Anti-C3, anti-C4, anti-C5, anti-factor B | Deposition of granular C3 in the mesangium and glomerular capillary walls, mesangial and endocapillary hypercellularity, tubular atrophy, interstitial fibrosis | Sethi et al. (2012) |
| NS (steroid-sensitive) | Anti-nephrin | Subtle dusting pattern of IgG at podocyte slit, focal segmental glomerulosclerosis, MCD | Doublier et al. (2001) |
| PMN | Anti-semaphorin 3B, anti-PLA2R, anti-PCDH7, anti-contactin 1, anti-TSHD7A, anti-netrin G1, anti-HTRA1, anti-NELL1 | Deposition of granular IgG1, IgG3, IgG4, and/or C3 along the GBM, thickened glomerular capillary wall, focal segmental glomerulosclerosis, tubular atrophy, interstitial fibrosis | Stangou et al. (2019), Tesar and Hruskova (2021) |
| MPGN | Anti-C1q, anti-C3 convertase, anti-factor B and H | Deposition of monoclonal IgG and C3 in glomerular capillary walls and mesangium, global glomerulosclerosis, tubular atrophy, interstitial fibrosis | Nasr et al. (2004) |
| HUS (atypical) | Anti-factor H | Over-activation of alternative complement pathway leading to complement deposition on endothelial cells resulting in endothelial damage and thrombosis in blood vessels | Raina et al. (2022) |
| CV | RF activity involving the binding of IgM to Fc region of IgG | Luminal deposition of cryoglobulin and IgG leading to glomerular vasculitis | Silva et al. (2019) |
This mechanism has been proposed based on evidence of kidney patients with serum antibodies against SIC, but has not been established yet.
Abbreviations: ANCA: antineutrophilic cytoplasmic antibody, SpeB: streptococcal pyrogenic exotoxin B, C1q: complement component C1q, C3: complement component 3, C3GN: complement 3 glomerulonephritis, C4: complement component 4, C5: complement component 5, CV: cryoglobulinemic vasculitis, dsDNA: double-stranded deoxyribonucleic acid, Fc region: fragment crystallizable region, GBM: glomerular basement membrane, Gd IgA1: galactose deficient IgA1, GN: glomerulonephritis, HTRA1: human high temperature requirement A1, HUS: haemolytic uremic syndrome, IC: immune complex, IgA: immunoglobulin A, IgG: immunoglobulin G, IgM: immunoglobulin M, LN: lupus nephritis, M: M protein, MCD: minimal change disease, MPGN: membranoproliferative glomerulonephritis, MPO: myeloperoxidase, NAPlr: nephritis-associated plasmin receptor, NELL1: NEL-like protein 1, NET: neutrophil extracellular traps, NS: nephrotic syndrome, PCDH7: protocadherin 7, PLA2R: phospholipase A2 receptor, PMN: primary membranous nephropathy, PR3: proteinase 3, PSGN: post-streptococcal glomerulonephritis, RF: rheumatoid factor, SIC: streptococcal inhibitor of complement, TSHD7A: thrombospondin type 1 domain-containing protein 7A.
Second, glomerular subepithelial immune deposits may cause the damage to the podocytes via three different mechanisms (Fig. 1b; immune complex-mediated injury), i.e. circulatory immune complex formation, in situ immune complex formation, and molecular mimicry. In case of circulatory immune complex formation (Fig. 1b1), antibodies bind the streptococcal antigens (e.g. NAPlr/SpeB/SIC) in the circulation to form complexes. During glomerular filtration, these immune complexes can deposit in the dense capillary tufts of the glomeruli (Satoskar et al. 2014, Balasubramanian and Marks 2017, Satoskar et al. 2020). In the event of in situ immune complex formation (Fig. 1b2), antibodies bind specifically to the streptococcal antigens (e.g. NAPlr/SpeB/SIC) already embedded in the glomerular capillary tufts and form complexes (Balasubramanian and Marks 2017, Satoskar et al. 2020). Additionally, antibodies against streptococci can cross-react (Fig. 1b3) with intrinsic glomerular matrix proteins (extracellular), such as laminin, collagen, and heparan sulphate proteoglycan, via molecular mimicry and form immune complexes (Kefalides et al. 1986, Satoskar et al. 2020).
The immune deposits, in the glomerular capillary tufts (specifically in the subepithelial region) through either of the above-mentioned pathways, represent the accumulation of IgG, IgM, and/or IgA, along with complement C3 (Sorger 1986, Mosquera and Pedreañez 2021). These result in the activation of complements leading to the release of chemotaxins C3a and C5a (Satoskar et al. 2020). This further causes the recruitment of neutrophils, monocytes/macrophages, and helper (CD4) or cytotoxic (CD8) T cells in the glomerulus (Mosquera and Pedreañez 2021). The infiltration of inflammatory cells results in the release of pro-inflammatory cytokines and reactive oxygen species, leading to glomerular inflammation and subsequent damage to the glomerular capillary tufts (Satoskar et al. 2014, Satoskar et al. 2020). The glomerular damage mainly involves injury to the podocyte cytoskeleton leading to podocyte detachment from the GBM, which initiates proteinuria (Mosquera and Pedreañez 2021, Bruno et al. 2023).
Third, SIC through multiple mechanisms may cause podocyte injury that can lead to glomerulonephritis. The SIC-mediated podocyte damage may occur when renal filtration facilitates the ingress of circulating SIC into the glomerular milieu. SIC may subsequently infiltrate podocytes, wherein it binds and co-localizes (intracellularly) with Ezrin (Fig. 1c). Since Ezrin-Radixin-Moesin are the structural proteins of cell membrane and functionally link the cytoskeleton to the plasma membrane, binding of SIC-Ezrin may cause cytoskeletal defects leading to podocytopathy and apoptosis of podocyte, resulting in podocyte detachment from the GBM, alterations in slit diaphragm, and proteinuria. This mechanism is based on the observation that SIC can penetrate intact epithelial cells (an uncommon characteristic for proteins) and bind mainly to the F-actin-binding site located at the carboxyl end of Ezrin (Hoe et al. 2002). This interaction resulted in the collapse of the cytoskeletal structure and the subsequent loss of cellular integrity (Hoe et al. 2002). Additionally, SIC is highly immunogenic, and SIC antibodies are persistent (Hoe et al. 2000). These properties of SIC and anti-SIC antibodies, may provide opportunity for immune complex-mediated damage to glomeruli (as shown in Fig. 1b1, b2).
Pathological features in PSGN
Patients with PSGN can be diagnosed (Fig. 2) based on histopathology, immunofluorescence, and electron microscopy findings (Rodríguez-Iturbe and Batsford 2007, Satoskar et al. 2020, Alhamoud et al. 2021). Using light microscopy, normal and abnormal glomeruli can be typically identified (Fig. 2a–c). Abnormal glomeruli are characterized by diffuse or focal endocapillary hypercellularity (Satoskar et al. 2014, Satoskar et al. 2020). In many cases, numerous polymorphonuclear leukocytes (neutrophils) are seen. Glomerular crescents are uncommon and are associated with adverse clinical outcomes (Balasubramanian and Marks 2017, Satoskar et al. 2020). Mesangial hypercellularity and mild expansion of mesangial matrix can also be detected (Fogo and Kashgarian 2016, Satoskar et al. 2020). Acute tubular necrosis may be evident, identifiable by irregular dilation of tubules and the presence of eosinophilic cast-like materials or sloughed semi-viable tubule epithelial cells within the renal tubules (Jennette et al. 2014, Satoskar et al. 2020). Additionally, there may be a variable degree of tubulointerstitial inflammation and oedema. Immunofluorescence (Fig. 2d) shows a classical granular ‘lumpy-bumpy’ C3 deposition (with or without IgG) along the glomerular capillary loops and mesangium variably described as garland pattern and starry sky pattern (Satoskar et al. 2014, Satoskar et al. 2020, Rawla et al. 2022). In later stages of the disease, only the mesangial staining is detectable. Electron microscopy (Fig. 2e) frequently shows large and scattered dome-shaped subepithelial electron-dense deposits (humps) (Satoskar et al. 2014, Rawla et al. 2022). These distinctive subepithelial deposits are usually detectable within the first few weeks of PSGN, later decreasing in number. Thus, sparse mesangial and subendothelial deposits are typically found in most cases of PSGN (Satoskar et al. 2020).
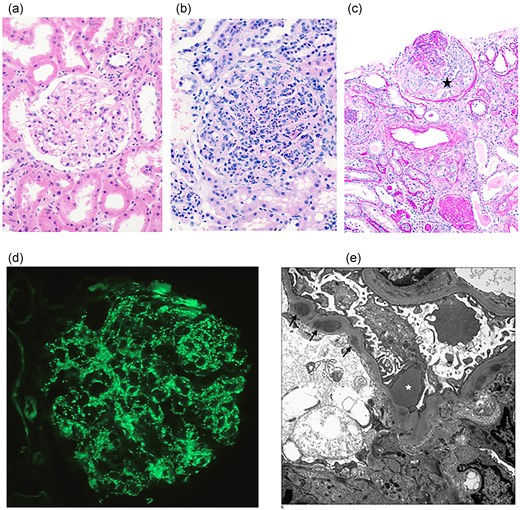
Histological features of kidney pathology. (a) Light microscopy demonstrating normal renal cortex (Haematoxylin and Eosin stain, 200 × magnification); (b) Light microscopy demonstrating abnormal glomerulus in PSGN with diffuse hypercellularity including numerous neutrophils and mesangial cell increase (Haematoxylin and Eosin stain, 200 × magnification); (c) In PSGN, a cellular crescent (black star) is observed in the glomerulus (Periodic acid-Schiff/diastase stain, 100 × magnification); (d) Immunofluorescence demonstrating granular staining of C3 along the glomerular capillary loops in PSGN (400 × magnification); (e) Electron microscopy demonstrating characteristic subepithelial deposit/hump (white star) and several smaller subendothelial deposits (arrows) in PSGN.
Major streptococcal antigens associated with PSGN
SpeB
SpeB is an extracellular, cationic cysteine protease secreted as a zymogen by GAS and is recognized as a potent nephritogen (Batsford et al. 2005, Satoskar et al. 2020). SpeB has an ability to convert the plasminogen into plasmin (Batsford et al. 2005, Satoskar et al. 2020). The activated plasmin causes proteolysis leading to glomerular damage, resulting in the development of PSGN. In addition to the interaction with plasminogen, SpeB cleaves C3 and subsequently activates the alternative complement pathway. Furthermore, like other superantigens, SpeB predominantly binds to the major histocompatibility complex class II molecules on the antigen-presenting cells and to specific Vβ chain of T cell receptors, which ultimately leads to hyper-stimulation and activation of T cells and initiates the inflammation of glomeruli (Satoskar et al. 2020). SpeB can form complexes with immunoglobulin and co-localize with complement in glomerular subepithelial deposits (humps), the hallmark of PSGN (Batsford et al. 2005). The localization of SpeB in the glomerulus is facilitated by the cationic charge of this protein, allowing interaction with the negatively charged GBM (Vogt et al. 1990). Glomerular deposits of SpeB have been detected in kidney biopsies of patients with PSGN and in a mouse model of glomerulonephritis (Batsford et al. 2005, Luo et al. 2007). Earlier research has demonstrated that majority of the convalescent sera from patients with kidney diseases were found with higher antibody titres of SpeB and its zymogen precursor (zSpeB) than those of other streptococcal antigens, i.e. streptolysin O, DNase B, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Parra et al. 1998, Batsford et al. 2005).
SpeB may also exhibit other distinct biological properties. For instance, SpeB and zSpeB have been found to induce apoptosis and proliferation in human leukocytes (Viera et al. 2001). Moreover, they can increase the production of cytokines in leukocytes (Viera et al. 2007). SpeB can also promote the production of angiotensin II, along with increasing oxidative stress, in mesangial cells and leukocytes (Viera et al. 2009). Increased angiotensin II and oxidative stress are predicted to play crucial roles as mediators of inflammatory processes in renal tissues and in the context of PSGN. It is interesting, however, that the gene encoding SpeB is absent in all strains of SDSE (Xie et al. 2024). This suggests that SpeB is not the sole contributor to the aetiology of PSGN.
NAPlr
The NAPlr is a cell surface protein and nephritogen of GAS, which shows distinct homology in the nucleotide sequence with streptococcal GAPDH and is regarded as structurally and functionally identical to GAPDH (Winram and Lottenberg 1996, Yoshizawa et al. 2004, Oda et al. 2010, Oda et al. 2012, Uchida and Oda 2020). Circulating NAPlr can deposit in the mesangium and GBM by adhesion. It can also be localized within the glomerular endocapillary neutrophils or on the endothelial cells of patients with PSGN (Yamakami et al. 2000, Oda et al. 2010). The NAPlr-positive neutrophils can cause damage to the mesangium and GBM by promoting the plasmin-catalyzed proteolysis. NAPlr activates plasmin by binding with high affinity and maintains its proteolytic activity (Lottenberg et al. 1987). Activated plasmin can directly damage glomerular extracellular matrix proteins fibronectin and laminin, or indirectly by activating pro-matrix metalloproteases (Vassalli et al. 1991, Uchida and Oda 2020). Plasmin can also initiate glomerular inflammation by attracting monocytes and neutrophils (Montrucchio et al. 1996, Burysek et al. 2002) and further promote immune complex deposition in the glomerular subepithelial spaces. The immune complexes can easily pass through the damaged GBM to form subepithelial deposits (humps) (Mosquera and Pedreañez 2021). It has been reported that NAPlr can also activate the alternative complement pathway and cause glomerular capillary damage (Yoshizawa et al. 2004).
Urinary plasmin was found to be higher in patients with PSGN than in healthy individuals (Oda et al. 2012, Satoskar et al. 2020). Glomerular deposition of NAPlr was observed in 100% of patients with PSGN within two weeks after the disease onset and in 84% of patients within a month after the disease onset (Oda et al. 2012). This suggests that NAPlr deposition in renal glomeruli decreases over time. A study from Japan showed anti-NAPlr antibody in >90% of convalescent serum samples from patients with PSGN, but only in 60% of patients with uncomplicated streptococcal infections (Yoshizawa et al. 2004). In addition, antibodies against NAPlr have been detected in a minor proportion (<5%) of patients with renal complications other than PSGN (Yamakami et al. 2000).
SIC
SIC is a negatively charged, extracellular protein secreted by M1 and M57 GAS strains (Akesson et al. 1996, Fernie-King et al. 2001, Binks et al. 2003). A SIC-like protein called ‘distantly related to SIC’ (DRS) that shares some biological properties with SIC has also been reported to be produced by M12 and M55 GAS and some strains of SDSE (Minami et al. 2011, Smyth et al. 2014).
SIC has a wide range of activities. It inhibits the function of the membrane attack complex (MAC) of complement by restricting the insertion of C5b67 into the bacterial cell membrane, thereby inhibiting complement-mediated lysis of bacteria (Akesson et al. 1996, Westman et al. 2018). However, inhibition of complement-mediated cell lysis may not be of significance for Gram-positive GAS. SIC binds to another MAC inhibitor clusterin and also to histidine-rich glycoproteins, inhibiting the complement system and potentially leading to the deposition of SIC in the affected glomeruli of patients with prior streptococcal infections (Akesson et al. 1996, Sriprakash et al. 2002). Furthermore, SIC can block the cytolytic properties of several antimicrobial peptides found on mucosal surfaces (Fernie-King et al. 2002, Frick et al. 2003, Fernie-King et al. 2004, Binks et al. 2005, Åkesson et al. 2010, Pence et al. 2010, Frick et al. 2011).
A higher proportion of subjects with recorded history of PSGN in a PSGN-endemic region of the Northern Territory of Australia, showed antibodies to SIC and DRS than those with no recorded history of PSGN (Martin and Sriprakash 1996). This suggests an association between SIC and DRS in the development of PSGN. Furthermore, past infection with SIC-positive GAS is a risk factor for CKD in the Mumbai area (Karmarkar et al. 2013). However, the exact mechanisms of how SIC and/or DRS may contribute to the pathogenesis of PSGN, CKD, and kidney failure have yet to be investigated. The availability of a preclinical animal model will enable interrogation of the early events following exposure to SIC and other proteins associated with PSGN.
M protein
The M protein is a cell-wall-associated protein and a major virulence factor of various GAS serotypes (Cunningham 2000). Cross-reactivity of M proteins with basement membrane and renal glomeruli has been documented in several studies. Sera from patients with PSGN have demonstrated cross-reactivity with basement membrane collagen, laminin, and heparan sulphate proteoglycan, while sera generated in rabbits against amino terminus of streptococcal M protein have shown cross-reactivity with human glomeruli (Fillit et al. 1985, Kefalides et al. 1986, Kraus and Beachey 1988). Additionally, streptococcal M protein has been found to contain an autoimmune epitope shared with human renal glomeruli and myocardium (Kraus et al. 1990).
M proteins have been shown to be associated with the pathogenesis of PSGN. Kaplan experimentally demonstrated that glomerular tufts in mice were one of the primary sites for the deposition of M proteins (Kaplan 1958). Furthermore, Kantor (1965) found that streptococcal M protein was effective in inducing glomerular lesions in rats and mice. These lesions were characterized by localized M protein-fibrinogen complexes, implicating M protein in the pathogenesis of PSGN. Furthermore, Humair et al. (1969) observed that M protein, either independently or in combination with fibrinogen, induced kidney lesions in mice. Whereas, Burova et al. (2013) induced experimental glomerulonephritis in rabbits with GAS M protein (type12), characterized by anti-IgG production, increased glomerular deposition of IgG, and immune complexes, with subsequent complement activation. In contrast, studies on renal specimens of patients with PSGN have indicated that factors other than M protein may contribute more significantly to the development of PSGN (Treser et al. 1971, Rodríguez-Iturbe et al. 1980).
Streptokinase
Streptokinase is an extracellular secreted protein of GAS (Johnston and Zabriskie 1986). It is non-protease in nature and can act as a plasminogen activator (Babashamsi et al. 2009). The association of streptokinase with PSGN has been reported in several studies. Streptokinase was found to be a mediator of PSGN in a mouse model, characterized by the glomerular deposition of streptokinase and subsequent activation of complement cascade (Nordstrand et al. 1998). However, Nordstrand et al. (1999) observed that renal interactions with certain nephritis-associated variants of streptokinase might be needed for the initiation of PSGN. To confirm this observation, allelic substitution of streptokinase gene was carried out, resulting in reduced nephritogenic capacity (Nordstrand et al. 2000). While some investigations suggest a correlation between streptokinase and the pathogenesis of PSGN, other studies have reported inconsistent associations (Villarreal et al. 1979, Mezzano et al. 1992, Tewodros et al. 1995).
Streptokinase can activate plasminogen, leading to subsequent plasmin-mediated degradation of extracellular matrix proteins and local activation of complement and coagulation pathways (Bajaj and Castellino 1977, Nordstrand et al. 1999). Streptokinase-mediated plasminogen activation has been shown to vary among animal species and humans (Wulf and Mertz 1969). Poon-King et al. (1993) identified another protein with human plasmin-binding property other than streptokinase, in the same nephritogenic strain of streptococcus, although did not further characterize this protein. To identify the association and/or specific roles of streptococcal proteins, either by themselves or in combination, in the pathogenesis of PSGN, robust animal models may be required.
Treatment and prevention of PSGN
Early diagnosis and subsequent antibiotic treatment (penicillin or erythromycin) of streptococcal infections can prevent the development of PSGN (Bateman et al. 2022). Protecting the glomeruli from persistent insults via the aforementioned pathways implicated in PSGN is a promising avenue to mitigate progression. Plasmin-mediated injury to the renal glomeruli leading to proteinuria can be inhibited by amiloride (Egerman et al. 2020). This agent can inhibit the plasminogen activation, leading to prevention of plasmin-mediated injuries to GBM and podocytes. Furthermore, this inhibitory effect can prevent plasmin-mediated stimulation of epithelial sodium channel activity and subsequent sodium retention contributing to hypervolemia and hypertension (Ray et al. 2018, Chen et al. 2019). Hence, plasminogen and proteinuria may serve as biomarkers for monitoring the progression of PSGN.
Therapeutic approaches targeting both the induction and inhibition of apoptosis to resolve hypercellularity and inflammation might be effective to prevent chronicity in patients with PSGN (Mosquera-Sulbaran et al. 2024). In cases of immune-mediated glomerulonephritis (IMGN) caused by circulating antibodies, plasmapheresis with endopeptidase imlifidase has been used to rapidly remove nephritogenic antibodies (Uhlin et al. 2022). In addition, glucocorticoids remain a treatment option for IMGN, although their uses are limited by their side effects. Monoclonal antibodies (belimumab/rituximab), complement inhibitors (C5aR inhibitors), and calcineurin inhibitors can be used for the treatment of IMGN as steroid-sparing agents (Anders et al. 2023).
Actin cytoskeleton stabilization is crucial for treating podocytopathies. In podocyte injury, various proteins (nephrin, podocin, CD80, synaptopodin, and α3 integrin) act as urinary biomarkers (Singh et al. 2015). In this context, rituximab, cyclosporine, and abatacept have been described as potential therapeutics (Singh et al. 2015). Additionally, podocyte replenishment using fibroblast growth factors, cyclin-dependent kinase inhibitors, or glycogen synthase kinase-3 inhibitors could be effective treatment options (Obligado et al. 2008, Singh et al. 2015). Therefore, therapeutic approaches potentially include immunosuppressive, immunomodulatory, and podocyte-stabilizing agents, as well as treatments targeting both systemic and intraglomerular hypertension. To evaluate the efficacy of these therapies in preventing the progression of PSGN, well-characterized preclinical models are required.
Currently several streptococcal vaccines, including streptococcal M protein-based vaccines are being developed to prevent streptococcal infections, which have the potential to significantly decrease the incidence of post-streptococcal complications (Dooley et al. 2021, Fan et al. 2024). While designing such vaccines, consideration should also be given to cover streptococcal strains that are known to produce proteins implicated in the pathogenesis of PSGN.
Necessity for preclinical models
Experimental models, i.e. in silico, in vitro, in vivo, and bioengineered models, can provide vital clues regarding the pathophysiology of various renal diseases (Gozalpour and Fenner 2018, Rizki-Safitri et al. 2021, Shi et al. 2021, Bichlmayer et al. 2022). Due to limited access to kidney biopsies and the ethical consideration of obtaining tissue samples from patients, animal models are ideal to investigate the early events in the pathogenesis of PSGN, CKD, and kidney failure (Sriprakash et al. 2002, Oda et al. 2010, Oda et al. 2012, Karmarkar et al. 2013). Induced (surgical/chemical) and spontaneous models have been reported for the studies of renal fibrosis and glomerular nephropathy (Hosszu et al. 2021, Liang and Liu 2023). In a review by Yang et al. (2010), various models (spontaneous glomerular and interstitial injury models, hypertensive vascular injury models, genetically engineered models, and acquired models) have been cited for investigating CKD.
Laboratory animals, such as rats, mice, and rabbits, have been used to study the pathogenesis of immune-mediated renal complications (Nordstrand et al. 1996, Nordstrand et al. 1998, Burova et al. 2013, Naka et al. 2020). Of the many described models to study glomerular complications and nephropathy, only a limited number of animal studies have investigated the role of SpeB in PSGN (Kuo et al. 1998, Kuo et al. 2004, Luo et al. 2007). To our knowledge, no animal studies have been conducted to determine the role of NAPlr or SIC, either independently or in combination with other streptococcal proteins, in the pathogenesis of PSGN.
Rodent models are the preferred models due to the convenience of animal management, alongside the animals’ rapid reproductive maturity and shorter life span (Ericsson et al. 2013, Rush et al. 2014). Rat strains (Wistar, Lewis, Fawn-hooded, Fisher, and Sprague–Dawley) have been most commonly used as models for renal and cardiovascular diseases (Grossman 2010). Lewis rats have been used successfully to model autoimmune complications (i.e. rheumatic heart disease and autoimmune encephalomyelitis) (Pitarokoili et al. 2017, Rafeek et al. 2021, Rafeek et al. 2022). In particular, the inbred male Lewis rats have an increased tendency to develop autoimmune complications compared to cross-bred rats. While age-related renal impairments (i.e. glomerulonephritis, renal fibrosis, and degeneration) are more common in male rats than in female rats (Yang et al. 2010), male rats have fewer hormonal fluctuations with age. Therefore, male Lewis rats may be a desirable preclinical model for the study of PSGN.
Conclusions
Undiagnosed or untreated PSGN leads to life-threatening complications, which require renal replacement therapy. Currently, kidney biopsy, along with haematobiochemical assays and urinalysis, remains the key to clinical diagnosis. SpeB and NAPlr have been established as major nephritogens of streptococci. Other streptococcal virulence factors, such as M protein and streptokinase, although associated with PSGN, are no longer considered primary nephritogens. There is also clinical evidence to suggest an association of another streptococcal virulence factor, SIC, with PSGN. Overall, our current understanding suggests that the aetiology of PSGN could be attributable to possibly multiple streptococcal factors operating by multiple mechanisms. For further understanding, a suitable animal model is needed, which may also facilitate the assessment of potential therapeutic approaches. Homing in on the streptococcal factors responsible for PSGN and exploring therapeutic modalities may offer us a hand in preventative measures.
Acknowledgements
Ethical approval is not required for this work/manuscript.
Author contributions
Mohammad Raguib Munif (Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing), Robert A. Hart (Conceptualization, Supervision, Validation, Writing – review & editing), Rukshan A.M. Rafeek (Conceptualization, Supervision, Validation, Writing – review & editing), Amali C. Mallawaarachchi (Methodology, Resources, Writing – review & editing), Lyndal Anderson (Methodology, Resources, Writing – review & editing), David J. McMillan (Conceptualization, Supervision, Validation, Writing – review & editing), Kadaba S. Sriprakash (Conceptualization, Supervision, Validation, Writing – review & editing), and Natkunam Ketheesan (Conceptualization, Supervision, Validation, Writing – review & editing)
Conflict of interest
None declared.
Funding
Mohammad Raguib Munif is supported by the University of New England Strategic Scholarship. Research conducted by Natkunam Ketheesan, Rukshan AM Rafeek, and David J McMillan is part funded by NHMRC Ideas Grant (APP 2010336).
Data Availability
All data are presented within this manuscript.

{kind=link}
{kind=link}