





Abstract
Epstein–Barr virus (EBV) may cause harm in immunocompromised conditions or on stress stimuli. Various chemical agents have been utilized to induce the lytic cycle in EBV-infected cells. However, apart from chemical agents and external stress stimuli, certain infectious agents may reactivate the EBV. In addition, the acute infection of other pathogens may provide suitable conditions for EBV to thrive more and planting the roots for EBV-associated pathologies. Various bacteria such as periodontal pathogens like Aggregatibacter, Helicobacter pylori, etc. have shown to induce EBV reactivation either by triggering host cells directly or indirectly. Viruses such as Human simplex virus-1 (HSV) induce EBV reactivation by HSV US3 kinase while other viruses such as HIV, hepatitis virus, and even novel SARS-CoV-2 have also been reported to cause EBV reactivation. The eukaryotic pathogens such as Plasmodium falciparum and Aspergillus flavus can also reactivate EBV either by surface protein interaction or as an impact of aflatoxin, respectively. To highlight the underexplored niche of EBV reactivation by biological agents, we have comprehensively presented the related information in this review. This may help to shedding the light on the research gaps as well as to unveil yet unexplored mechanisms of EBV reactivation.
Introduction
Epstein–Barr virus (EBV) is an ubiquitous human virus with prevalence in 90% of the adult population. It was the first human oncogenic virus to be identified in Burkitt lymphoma cells (Young and Rickinson 2004). The virus is primarily the causative agent of infectious mononucleosis (Cohen 2000). After acute infection, the virus can remain indefinitely latent in cells. However, under immunologically challenging conditions, such as infections, cancers, or autoimmune diseases, the virus can reactivate in the body and further establish a chronic infection. Chronic EBV infection has been associated with various pathologies, such as Burkitt’s lymphoma, Hodgkin’s disease, nasopharyngeal carcinoma, gastric adenocarcinoma, post-transplant lymphoproliferative disease, nasal T-cell/NK cell lymphoma, primary nervous system lymphoma, and AIDS-associated lymphoblastoma (Farrell 2019). Furthermore, it has also been reported to be possibly associated with neurological diseases, such as Alzheimer’s disease, multiple sclerosis, Sjorgren’s syndrome, rheumatoid arthritis, inflammatory bowel syndrome, and so on (Indari et al. 2022b,e, Zhang et al. 2022).
EBV possesses a ∼184-kb long genome consisting of a linear double-stranded DNA molecule that encodes ∼100 proteins. These proteins play roles in controlling viral gene expression, viral DNA replication, production of virion structural components, and regulation of the host immune response. This virus primarily infects epithelial cells and B lymphocytes but can also target other types of cells (Indari et al. 2021b, 2022d). The route of transmission comes into close contact with the oropharyngeal secretions of an infected individual. After contact, the virus present in the secretion can infect the host epithelial cells and further spread to other nearby epithelial cells, as well as B lymphocytes, following lytic infection (Cohen 2000). The presence of the virus in peripheral blood lymphocytes may facilitate the spread of the infection through blood transfusions and donated organs. Although it is not well documented, transmission of infection can also occur through sexual contact (Allen 2005). The life cycle of EBV is divided into two phases: the latent phase, in which the virus remains asymptomatic and produces the least number of proteins, and the lytic phase, in which most virulent viral proteins are produced. Many of the abovementioned disease pathologies are accelerated by the ability of the virus to be in the latent and lytic stages interchangeably. While EBV generally remains latent in cancerous cells, some studies have suggested that its lytic cycle may be involved in cancer onset and progression (Xue et al. 2002, Zheng et al. 2015) because lytic replication promotes viral or host cytokine and growth factor secretion, which contributes to inflammation and angiogenesis (Ye et al. 2021, Yokoe et al. 2022).
Out of almost a 100 genes expressed during active replication, only a few are expressed in latently infected B lymphocytes (Münz 2019). The latent genes produce proteins that aid in the maintenance of the viral genomic DNA as an episome, which replicates during the S-phase of cell division and transit to both daughter cells in mitosis. Because of the restricted number of proteins that can be targeted by the host immune system, this manner of infection is particularly favorable to the virus, since it allows it to persist for extended periods of time (Murata and Tsurumi 2014).
Latently persisting EBV can be reactivated under certain conditions, such as stress, pharmacological stimulation, and immunosuppression. Certain chemical or biological inducers, such as calcium ionophores, DNA methyltransferases, and tumor growth factor (TGF)-β inhibitors, can also cause this effect. In addition, chemical agents, such as 12-O-tetracanoylphorbol-13-acetate (TPA) and histone deacetylase inhibitors, may reactivate the virus. During the lytic phase of EBV, all lytic genes are expressed in a timely manner based on their regulatory role in the viral life cycle (Münz 2019). This latent-to-lytic switch, which is known as reactivation, is divided into three main stages. In the first stage, the immediate early genes like BZLF1 (protein ZEBRA or Zta) and BRLF1 (protein Rta) are transcribed immediately after the viral entry within host cells or receiving an inducing signal. In particular, the immediate-early genes encoded during the initial period of infection play a more prominent role in the reactivation of EBV as well. These genes not only activate the viral lytic cascade but also synergistically activate their own and each other’s promoters, resulting in a significant upregulation in the expression of the viral lytic phase. Among other factors, this synergistic action is required for EBV reactivation. These are activators of transcription and lead to the expression of early stage genes, such as DNA polymerase catalytic subunits BALF1, BMRF1, and BALF2, which are single-stranded DNA-binding proteins. Following the formation of the components, replication of the viral genome occurs. In the third stage, i.e. the late stage, the late genes are expressed once the replication process is completed. The proteins encoded by these genes are responsible for the formation of the structural components of the virus, including capsid proteins, viral antigens, and glycoproteins. Once the structural components are formed, the assembly of the components and genome occurs following the enveloped viral assembly pathway. After the assembly of all viral parts, the virus buds out from the host cell (Young et al. 2007).
The mechanism of reactivation at the cellular level involves the induction of cellular components and the downstream interplay of molecular signaling pathways (Fig. 1). Several studies have shown that the pathways, such as Phosphoinositide 3-kinase (PI3K)/Akt serine/threonine kinase (AKT) pathway, c-Jun N-terminal Kinase (JNK)/Signal transducer and activator of transcription (STAT) pathway, and mitogen-activated protein kinase (MAPK) pathway are found to be involved in the reactivation mechanism of the virus (Luo et al. 2021). In case, if the Protein kinase C (PKC) pathway is triggered, the transcription factor Activator protein 1 (AP-1) is activated. This further binds with the BZLF1 promoter (Gradoville et al. 2002). Similarly, if the MAPK pathway is activated, downstream proteins such as JNK, P38, or ERK are activated (Li et al. 2016). The expression of these proteins may lead to the activation of transcription factors, such as Activating transcription factor (ATF)/Transcription factor Jun (cJUN)/cAMP Response Element-Binding Protein (CREB). Additionally, activation of the PI3K pathway triggers proteins such as p38 or Extracellular signal-regulated kinase (ERK), which is followed by the activation of several transcription factors, resulting in EBV reactivation (Li et al. 2016). Several intrinsic cellular factors can also be responsible for the virus reactivation. These factors may induce cellular damage such as double-stranded DNA breaks leading to DNA damage (Fig. 1) (Cao et al. 2018). When this happens, ataxia telangiectasia mutated (ATM) protein kinase is activated for the maintenance and repair of DNA by downstream activation of AKT. This AKT protein then activates either of the transcription factors, such as Myocyte Enhancer Factor 2D (MEF2D) or Specific protein 1 (Sp1), which bind to the ZI motif of the BZLF1 promoter, ultimately resulting in EBV reactivation.
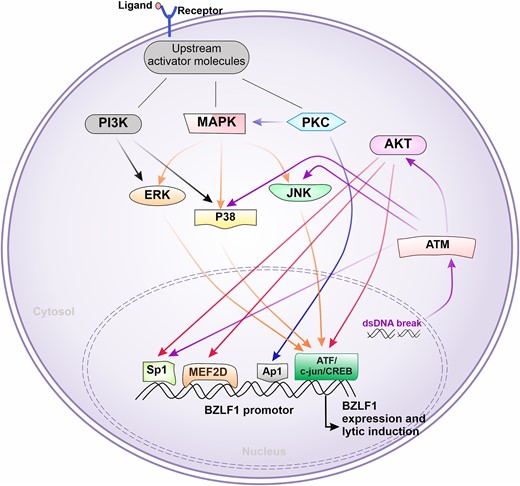
Molecular pathways commonly involved in EBV reactivation. After receiving stimulus from external factors such as binding of ligand to receptor (e.g. pathogen-associated molecular pattern to pattern recognition receptor), the downstream molecular pathways can be initiated. Initially, either one of the proteins PI3K MAPK or PKC can get triggered or multiple pathways can be triggered. This, in turn, ends up inducing the transcription factors such as Sp1, MEF2D, AP1, or ATF/c-jun/CREB, which can go and bind BZLF1-promotor region. This process initiates the BZLF1 transcriptional activation, a start point of the EBV lytic cycle. Sometimes, the pathway can also be triggered by internal factors such as double-stranded DNA derived by damaged mitochondria or nucleus, which further activates ATM. This further can drive Sp1 induction or may move forward with AKT activation cascade as shown in the figure.
Although the specific physiological triggers that cause EBV to reactivate are not clearly understood, various factors or compounds secreted by biological entities, such as bacteria, protozoans, helminths, fungi, and other viruses may induce the EBV reactivation. During infection by other pathogens, an active immune response and subsequently modified intercellular crosstalk can lead to EBV reactivation. However, this may not necessarily occur in the case of every pathogenic infection in EBV-infected individuals. It has been observed that during infection with specific pathogens, the molecular machinery of EBV is prominently activated, leading to an increase in its expression and ultimately reactivation (Daud et al. 2015, Hirsiger et al. 2019, Dávila-Collado et al. 2020). This can result in increasing health complications, disease severity, and even changes in disease symptoms, which may lead to misdiagnosis. If the mechanism of EBV reactivation by such pathogens is understood, it can be targeted for therapeutic interventions to avoid the above-mentioned complications. There are few studies, exploring the mechanism of EBV reactivation induced by other pathogens, which are scattered in different reports. In this review, we comprehensively summarize the information available regarding pathogens that have been reported or may possibly induce EBV reactivation. This review highlights the gaps that need to be studied further at the experimental and clinical levels with respect to EBV reactivation in other infections. This may further help in developing strategies to combat complications associated with EBV reactivation in diseased individuals.
EBV reactivation by bacteria
Several bacterial infections have been reported to induce EBV reactivation (Fig. 2). Some of the studies have elucidated the molecular mechanism of EBV reactivation by these bacterial infections, which are explained below.
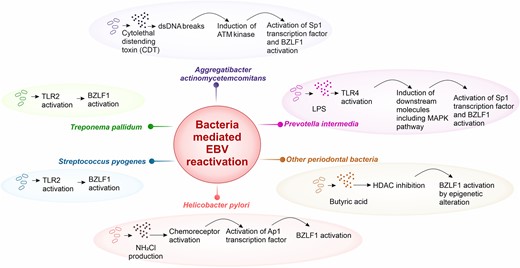
Schematic representation of bacteria-mediated EBV reaction. Various bacteria can induce EBV reactivation through inducing host proteins playing role in lytic cycle activation. Aggregatibacter actinomycetemcomitans produces cytolethal distending toxin (CDT), which causes double-stranded DNA breaks. This further activates the ATM protein cascade inducing Specific protein 1 (Sp1) and BZLF1 activation. Prevotella intermedia triggers MAPK pathway through toll-like receptor (TLR) 4 and ultimately, stimulates Sp1, which binds BZLF1. Other periodontal bacteria, such as Porphyromonas gingivalis secrets butyric acid, which inhibits the histone deacetylase (HDAC), this drives the epigenetic alteration leading to expose BZLF1 promoter region and its activation. Helicobacter pylori changes the cellular microenvironment at the site of infection and can lead to generation of NH2Cl. This triggers the TlpD chemoreceptor and could cause BZLF1 induction via Ap1 transcription factor activation. Some other bacteria such as Streptococcus pyogenes and Treponema pallidum have also shown to reactivate EBV. Their tendency to activate TLR2 could further initiate downstream pathway for BZLF1 activation and lytic induction.
Aggregatibacter actinomycetemcomitans
Aggregatibacter actinomycetemcomitans is a common oral pathogenic Gram-negative anaerobic bacterium closely associated with periodontitis (Fine et al. 2019). Periodontitis is a serious disease in which destruction of the gums and jaw bones of positive patients occurs due to bacterial infection (Fine et al. 2019). Studies have shown the involvement of EBV and periodontal pathogens in causing chronic periodontitis (Imai et al. 2012a, Maulani et al. 2021). The periodontal bacterium, A. actinomycetemcomitans, was found to reactivate EBV in its productive viral cycle when infected with EBV-carrying epithelial cells (Frisan et al. 2019). Observational studies in some strains of the bacterium have shown its potential to secrete CDT (Bezine et al. 2014). This heat-labile toxin, which comprises three protein subunits, two CdtA subunits, and one CdtB subunit, plays a vital role in causing double-stranded DNA damage (by the action of CdtB) (Bezine et al. 2014). In particular, it activates ATM kinase, which plays a significant role in the DNA double-strand break response (Frisan et al. 2019). This process involves phosphorylation of (Kruppel-associated box)-associated protein 1 (KAP1) and H2A histone family member X (H2AX). These two factors act as sensors of DNA damage response amplifiers and also play a role in the recruitment of DNA repair effectors, such as 53BP1, cell cycle arrest effectors, such as Checkpoint kinase 2 (CHK2) and p53, several other gene transcription factors, and chromatin remodeling. SP1, one of the major targets of ATM, has been shown to activate BZLF1 transcriptionally. The attachment of the transcription factor to the promoter of BZLF1 further leads to the expression of the lytic genes of the EBV, denoting its reactivation (Frisan et al. 2019).
Other periodontal pathogens
Porphyromonas gingivalis, Fusobacterium nucleatum, and P. endodontalis significantly contribute to periodontitis (Popova et al. 2013). According to several studies, the viral ZEBRA protein acts as an activator of the BZLF1 promoter and is a master regulator of the reactivation switch from the latent to a lytic phase of EBV. Studies have shown that BZLF1 is transcriptionally activated by hyperacetylation of histone core proteins near the BZLF1 promoter (Murata et al. 2012). Imai et al. (2012a) examined the effect of P. gingivalis on EBV reactivation in Burkitt lymphoma cells. In this study, the level of ZEBRA protein expression was observed when the culture supernatant of P. gingivalis W83 was added to the cell line culture. Moreover, EBV early antigen diffuse protein (EA-D) expression was also induced by the supernatant. On further testing, the supernatant was found to be composed of short chain fatty acids (SCFAs) and a portion of butyric acid. According to several studies, butyric acid was found to have the capability to inhibit the function of histone deacetylase (HDAC) resulting in increased histone acetylation. This hike in the histone acetylation may result in the transcriptional activation of BZLF1 promoter (Imai et al. 2012a). Similar mechanisms can be followed by butyric acid producing anaerobic bacteria found in the gastrointestinal tract such as P. endodontalis, F. nucleatum, and Clostridium, Eubacterium (Imai et al. 2012b, Makino et al. 2018, Himi et al. 2020).
Prevotella intermedia
Gram-negative, anaerobic, rod-shaped Pr. intermedia is also a periodontal pathogen that causes periodontitis (Popova et al. 2013). The oral epithelium and connective tissues are the major sites of bacterial colonization. Like other periodontal pathogens, these bacteria also secrete butyric acid but at very low concentrations. Therefore, it is assumed that this bacterium does not follow the HDAC inhibition pathway, but mediates the reactivation of EBV through alternate pathways (Himi et al. 2020).
The main structural component of the cell wall of this bacterium is lipopolysaccharide (LPS). Studies have shown that LPS can stimulate inflammatory responses during periodontitis, including triggering mononuclear phagocytes to release interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α (Morrison and Ryan 1987). It has been reported that the LPS of Pr. intermedia has unique chemical and immunobiological properties in comparison with the LPS of the family Enterobacteriaceae (Hamada et al. 1990). In 2003, Hashimoto et al. (2003) demonstrated that LPS from Pr. intermedia can activate murine cells through a TLR4-mediated signaling pathway. TLR4 is a cell-surface receptor. It activates both myeloid differentiation primary response protein (MyD)88-dependent pathway and TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathways. After the activation of TLR4, the signal is conducted downstream via Tumor necrosis factor receptor-associated factor (TRAF) to TRIF. Studies have shown that TRIF interacts with TRAF6, which recruits receptor-interacting protein (RIP)-1 kinase. This kinase interacts with the transforming growth factor-β-activated kinase 1 (TAK-1) complex. This interaction results in the activation of the complex and leads to the activation of the nuclear factor kappa B (NF-κB) and MAPK signaling pathways. This activation induces cytokine release. Activation of the TAK-1 complex leads to switching of MAPK into its active form (Kawasaki and Kawai 2014). As a result of this activation, transcription factor AP-1 is converted into the active form, which, in turn, binds to the BZLF1 promoter, leading to the expression of lytic genes and the reactivation of EBV.
Helicobacter pylori
Helicobacter pylori is a spiral-shaped bacterium that grows in the mucus layer and coats the inside of the human stomach (Kashyap et al. 2023). This bacterium is associated with gastritis, intestinal metaplasia, gastric mucosal atrophy, and other gastric infections (Kashyap et al. 2023). Infection with H. pylori was found to be significantly related to gastric cancer, but the presence of H. pylori infection along with EBV DNA was associated with both gastric cancer and peptic ulcer disease (Shukla et al. 2011). The EBV DNA viral load was higher in H. pylori-positive patients, suggesting a role for the bacterium in the reactivation of EBV (Shukla et al. 2011). The gastric mucosa colonized by H. pylori has a characteristic feature of accumulation of neutrophils and lymphocytes, which are dominantly seen in the gastric mucosa. Activated neutrophils produce superoxide (O2−) and hydrogen peroxide (H2O2), which are further catalyzed by myeloperoxidase from neutrophils, yielding hypochlorous acid. The ammonia resulted from the urease activity of H. pylori and the hypochlorous acid reacts with each other yielding monochloramine (NH2Cl), a toxic and reactive molecule, which has the potential to generate gastric lining injury through double-stranded DNA damage and condensation of chromatin fibers due to the lipophilicity and low molecular mass of the molecule (Minoura-Etoh et al. 2006). This neutrophil-derived oxidant, NH2Cl, induces detachment of cell cultures and changes in morphological characteristics owing to the dephosphorylation of p-Tyr and the increase in intracellular Ca2+ (Patekar et al. 2015). This intracellular Ca2+ mobilization is correlated with the induction of the immediate early gene product encoded by BZLF1 and ZEBRA protein. The conversion of the latent phase to the lytic phase of the virus takes place rapidly, which is mediated through cell detachment, ballooning, and hypercontraction when exposed to high concentration of NH2Cl (>100 mM) (Minoura-Etoh et al. 2006). NH2Cl also acts as a ligand, binding to the chemoreceptors on the EBV-infected B cells, triggering the activation of AP-1 transcription factor, which plays a key role in the reactivation of EBV (Minoura-Etoh et al. 2006). Some in vitro studies have also suggested that the reactivation of the virus occurs by Phospholipase C, gamma 1 (PLCγ) pathway due to some carcinogenic secretions from H. pylori, such as cytotoxin-associated gene A (CagA) and vacuolating toxin A (VacA), which activates PLCγ and other associated kinases for the viral reactivation (Singh and Jha 2017).
Streptococcus pyogenes
Streptococcus pyogenes is also known as a Group A Streptococci (GAS). It is a Gram-positive bacterium (Ferretti et al. 2016). It causes a variety of infections, including skin infections, pharyngitis, toxic shock-like syndrome, postinfectious sequelae, acute rheumatic fever, and poststreptococcal glomerulonephritis (Ferretti et al. 2016). Most of these infections are highly contagious. These bacteria colonize the pharynx, anus, and genital mucosa. The mode of transmission of bacteria occurs through airborne droplets, physical contact with skin lesions due to bacterial infection, or on flat surfaces with droplets of nasal discharges. It has been found that coinfection of EBV and GAS is present in acute pharyngitis and further leading to acute glomerulonephritis, which causes decreased glomerular filtration (Hashimoto et al. 2003). This coinfection is more frequent in children than in adolescents. Studies have shown that ∼20% of healthy kids are GAS infected in tonsils (Roberts et al. 2012).
According to Ueda et al. (2014), the saliva of GAS-colonized individuals was found to have a higher amount of EBV DNA than those with no GAS colonization. Additionally, they observed the same result for BZLF1 mRNA expression. Moreover, the heat-killed strain of GAS (M1T1 strain 5448) was found to induce EBV reactivation in vitro. Heat-killed GAS was also found to induce lytic phase in latently infected lymphoblastoid cell lines. GAS peptidoglycans and heat-killed GAS upregulate the expression of BZLF1 by triggering TLR2. TLR2 interrupts the EBV default latent phase (Ueda et al. 2014). Coinfection with GAS and EBV has also been reported in pharyngitis. GAS produces superantigens. These antigens stimulate T cells in a nonspecific manner, causing inflammatory reactions. This, in turn, leads to the activation of B cells, which are latently positive for EBV, resulting in the reactivation of EBV into the lytic form and the release of EBV DNA into saliva (Skovbjerg et al. 2015). There are also reports of cases in which coinfection with EBV and GAS resulted in the development of guttate psoriasis. In such immunosuppressed cases, the chances of switching from the latent to lytic state are high (Skovbjerg et al. 2015).
Treponema pallidum
Treponema pallidum is a spiral-shaped, Gram-negative bacterium that causes infectious diseases, such as bejels and yaws (Mitjà et al. 2013). This organism is also responsible for causing syphilis, a sexually transmitted disease. The spread of this bacterium occurs mainly through direct contact with active lesions or sexual contact. The infection is also known to cross the placenta and infect the fetus during pregnancy. Some cases have also been reported in the development of hepatitis during secondary infections (Alemam et al. 2021). Fatal hepatitis has been reported in EBV-positive immunocompromised patients and, in some cases, immunocompetent individuals.
In a study conducted by Hirsiger et al. (2019), EBV hepatitis was found in an immunocompetent patient with syphilis. Observations were made showing the in vitro reactivation of EBV by T. pallidum. The proteins of T. pallidum induced dose-dependent reactivation in vitro, which resulted in an increase in the transcription of the early lytic gene BZLF1. Reactivation is mediated by TLR2. The stimulation of TLR2 results in the direct activation of TLR2-reporter cell lines, which leads to the activation of B cells and an increase in cell number. It is found that T cell activity is inhibited during pregnancy. A similar inhibition of T cell proliferation was observed during syphilis inhibition. Hence, the report also suggests that during T. pallidum infection, the inhibited T cell activity and proliferation may have co-acted to stimulate B cell activation via TLR2 and further reactivate the virus, leading to lymphoproliferation.
Other bacteria
It is to be noted that the reactivation of EBV may take place in case of sepsis (Walton et al. 2014). This condition is mainly caused by bacterial infections. Sepsis is a state in which the host inflammatory mechanism is unresolved, leading to organ destruction. During sepsis, the body is in an immunosuppressed state. Moreover, latently infected viruses, such as EBV, may be reactivated if impairment of cellular and immune surveillance mechanisms occurs due to sepsis. It is also found that septic conditions regulate the innate and adaptive immunity by several mechanisms. These mechanisms involve the depletion of immune effector cells via apoptosis, resulting in T cell exhaustion. This leads to a decrease in host immunity, which aids in virus reactivation. In some reports, it was found that septic patients in whom viral reactivation took place had a high infection rate with bacteria that are not generally found to cause diseases in a competent host. These bacteria may include Stenotrophomonas maltophilia, some species of E nterococcus and Acinetobacter (Walton et al. 2014).
EBV reactivation by viruses
Viruses modulate the infected cells along with nearby cells for an establishment of successful infection. Many viral proteins have been shown to interact with a variety of host proteins to modulate the cellular microenvironment. There have been reports that during some viral infections, EBV reactivation could occur (Fig. 3); however, the detailed molecular mechanism associated with this is seldom explored to date.
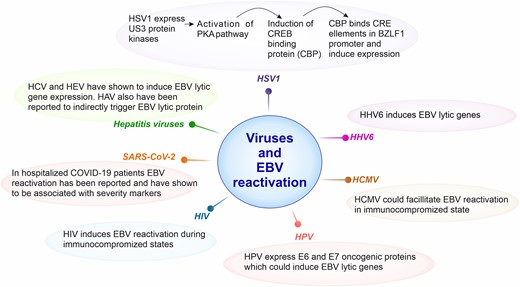
Schematic representation of information regarding viruses reported to reactivate EBV. Human Simplex Virus 1 (HSV-1) encodes US3 protein kinase, which activates protein kinase A (PKA) pathway. This further induces cAMP-response element binding (CREB) protein (CBP), which binds CRE elements in BZLF1 promoter leading to its expression and lytic activation. Human herpesvirus type 6 (HHV6) has been shown to elevate levels of EBV lytic gene expression. Based on few reports, human cytomegalovirus (HCMV) could also facilitate EBV reactivation leading to generation of coinfection scenario of both of these viruses. Human immunodeficiency virus (HIV) has been widely known to develop immunocompromised states, during such scenario’s reports have shown that it could also induce EBV reaction. Clinical studies including hospitalized SARS-CoV-2 patients have shown the EBV reactivation, in addition, it was prominently correlated with severity markers. Hepatitis causing viruses (HCV and HEV) have also shown to elevate the EBV lytic gene expression in vitro.
Herpes Simplex Virus Type 1
Herpes Simplex Virus Type 1 (HSV-1) is one of two types of HSVs belonging to the Herpesviridae family. The main mode of HSV-1 transmission is saliva. Blood-borne, sexual, and transplacental modes of transmission can also occur; however, they are less prevalent than salivary transmission (Arduino and Porter 2007). HSV-1 is also known as Human Herpesvirus 1 (HHV-1). HSV-1 is the most prevalent cause of oral herpes, and rarely causes genital herpes (Arduino and Porter 2007). HSV-2 (also known as HHV-2) is predominantly associated with genital herpes. HSV-1 is the most common cause of gingivostomatitis, herpes labialis, and herpes keratitis (Arduino and Porter 2007). The coinfection of EBV and other herpesviruses has been observed in semen samples of patients from infertility clinics (Kapranos et al. 2003, Neofytou et al. 2009). The possibility of EBV reactivation due to its genetic relatives, the other herpesviruses, could not be neglected in such cases.
Despite the fact that the specific mechanism behind EBV reactivation is unknown, Wu et al. (2012) found that HSV-1 can trigger EBV reactivation with the help of BZLF1. Both HSV-1 infection and chemical inducer TPA, which is known to reactivate EBV virus from latency, were found to upregulate the expression of BZLF1 protein in EBV transformed B cells, particularly Akata cells. Even HEK293T human cell lines with a bioluminescent enzyme, luciferase, when infected with HSV-1, also showed BZLF1 promoter activation. Furthermore, after infecting EBV-positive Burkitt’s lymphoma cells (Raji and Namalwa) with HSV-1, BZLF1 expression consistently increased 48 h later. After HSV-1 infection, all types of cell lines showed elevated expression of the EBV early gene, BALF5 (Wu et al. 2012). The cell entry mechanism of HSV-1 is through receptor-mediated endocytosis. After entering the cell, this virus encodes a serine/threonine kinase called US3 protein kinase. Then, the tegument protein activates a host protein kinase A known as PKA, by converting cyclic AMP (cAMP) to ATP in the presence of the Adenylate Cyclase enzyme found in the host cell membrane. PKA then phosphorylates cAMP response element-binding protein (CREB), which binds to CREB-binding protein (CBP). These two proteins bind to the cAMP response element (CRE) found within the BZLF1 promoter, causing viral reactivation. It is worth mentioning that out of three cis-regulating elements, the ZII domain of Zp is crucial for HSV-1-induced Zp activation (Wu et al. 2012).
Human Herpesvirus Type 6
Similar to EBV, >90% of adults are seropositive for HHV-6, with infection peaks occurring at 9 to 21 months of age (Cohen 2015). HHV-6 belongs to the Roseolovirus genus of the betaherpesvirus family and shares resemblance with cytomegalovirus. The mode of transmission of HHV-6 is probably by coming in close contact with the saliva of the infected person (Cohen 2015). As stated earlier, the coinfection of these viruses with EBV has been found in semen of infertility patient samples (Kapranos et al. 2003, Neofytou et al. 2009) as well as in some other conditions (Brooks et al. 2022, Chen et al. 2023).
During simultaneous presence of both the viruses during a disease, it is challenging to establish which virus reactivated in which human systems. However, results from in vitro studies provide the insights of possible mechanisms involved in such scenarios. In a previous study (Flamand et al. 1993), when EBV-positive Burkitt lymphoma cells (Akata and Raji cells) were treated with HHV-6, it was observed that HHV-6 elevated the expression of ZEBRA and EA-D by 5-fold and 10-fold, respectively. Even the expression of early antigen-restricted protein (EA-R) was upregulated in these Burkitt lymphoma cells. Expression of EBV viral capsid protein gp350 was also enhanced in EBV-positive Burkitt’s lymphoma cell lines, suggesting that HHV-6 is capable of inducing the full lytic cycle of EBV. To prove this, researchers found that the majority of cells expressing glycoprotein (gp) 350 were also positive for the HHV-6 antigen (Flamand et al. 1993). Furthermore, to determine whether infectious HHV-6 is required for EBV reactivation, HHV-6 cells were UV- or heat-treated. No ZEBRA protein or EA-D titer was observed in Raji cells when UV or heat-treated HHV-6 cells were used. This hints toward the requirement of active HHV-6 protein machinery for EBV reactivation (Flamand et al. 1993). Another single cell RNAseq study suggested that the rare instances of HHV-6 expression appear enriched with EBV reactivation (Miranda 2023). Based on these findings, it can be concluded that EBV reactivation can be influenced directly by HHV-6 replication. However, the exact mechanism behind such reactivation is still being studied (Flamand et al. 1993). Additionally, it would also be interested to look into other aspects of these viral interplay, such as if EBV can reactivate HHV-6 or other herpesviruses as well.
Cytomegalovirus
HCMV is a double-stranded DNA virus that belongs to the Herpesviridae family and has one of the longest viral genomes (Dioverti and Razonable 2016). HCMV is linked to a variety of clinical manifestations that are dependent on the immune system status of the host, ranging from asymptomatic infections in healthy individuals to severe infections in immunocompromised patients. However, healthy HCMV-seropositive patients can develop infectious mononucleosis-like syndrome. HCMV is most typically transmitted by contact with infected individuals’ bodily fluids and secretions. It can also be spread by organ and tissue transplantation, blood transfusion as well as through infected mothers via the placenta, breast milk, and vaginal secretions (Dioverti and Razonable 2016).
HCMV and EBV can be reactivated in immunocompromised patients, such as those with HIV infection, organ transplant recipients, or those receiving immunosuppressive medications. This was observed in ICU patients in a study by Coskun et al. (2017). Twelve patients had EBV reactivation, while only two patients had both EBV and CMV reactivation. As for the reactivation of latent EBV by CMV, Aalto et al. reported that CMV might facilitate a favorable environment for the reactivation of EBV by increasing immunosuppressive status in immunocompromised patients. It was also observed that sore throat was more frequently associated with CMV-induced EBV immunoreactivity than CMV mono-infection (Aalto et al. 1998). A correlation between symptomatic CMV infection and EBV reactivation was found in a study on renal transplant patients, and serological indications of both viral infection and reactivation appeared within a small interval of 20 days. EBV reactivation was found to be six times more likely in patients with symptomatic CMV infection than in asymptomatic CMV patients and in patients without any evidence of CMV infection. In addition, two-thirds of these patients exhibited serological indications of EBV reactivation after CMV infection, whereas one-third of these patients developed them before CMV infection (Hornef et al. 1995). In fact, CMV reactivation was found to be the only independent variable linked with EBV reactivation and the risk of PTLD in individuals who underwent hematopoietic stem cell transplantation (HSCT) (Zallio et al. 2013). Loutfy et al. (2017) showed that coinfection with EBV and CMV results in the worst overall survival of immunocompromised pediatric leukemic patients. Another study that backs up the previous finding states that HIV seropositive patients with high immunoglobin (Ig) G antibody titers against EBV and CMV had lower CD4 cell count, and oral manifestations such as specific gingivitis and periodontitis were more common in people with lower CD4+ cell count (Patekar et al. 2015, de Melo Silva et al. 2020).
The exact involvement of CMV in the reactivation of latent EBV is still a matter of debate. However, it can be concluded from these studies that the presence of both EBV and CMV can exacerbate disease outcomes and, if not properly treated, can have a significant impact on patient morbidity.
Human papillomavirus
Human papillomavirus (HPV) is known to infect the squamous epithelia and generate warts on the skin of infected individuals. HPV belongs to the Papillomaviridae family and there are ∼200 different types of HPVs. HPV-16 and HPV-18 are responsible for ∼54% and 17% of invasive cervical malignancies, respectively (Muñoz et al. 2003), whereas HPV-6 or HPV-11 is responsible for ∼90% of genital warts (Greer et al. 1995). The prevalence of HPV in women worldwide is estimated to range from 2% to 44%. HPV is also common in men but not studied as thoroughly as in women (Baseman and Koutsky 2005).
Various reports have shown the coinfection of HPV and EBV in human tumorigenesis (Shi et al. 2016). As EBV is more prevalent in population and HPV acute infection is generally acquired in later stages of life, the chance of HPV-mediated EBV reactivation in such coinfection cases cannot be neglected. EBV and HPV can infect and proliferate in the epithelial lining of the upper aerodigestive tract. EBV infects the oral epithelia, whereas HPV infects the oral keratinocytes. HPV encodes the E6 and E7 oncogenes. Scientists suggested that high-risk HPV promotes the lytic cycle of EBV in differentiating epithelial cells, and E6 and E7 are essential to promote HPV-induced EBV reactivation (Makielski et al. 2016). They used the double digestion procedure to infect normal oral keratinocytes (NOK) with the HPV genome in an in vitro experiment. Subsequently, they infected NOK cells with EBV. Interestingly, the two oncogenes, E6 and E7, were found to induce HPV-mediated carcinogenesis in the same stratified epithelial compartment where EBV was also reactivated. EBV lytic reactivation occurs in the suprabasal compartment of the stratified epithelium, where the HPV-productive phase occurs. Additionally, NOKs raft cultures with EBV and null mutations in the E6 or E7 oncogenes showed the same ZEBRA protein levels as cultures that had not been transfected with HPV, suggesting that both of them are necessary for HPV-mediated EBV reactivation (Makielski et al. 2016). However, in contrast, it was seen that HPV inhibits the EBV lytic cycle and facilitates the latent cycle. The E7 oncogene was found to be sufficient to halt the lytic cycle, and unlike E7, E6 did not influence the lytic cycle of EBV (Guidry et al. 2019). Currently, it is unclear how HPV affects EBV’s lytic cycle of EBV.
Human immunodeficiency virus
HIV causes the life-threatening condition of acquired immunodeficiency syndrome (AIDS). HIV can infect and weaken the immune system, making the host body vulnerable to opportunistic pathogens. HIV is spread through contaminated blood transfusions, sexual contact, and prenatal transmission by an infected mother (Fauci 1988). HIV and EBV coinfection has been long reported in context of EBV-associated lymphomas in HIV-infected individuals (Bibas and Antinori 2009).
It has been observed that HIV-induced immunosuppression reactivates EBV. EBV is present in those who have AIDS or are at risk of developing it. This was observed in a study that HIV-1 infection of EBV-positive B cells transiently activated the replicative cycle of EBV (Lai et al. 1989). EBV can be similarly reactivated by HIV-1 or TPA, a chemical inducer. EBV-antigen positive cells were considerably higher in the presence of both than in the absence of either HIV-1 or TPA. This finding indicates that a particular stimulus threshold is required for EBV reactivation. Interestingly, EBV can be reactivated each time HIV-1 reinfects EBV/HIV-1-infected B cells (Lai et al. 1989). Consistent with a previous study, a clinical study showed that all HIV seroconverters and the majority of HIV nonseroconverters were seropositive for EBV at the beginning of the study on the basis of IgG titer for EBV viral capsid antigen (VCA). (Rahman et al. 1991). Increased levels of EBV DNA in peripheral blood mononuclear cells were seen in the blood of HIV-infected individuals as compared to healthy individuals (Fellner et al. 2007). In contrast, Dehee et al. (2001) showed that there is no relationship between HIV infection markers, such as CD4+ or CD8+ levels, and EBV DNA in HIV-infected patients. An in vivo study demonstrated that HIV can induce EBV reactivation and augments EBV-induced tumorigenesis (Whitehurst et al. 2022). EBV has been shown to be associated in lymphomagenesis in HIV-infected individuals (Bibas and Antinori 2009). Introduction of highly active antiretroviral therapy (ART) has been shown to reduce occurrences on EBV associated lymphomas in HIV-infected individuals. This suggests that ART can impact EBV biology and its outcome in HIV-infected individuals (da Silva and de Oliveira 2011). Both viruses may have exploited stimulation of the immune system during HIV infection. EBV reactivation is most likely induced by transitory weakening of the immune system triggered by primary HIV infection. The deterioration of the immune system described here may be due to the diminished function of EBV-specific T cells, potentially due to a decrease in the CD4+ T cell count. Perhaps, the CD4+ cell count plays an indirect role in EBV reactivation in HIV-infected patients.
The repercussions of EBV reactivation in HIV-infected patients were seen in a rare case of a 70-year-old man with hemophagocytic lymphohistiocytosis (HLH) (Thoden et al. 2012). Similarly, a 46-year-old HIV-positive woman died of acute liver failure (ALF). Serological evidence revealed a past EBV infection, and since the viral load was high, it was determined to be reactivation rather than primary infection (Ahmed et al. 2021). Thus, it can be concluded that HIV may reactivate latent EBV infection, and reactivated EBV can cause a worse prognosis in immunocompromised patients with HIV. However, more research could provide important details of major factors responsible for HIV mediated EBV reactivation.
Hepatitis C virus
Hepatitis C virus (HCV) is estimated to infect 170 million individuals globally and is five times as widespread as HIV-1. In the Flaviviridae family, HCV is a distinct genus composed of single-stranded RNA. It induces chronic infection of the liver, resulting in inflammation. Intravenous drug use, prenatal transmission, and sexual interactions are all clinical risk factors for HCV infection (Lauer and Walker 2001). B-cell infection in HCV-positive patients is said to be a predictor of lymphoproliferative disease, as HCV infection causes clonal expansion of B cells, which can progress to lymphoproliferative diseases. EBV is also associated with B-cell transformation and the development of lymphoproliferative diseases. Clinical trials on EBV-positive patients chronically infected with HCV revealed that the virus can also elevate the production of EBV lytic genes. HCV has been found to bind to CD81 receptors on B lymphocytes, causing them to clonally proliferate, and increased BZLF-1 gene expression in these patients indicates EBV reactivation (Shimozuma et al. 2010). Another interesting point that should be noted is that during this study, patients who received interferon therapy were tested for BZLF1 mRNA expression to determine the correlation between HCV infection and EBV reactivation. BZLF1 mRNA expression was decreased in three patients, but EBER1 RNA expression was observed in these patients. There were no signs of EBV reactivation even three months after the end of therapy, suggesting that this therapy might play a role in the reactivation of EBV. However, further research is required in this area (Shimozuma et al. 2010). Interestingly, it was observed that chronic HCV patients with reactivated EBV had higher mean values of serum HCV RNA than EBV-seropositive patients without reactivation. The hypothesis behind this is that when EBV-specific T cells are reactivated, cytokines, including interferon (IFN)-γ, IL-1, IL-2, and IL-10, are produced. HCV replication is boosted by EBV BCRF1 gene product viral IL-10, which mimics the function of human IL-10 (Petrova et al. 2010).
Hepatitis E virus
Hepatitis E is generally a self-limiting disease but it can cause chronic infection in organ and stem cell transplant recipients, HIV-positive patients, and patients receiving chemotherapy for hematological malignancies (Kamar et al. 2015). Hepatitis E virus (HEV) was previously classified as a member of the Caliciviridae family but was later removed due to lack of similarity. It is now classified as a member of the Hepeviridae family. Orthohepevirus is one of the two genera in this family, which is further divided into four different species (A–D). The human HEV belongs to the Orthohepevirus A species, which has four genotypes (GT1–GT4), with GT1 and GT2 infecting only humans and GT3 and GT4 infecting both humans and animals (Doceul et al. 2016). Consumption of contaminated meat and feces-contaminated water accounts for most HEV transmission. However, additional modes of transmission are blood-borne, human-to-human, and perinatal (Mirazo et al. 2014). EBV reactivation was observed in a liver transplant patient with a chronic HEV infection. Clearance of HEV was achieved by 9-week ribavirin therapy. During this period, the patient developed PTLD in the form of Burkitt lymphoma, which is a non-Hodgkin B-cell disorder associated with EBV. It is worth mentioning that EBV reactivation occurred after chronic HEV infection when the patient was receiving ribavirin therapy and was under minimal immunosuppressive therapy (Mainardi et al. 2019). Possible HEV-induced EBV reactivation was observed in a patient with rheumatoid arthritis (RA). Viral load testing revealed low viral loads of EBV DNA and HEV RNA. EBV VCA-IgG, EBV VCA-IgM, and EBNA-1 IgG antibodies were also positive in the patient. The presence of these antibodies in the patient can indicate prior infection, but it can also indicate EBV reactivation (Nagata and Hayashi 2020). Tests revealed that the patient had a recent HEV infection with concurrent reactivation of the latent EBV. Finally, after discontinuation of methotrexate and prednisolone therapy in a patient with RA for 1 month, neither HEV RNA nor EBV RNA was detected (Schultze et al. 2015). In this report, the reactivated EBV could be an impact of immunosuppressive therapy. However, the presence of HEV raises valid possibility of its involvement in EBV reactivation. Overall, these studies suggest that EBV may be reactivated by HEV infections. However, further research is required to validate these results.
Hepatitis A virus
Hepatitis A virus (HAV) is another type of virus responsible for hepatitis, a highly contagious disease that causes liver inflammation. HAV is a single-stranded RNA virus that belongs to the Picornaviridae family. It is primarily transmitted via the fecal-oral route, and despite the development of a vaccine for HAV in the early 1990s, it remains a major cause of acute hepatitis worldwide. Similar to Hepatitis E, Hepatitis A is usually a self-limiting disease, but the severity of the disease depends on age. More than 70% of infants under the age of six are asymptomatically infected, whereas >70% of adults have a symptomatic infection with symptoms, such as jaundice and elevated serum aminotransferase (Jeong and Lee 2010).
In a clinical trial (Ritter et al. 1996) involving 134 patients infected with HAV, the role of reactivation of EBV during the progression of HAV infection as well as its potential role in hepatocellular illness was studied. Autoantibodies of immunoglobulin M against triosephosphate isomerase (IgM anti-TPI) were detected in the serum of the HAV-positive patients. All patients were previously infected with EBV. IgM anti-TPI was sufficient to trigger hemolysis. The presence of both IgM anti-TPI titer and hemolysis are not characteristic features of acute hepatitis A. The detection of IgM anti-TPI and its link to reactivation of EBV infection, which is a unique characteristic of HAV infection, was further confirmed by the detection of anti-EA in the sera of 27 patients who were positive for IgM anti-TPI. However, further studies are required to establish the mechanisms involved. Furthermore, in a study focusing on EBV reactivation in individuals with HAV infection, researchers observed that reactivation of EBV affects the period of full recovery from HAV infection and causes severe liver damage compared to nonreactivated patients. Authors speculated that due to the HAV’s cytotoxic action and consequent immunological-mediated damage, immune dysfunction occurs, leading to the reactivation of EBV and worsening the HAV patients’ condition (Kim 2020).
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
The highly infectious novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome 2), which causes coronavirus disease 19 (COVID-19), belongs to the subfamily Coronavirinae of the Coronaviridae family. Its genome is a single-stranded positive-sense RNA (+ssRNA), which is larger than that of other RNA viruses (Wang et al. 2020). This pandemic associated virus has raised various concerns in the recent year (Indari et al. 2021c, 2022a) and many of its impacts on human health are yet to be explored such as its coinfection with other pathogens (Indari et al. 2021a, 2022c). In a clinical study, two-thirds of long-COVID patients had EBV reactivation, as assessed by EBV EA-D IgG (ranging from 10 to >150 U/ml) or EBV VCA IgM titers (ranging from 67 to 82 U/ml) (Gold et al. 2021). Long-COVID is a condition that can afflict persons of all ages, in which COVID symptoms linger or reappear after the initial COVID infection has cleared up. Tinnitus and hearing loss were also observed in some of these patients, which has been linked with EBV. Hence, it is plausible that COVID-induced EBV reactivation may be responsible for a fraction of the long-COVID symptoms. Because EBV reactivation has been observed in both long-term and short-term long-COVID patients, it is likely that reactivation occurs early in coronavirus disease (Gold et al. 2021). This conclusion is supported by a study of COVID-positive ICU patients who experienced EBV reactivation shortly after admission (Simonnet et al. 2021). In another study (Paolucci et al. 2021) on COVID-positive intensive care unit (ICU) patients, the prevalence of positive EBV DNA and median EBV DNA levels were found to be substantially greater than that in COVID-positive sub-intensive care unit (SICU) patients. In addition, when compared to SICU patients, CD8+ T cells and NK cells were considerably lower in ICU patients, but ICU patients had a much higher B-cell count. Despite the lack of a linear relationship between EBV DNA load and NK, CD8+ T, and B cells, it is possible that the increase in B cell count in immunocompromised patients is attributable to increased EBV DNA load. And since the activities of CD8+ T cells and NK cells were inhibited, which are crucial for the suppression of viral reactivation, EBV could be reactivated (Paolucci et al. 2021). Xie et al. (2021) showed that critically ill COVID-19 patients who also had EBV reactivation had considerably lower lymphocyte and albumin levels than EBV-negative COVID-19 patients. Lower levels of lymphocytes and albumin lead to a worse prognosis. They also had higher D-dimer, serum calcium, and C-reactive protein (CRP). Higher D-dimer and CRP levels are associated with severe EBV infection. The incidences of tachypnea, hypoproteinemia, and respiratory failure were much higher in patients with reactivated EBV. Additionally, 28- and 14-day mortality rates were higher in these patients (Xie et al. 2021). Chen et al. (2021) found that COVID-19 patients who were seropositive for anti-VCA IgM were associated with three times more risk of having fever, higher CRP, and higher AST. They are also more likely to receive corticosteroid therapy than patients with COVID-19 alone (Chen et al. 2021). An interesting meta-analysis by Manoharan et al, mentioned EBV reactivation during COVID-19 showed increased mortality in the hospitalized patients (Manoharan and Ying 2023). A recent study reported increased EBV genome load in COVID-19 patients compared to COVID-19 negative individuals (Bernal and Whitehurst 2023). These studies suggest that COVID-19 patients with reactivated EBV exhibit more severe symptoms and increased mortality.
Recent reports have shown the interaction between various SARS-CoV-2 and host proteins. Considering the concern of rising variants of this pandemic-causing virus, we sought to further investigate the connection between SARS-CoV-2 and EBV reactivation. It is possible that SARS-CoV-2 induces a host protein that may play a vital role in EBV reactivation or maintenance of EBV inside cells. To identify such proteins, we first investigated the possible host proteins interacting with any of the SARS-CoV-2 proteins using an online human–virus interaction database (Yang et al. 2021). Each of the SARS-CoV-2-interacting host proteins was thoroughly searched in the literature in the context of their role in EBV reactivation or maintenance. All proteins that were found to interact with SARS-CoV-2, as well as having any role in EBV reactivation or maintenance, are listed in Table 1 along with their particular roles and references. This information will aid in understanding the SARS-CoV-2-mediated EBV reactivation at the molecular level.
List of SARS-CoV-2 and their possible interactive host proteins, which have earlier been shown in the context of EBV infection.
| SARS-CoV-2 protein | Possible interactive host protein | Relation of host protein with EBV | Reference for host protein role in EBV infection/reactivation |
|---|---|---|---|
| E | BRD4 BRD2 | BRD4 and BRD2 plays an important role in EBV lytic reactivation. Both of them bind to the lytic origins of replication and inhibition of BRD4 and BRD2 by JQ1 inhibits the lytic cycle at two different steps. | Keck et al. (2017) |
| N | UPF1 | Knockdown of UPF1 upregulated the expression of EBV lytic genes BZLF1, BMRF1, BLLF1, and BcLF1 as well as elevated levels of lytic proteins ZEBRA and EA-R. | van Gent et al. (2021) |
| CSK2B CSK22 | Found similar CK2 proteomic interactions with EBNA1 in latent and lytic EBV infections. | Malik-Soni and Frappier (2012) | |
| DDX21 | Helicase protein DHX9, which also functionally similar to DDX21, interacts with EBV protein SM and restricts EBV lytic replication. | Fu et al. (2019) | |
| PABP1 PABP4 | Interactor of EBV lytic cycle activator ZEBRA protein | Zhou et al. (2020) | |
| SNIP1 | Possibly found its related proteins Smad1 and Smad5_along with BMP signaling and BMP-mediated EBV reactivation that is inhibited by miR-155. | Yin et al. (2010) | |
| M | ANO6 | Anoctamin-2 shows molecular mimicry with EBV nuclear antigen 1 and associates with multiple sclerosis risk. | Tengvall et al. (2019) |
| FAKD5 MPPA INT4 | Mitochondrion-enriched clusters and their components amongst upregulated proteins, maybe affecting the remodeling of B-cells during EBV infection. Integrator complex 11 (INT11) was among the cell proteins in the ZEBRA (Zta)-interactome undergoing EBV lytic replication. | Wang et al. (2019) Zhou et al. (2020) | |
| PSMD8 | Was seen to be in the list of genes and gene categories induced by LMP2A, a protein of EBV. | Portis et al. (2003) | |
| NSP1 | DPOLA | POLA2 [Polymerase (DNA-directed) alpha 2] was found in the list of genes and gene categories induced by LMP2A. And LMP2A is the viral transcript of EBV. | Portis et al. (2003) |
| Nsp2 | RAP1GDS1 | Downregulated in the presence of EBNA1 | Canaan et al. (2009) |
| Nsp4 | IDE | Major protein modulated by EBV dUTPase in human primary dendritic cells | Williams PhD et al. (2019) |
| NSP5 | HDAC2 | Histone deacetylase inhibitor-mediated EBV reactivation. | Tsai et al. (2011) |
| NSP6 | VAS1 | Relation of V-ATPase was observed with LMP1 and LMP2A that are the onco-proteins of EBV. | DeKroon et al. (2018) |
| NSP7 | PGES2 | PGES2 is responsible for the biosynthesis of PGE2, and it have a either an inhibitory, stimulatory role in there viral replication cycles of EBV. PGES2 undergoes synthesis and produce PGE2, and COX-2 induces lytic reactivation through PGE2 by modulating the EP receptor signaling pathway. | Gandhi et al. (2015) |
| SCPDL | Present in the list of K15-interacting proteins identified by LC-MSMS from latent infection in KSHV. K15 was seen to be related to EBV. | Abere et al. (2018) | |
| RAB10 | In cells provided with EBV lytic induction, Rab10 was found to be colocalized with gp350–gp220. | Nanbo (2020) | |
| NSP8 | DDX10 | Present in the list of genes enriched in the biological processes for cellular genes downregulated by DHX9 depletion (that restricts EBV replication). | Fu et al. (2019) |
| EXOSC3 | Present under the major pathways/genes modulated by EBV-encoded dUTPase in human dendritic cells. | Ariza et al. (2013) | |
| MRPS27 | Present in the list of genes and gene categories induced by LMP2A, i.e. a viral protein of EBV. | Portis et al. (2003) | |
| NSP9 | EIF4H | EBV LMP1 protein stimulates transcription of eIF4E to promote proliferation of EBV-infected cells. | Zhao et al. (2014) |
| NUP88 NUP62 | BGLF4 targets the nucleus through an unconventional interaction with nucleoporins, with the help of EBV lytic replication. | Lee and Chen (2021) | |
| NUP214 ZNF503 | Newly-infected EBNA2hi blast-like B lymphocytes codes high levels of candidate ZFPs (that includes ZNF503 out of the three different ZFPs considered). | Xu et al. (2019) | |
| Nsp12 | RIPK1 | EBV-encoded LMP1 targets RIPK1 ubiquitination and LMP1 is involved in EBV reactivation | Liu et al. (2018), Nawandar et al. (2017) |
| TCF12 | LMP2 promoter binds to TCF12 and LMP2 blocks reactivation of EBV | Arvey et al. (2013) | |
| IMPDH2 | Differentially expressed during EBV reactivation | Chen et al. (2008) | |
| ORF3a | SUN2 | During EBV reactivation, BGLF4 protein modifies the nuclear envelope-associated proteins of the infected cell, including SUN1 and SUN2 for further viral replication of EBV. | Chang et al. (2015) |
| TRIM59 | EBV circLMP2A promotes cancer stemness properties of cells via circLMP2A/miR-3908/TRIM59/p53 axis | Tan and Lim (2021) | |
| ORF10 | ELOB and ELOC | EBV ZEBRA protein interaction with Cul2 and Cul5 aid in building the multimolecular ECS complex (Elongin B/C-Cul2/5-SOCS-box protein), which further targets proteosomal breakdown of p53. | Germini et al. (2020) |
| CUL2 | EBV ZEBRA proteins forms complex with Cul2 and Cul5, and identifies p53 with C-terminal phosphorylation and drives degradation of p53 for promoting viral propagation. | Cai and Yang (2016) |
| SARS-CoV-2 protein | Possible interactive host protein | Relation of host protein with EBV | Reference for host protein role in EBV infection/reactivation |
|---|---|---|---|
| E | BRD4 BRD2 | BRD4 and BRD2 plays an important role in EBV lytic reactivation. Both of them bind to the lytic origins of replication and inhibition of BRD4 and BRD2 by JQ1 inhibits the lytic cycle at two different steps. | Keck et al. (2017) |
| N | UPF1 | Knockdown of UPF1 upregulated the expression of EBV lytic genes BZLF1, BMRF1, BLLF1, and BcLF1 as well as elevated levels of lytic proteins ZEBRA and EA-R. | van Gent et al. (2021) |
| CSK2B CSK22 | Found similar CK2 proteomic interactions with EBNA1 in latent and lytic EBV infections. | Malik-Soni and Frappier (2012) | |
| DDX21 | Helicase protein DHX9, which also functionally similar to DDX21, interacts with EBV protein SM and restricts EBV lytic replication. | Fu et al. (2019) | |
| PABP1 PABP4 | Interactor of EBV lytic cycle activator ZEBRA protein | Zhou et al. (2020) | |
| SNIP1 | Possibly found its related proteins Smad1 and Smad5_along with BMP signaling and BMP-mediated EBV reactivation that is inhibited by miR-155. | Yin et al. (2010) | |
| M | ANO6 | Anoctamin-2 shows molecular mimicry with EBV nuclear antigen 1 and associates with multiple sclerosis risk. | Tengvall et al. (2019) |
| FAKD5 MPPA INT4 | Mitochondrion-enriched clusters and their components amongst upregulated proteins, maybe affecting the remodeling of B-cells during EBV infection. Integrator complex 11 (INT11) was among the cell proteins in the ZEBRA (Zta)-interactome undergoing EBV lytic replication. | Wang et al. (2019) Zhou et al. (2020) | |
| PSMD8 | Was seen to be in the list of genes and gene categories induced by LMP2A, a protein of EBV. | Portis et al. (2003) | |
| NSP1 | DPOLA | POLA2 [Polymerase (DNA-directed) alpha 2] was found in the list of genes and gene categories induced by LMP2A. And LMP2A is the viral transcript of EBV. | Portis et al. (2003) |
| Nsp2 | RAP1GDS1 | Downregulated in the presence of EBNA1 | Canaan et al. (2009) |
| Nsp4 | IDE | Major protein modulated by EBV dUTPase in human primary dendritic cells | Williams PhD et al. (2019) |
| NSP5 | HDAC2 | Histone deacetylase inhibitor-mediated EBV reactivation. | Tsai et al. (2011) |
| NSP6 | VAS1 | Relation of V-ATPase was observed with LMP1 and LMP2A that are the onco-proteins of EBV. | DeKroon et al. (2018) |
| NSP7 | PGES2 | PGES2 is responsible for the biosynthesis of PGE2, and it have a either an inhibitory, stimulatory role in there viral replication cycles of EBV. PGES2 undergoes synthesis and produce PGE2, and COX-2 induces lytic reactivation through PGE2 by modulating the EP receptor signaling pathway. | Gandhi et al. (2015) |
| SCPDL | Present in the list of K15-interacting proteins identified by LC-MSMS from latent infection in KSHV. K15 was seen to be related to EBV. | Abere et al. (2018) | |
| RAB10 | In cells provided with EBV lytic induction, Rab10 was found to be colocalized with gp350–gp220. | Nanbo (2020) | |
| NSP8 | DDX10 | Present in the list of genes enriched in the biological processes for cellular genes downregulated by DHX9 depletion (that restricts EBV replication). | Fu et al. (2019) |
| EXOSC3 | Present under the major pathways/genes modulated by EBV-encoded dUTPase in human dendritic cells. | Ariza et al. (2013) | |
| MRPS27 | Present in the list of genes and gene categories induced by LMP2A, i.e. a viral protein of EBV. | Portis et al. (2003) | |
| NSP9 | EIF4H | EBV LMP1 protein stimulates transcription of eIF4E to promote proliferation of EBV-infected cells. | Zhao et al. (2014) |
| NUP88 NUP62 | BGLF4 targets the nucleus through an unconventional interaction with nucleoporins, with the help of EBV lytic replication. | Lee and Chen (2021) | |
| NUP214 ZNF503 | Newly-infected EBNA2hi blast-like B lymphocytes codes high levels of candidate ZFPs (that includes ZNF503 out of the three different ZFPs considered). | Xu et al. (2019) | |
| Nsp12 | RIPK1 | EBV-encoded LMP1 targets RIPK1 ubiquitination and LMP1 is involved in EBV reactivation | Liu et al. (2018), Nawandar et al. (2017) |
| TCF12 | LMP2 promoter binds to TCF12 and LMP2 blocks reactivation of EBV | Arvey et al. (2013) | |
| IMPDH2 | Differentially expressed during EBV reactivation | Chen et al. (2008) | |
| ORF3a | SUN2 | During EBV reactivation, BGLF4 protein modifies the nuclear envelope-associated proteins of the infected cell, including SUN1 and SUN2 for further viral replication of EBV. | Chang et al. (2015) |
| TRIM59 | EBV circLMP2A promotes cancer stemness properties of cells via circLMP2A/miR-3908/TRIM59/p53 axis | Tan and Lim (2021) | |
| ORF10 | ELOB and ELOC | EBV ZEBRA protein interaction with Cul2 and Cul5 aid in building the multimolecular ECS complex (Elongin B/C-Cul2/5-SOCS-box protein), which further targets proteosomal breakdown of p53. | Germini et al. (2020) |
| CUL2 | EBV ZEBRA proteins forms complex with Cul2 and Cul5, and identifies p53 with C-terminal phosphorylation and drives degradation of p53 for promoting viral propagation. | Cai and Yang (2016) |
List of SARS-CoV-2 and their possible interactive host proteins, which have earlier been shown in the context of EBV infection.
| SARS-CoV-2 protein | Possible interactive host protein | Relation of host protein with EBV | Reference for host protein role in EBV infection/reactivation |
|---|---|---|---|
| E | BRD4 BRD2 | BRD4 and BRD2 plays an important role in EBV lytic reactivation. Both of them bind to the lytic origins of replication and inhibition of BRD4 and BRD2 by JQ1 inhibits the lytic cycle at two different steps. | Keck et al. (2017) |
| N | UPF1 | Knockdown of UPF1 upregulated the expression of EBV lytic genes BZLF1, BMRF1, BLLF1, and BcLF1 as well as elevated levels of lytic proteins ZEBRA and EA-R. | van Gent et al. (2021) |
| CSK2B CSK22 | Found similar CK2 proteomic interactions with EBNA1 in latent and lytic EBV infections. | Malik-Soni and Frappier (2012) | |
| DDX21 | Helicase protein DHX9, which also functionally similar to DDX21, interacts with EBV protein SM and restricts EBV lytic replication. | Fu et al. (2019) | |
| PABP1 PABP4 | Interactor of EBV lytic cycle activator ZEBRA protein | Zhou et al. (2020) | |
| SNIP1 | Possibly found its related proteins Smad1 and Smad5_along with BMP signaling and BMP-mediated EBV reactivation that is inhibited by miR-155. | Yin et al. (2010) | |
| M | ANO6 | Anoctamin-2 shows molecular mimicry with EBV nuclear antigen 1 and associates with multiple sclerosis risk. | Tengvall et al. (2019) |
| FAKD5 MPPA INT4 | Mitochondrion-enriched clusters and their components amongst upregulated proteins, maybe affecting the remodeling of B-cells during EBV infection. Integrator complex 11 (INT11) was among the cell proteins in the ZEBRA (Zta)-interactome undergoing EBV lytic replication. | Wang et al. (2019) Zhou et al. (2020) | |
| PSMD8 | Was seen to be in the list of genes and gene categories induced by LMP2A, a protein of EBV. | Portis et al. (2003) | |
| NSP1 | DPOLA | POLA2 [Polymerase (DNA-directed) alpha 2] was found in the list of genes and gene categories induced by LMP2A. And LMP2A is the viral transcript of EBV. | Portis et al. (2003) |
| Nsp2 | RAP1GDS1 | Downregulated in the presence of EBNA1 | Canaan et al. (2009) |
| Nsp4 | IDE | Major protein modulated by EBV dUTPase in human primary dendritic cells | Williams PhD et al. (2019) |
| NSP5 | HDAC2 | Histone deacetylase inhibitor-mediated EBV reactivation. | Tsai et al. (2011) |
| NSP6 | VAS1 | Relation of V-ATPase was observed with LMP1 and LMP2A that are the onco-proteins of EBV. | DeKroon et al. (2018) |
| NSP7 | PGES2 | PGES2 is responsible for the biosynthesis of PGE2, and it have a either an inhibitory, stimulatory role in there viral replication cycles of EBV. PGES2 undergoes synthesis and produce PGE2, and COX-2 induces lytic reactivation through PGE2 by modulating the EP receptor signaling pathway. | Gandhi et al. (2015) |
| SCPDL | Present in the list of K15-interacting proteins identified by LC-MSMS from latent infection in KSHV. K15 was seen to be related to EBV. | Abere et al. (2018) | |
| RAB10 | In cells provided with EBV lytic induction, Rab10 was found to be colocalized with gp350–gp220. | Nanbo (2020) | |
| NSP8 | DDX10 | Present in the list of genes enriched in the biological processes for cellular genes downregulated by DHX9 depletion (that restricts EBV replication). | Fu et al. (2019) |
| EXOSC3 | Present under the major pathways/genes modulated by EBV-encoded dUTPase in human dendritic cells. | Ariza et al. (2013) | |
| MRPS27 | Present in the list of genes and gene categories induced by LMP2A, i.e. a viral protein of EBV. | Portis et al. (2003) | |
| NSP9 | EIF4H | EBV LMP1 protein stimulates transcription of eIF4E to promote proliferation of EBV-infected cells. | Zhao et al. (2014) |
| NUP88 NUP62 | BGLF4 targets the nucleus through an unconventional interaction with nucleoporins, with the help of EBV lytic replication. | Lee and Chen (2021) | |
| NUP214 ZNF503 | Newly-infected EBNA2hi blast-like B lymphocytes codes high levels of candidate ZFPs (that includes ZNF503 out of the three different ZFPs considered). | Xu et al. (2019) | |
| Nsp12 | RIPK1 | EBV-encoded LMP1 targets RIPK1 ubiquitination and LMP1 is involved in EBV reactivation | Liu et al. (2018), Nawandar et al. (2017) |
| TCF12 | LMP2 promoter binds to TCF12 and LMP2 blocks reactivation of EBV | Arvey et al. (2013) | |
| IMPDH2 | Differentially expressed during EBV reactivation | Chen et al. (2008) | |
| ORF3a | SUN2 | During EBV reactivation, BGLF4 protein modifies the nuclear envelope-associated proteins of the infected cell, including SUN1 and SUN2 for further viral replication of EBV. | Chang et al. (2015) |
| TRIM59 | EBV circLMP2A promotes cancer stemness properties of cells via circLMP2A/miR-3908/TRIM59/p53 axis | Tan and Lim (2021) | |
| ORF10 | ELOB and ELOC | EBV ZEBRA protein interaction with Cul2 and Cul5 aid in building the multimolecular ECS complex (Elongin B/C-Cul2/5-SOCS-box protein), which further targets proteosomal breakdown of p53. | Germini et al. (2020) |
| CUL2 | EBV ZEBRA proteins forms complex with Cul2 and Cul5, and identifies p53 with C-terminal phosphorylation and drives degradation of p53 for promoting viral propagation. | Cai and Yang (2016) |
| SARS-CoV-2 protein | Possible interactive host protein | Relation of host protein with EBV | Reference for host protein role in EBV infection/reactivation |
|---|---|---|---|
| E | BRD4 BRD2 | BRD4 and BRD2 plays an important role in EBV lytic reactivation. Both of them bind to the lytic origins of replication and inhibition of BRD4 and BRD2 by JQ1 inhibits the lytic cycle at two different steps. | Keck et al. (2017) |
| N | UPF1 | Knockdown of UPF1 upregulated the expression of EBV lytic genes BZLF1, BMRF1, BLLF1, and BcLF1 as well as elevated levels of lytic proteins ZEBRA and EA-R. | van Gent et al. (2021) |
| CSK2B CSK22 | Found similar CK2 proteomic interactions with EBNA1 in latent and lytic EBV infections. | Malik-Soni and Frappier (2012) | |
| DDX21 | Helicase protein DHX9, which also functionally similar to DDX21, interacts with EBV protein SM and restricts EBV lytic replication. | Fu et al. (2019) | |
| PABP1 PABP4 | Interactor of EBV lytic cycle activator ZEBRA protein | Zhou et al. (2020) | |
| SNIP1 | Possibly found its related proteins Smad1 and Smad5_along with BMP signaling and BMP-mediated EBV reactivation that is inhibited by miR-155. | Yin et al. (2010) | |
| M | ANO6 | Anoctamin-2 shows molecular mimicry with EBV nuclear antigen 1 and associates with multiple sclerosis risk. | Tengvall et al. (2019) |
| FAKD5 MPPA INT4 | Mitochondrion-enriched clusters and their components amongst upregulated proteins, maybe affecting the remodeling of B-cells during EBV infection. Integrator complex 11 (INT11) was among the cell proteins in the ZEBRA (Zta)-interactome undergoing EBV lytic replication. | Wang et al. (2019) Zhou et al. (2020) | |
| PSMD8 | Was seen to be in the list of genes and gene categories induced by LMP2A, a protein of EBV. | Portis et al. (2003) | |
| NSP1 | DPOLA | POLA2 [Polymerase (DNA-directed) alpha 2] was found in the list of genes and gene categories induced by LMP2A. And LMP2A is the viral transcript of EBV. | Portis et al. (2003) |
| Nsp2 | RAP1GDS1 | Downregulated in the presence of EBNA1 | Canaan et al. (2009) |
| Nsp4 | IDE | Major protein modulated by EBV dUTPase in human primary dendritic cells | Williams PhD et al. (2019) |
| NSP5 | HDAC2 | Histone deacetylase inhibitor-mediated EBV reactivation. | Tsai et al. (2011) |
| NSP6 | VAS1 | Relation of V-ATPase was observed with LMP1 and LMP2A that are the onco-proteins of EBV. | DeKroon et al. (2018) |
| NSP7 | PGES2 | PGES2 is responsible for the biosynthesis of PGE2, and it have a either an inhibitory, stimulatory role in there viral replication cycles of EBV. PGES2 undergoes synthesis and produce PGE2, and COX-2 induces lytic reactivation through PGE2 by modulating the EP receptor signaling pathway. | Gandhi et al. (2015) |
| SCPDL | Present in the list of K15-interacting proteins identified by LC-MSMS from latent infection in KSHV. K15 was seen to be related to EBV. | Abere et al. (2018) | |
| RAB10 | In cells provided with EBV lytic induction, Rab10 was found to be colocalized with gp350–gp220. | Nanbo (2020) | |
| NSP8 | DDX10 | Present in the list of genes enriched in the biological processes for cellular genes downregulated by DHX9 depletion (that restricts EBV replication). | Fu et al. (2019) |
| EXOSC3 | Present under the major pathways/genes modulated by EBV-encoded dUTPase in human dendritic cells. | Ariza et al. (2013) | |
| MRPS27 | Present in the list of genes and gene categories induced by LMP2A, i.e. a viral protein of EBV. | Portis et al. (2003) | |
| NSP9 | EIF4H | EBV LMP1 protein stimulates transcription of eIF4E to promote proliferation of EBV-infected cells. | Zhao et al. (2014) |
| NUP88 NUP62 | BGLF4 targets the nucleus through an unconventional interaction with nucleoporins, with the help of EBV lytic replication. | Lee and Chen (2021) | |
| NUP214 ZNF503 | Newly-infected EBNA2hi blast-like B lymphocytes codes high levels of candidate ZFPs (that includes ZNF503 out of the three different ZFPs considered). | Xu et al. (2019) | |
| Nsp12 | RIPK1 | EBV-encoded LMP1 targets RIPK1 ubiquitination and LMP1 is involved in EBV reactivation | Liu et al. (2018), Nawandar et al. (2017) |
| TCF12 | LMP2 promoter binds to TCF12 and LMP2 blocks reactivation of EBV | Arvey et al. (2013) | |
| IMPDH2 | Differentially expressed during EBV reactivation | Chen et al. (2008) | |
| ORF3a | SUN2 | During EBV reactivation, BGLF4 protein modifies the nuclear envelope-associated proteins of the infected cell, including SUN1 and SUN2 for further viral replication of EBV. | Chang et al. (2015) |
| TRIM59 | EBV circLMP2A promotes cancer stemness properties of cells via circLMP2A/miR-3908/TRIM59/p53 axis | Tan and Lim (2021) | |
| ORF10 | ELOB and ELOC | EBV ZEBRA protein interaction with Cul2 and Cul5 aid in building the multimolecular ECS complex (Elongin B/C-Cul2/5-SOCS-box protein), which further targets proteosomal breakdown of p53. | Germini et al. (2020) |
| CUL2 | EBV ZEBRA proteins forms complex with Cul2 and Cul5, and identifies p53 with C-terminal phosphorylation and drives degradation of p53 for promoting viral propagation. | Cai and Yang (2016) |
EBV reactivation by protozoa
Plasmodium falciparum
The erythrocytic parasite Pl. falciparum is responsible for the most world-wide morbidity and mortality due to malaria (Indari et al. 2021a, 2021b ). It is one of the main causes of juvenile death in the malaria-prone regions. Pathogenesis of Burkitt lymphoma due to EBV infection and high exposure to Pl. falciparum in the holoendemic malarial region has been long known (Thorley-Lawson et al. 2016). According to this study, BL is the most common cancer affecting children (74% of malignant disorders) in the equatorial countries of Africa.
The reactivation of EBV by Pl. falciparum and associated concerns are widely reported in multiple studies (Moormann et al. 2011, Indari et al. 2021b, 2022c). In 2007, Chene et al. (2007) deciphered the molecular link between malaria and EBV reactivation. Cysteine-rich interdomain 1α (CIDR1α) is part of the parasite-encoded erythrocytic surface protein Pl. falciparum erythrocyte membrane protein 1 (PfEMP1) (Fig. 4). According to a previous study, the interaction of this antigen with B lymphocytes leads to the activation of memory B cells. Since memory B cells are the latent bank of EBV, this activation results in elevated synthesis of EBV in positive cell lines (Chêne et al. 2007). The host immune system is simultaneously compromised and hyperactivated during Pl. falciparum infection, which is reflected in the impaired function of macrophages and presentation of antigens, diminished specific T cell responses, and induction of regulatory T cells. The activation of polyclonal B cells leads to hypergamma-immunoglobulinemia and autoantibody production. The accompaniment of pro-inflammatory and regulatory cytokines due to infection by the pathogen adds to the creation of a suitable immunological state for the reactivation of the virus (Thorley-Lawson et al. 2016).
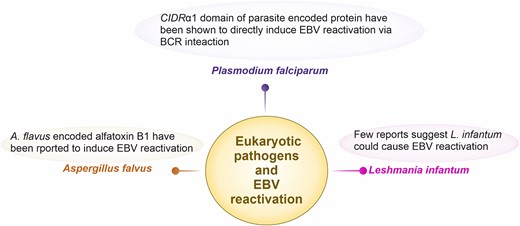
Eukaryotic pathogens that are reported or could possibly induce EBV reactivation. Malaria-mediated Pl. falciparum has widely been studied as it has further shown to generate Burkitt lymphoma in malaria endemic regions. In particular, the cysteine-rich interdomain region (CIDR) α1 domain of parasite-encoded protein Pl. falciparum erythrocytic membrane protein 1 has shown to induce EBV reactivation via B cell receptor activation. Few reports suggest that another protozoa Leshmania infantum could also cause EBV reactivation. A fungal pathogen Aspergillus flavus have been shown to reactivate EBV due to stimulation of cells via fungal alfatoxin B1.
Leishmania infantum
Humans and dogs are commonly affected by zoonotic visceral leishmaniasis, caused by L. infantum. Visceral leishmaniasis (VL) is an opportunistic disease. It may treat patients with low immunity, such as those with HIV infection. The spread of infection by this organism may also occur during organ and stem cell transplantation. A study conducted on a 59-year-old patient was diagnosed with VL secondary to allogeneic hematopoietic stem cell transplantation. During this study, it was observed that the reactivation of EBV occurred and displayed a high load of 3000 genome equivalents per ml (geq/ml), which was further reduced upon treatment with rituximab (Agteresch et al. 2007). Several studies have shown that tumor generation occurs in Leishmania-infected individuals (Schwing et al. 2019, Ouattassi et al. 2022, Vega-Benedetti et al. 2022). However, oncogenic EBV involvement remain unexplored in these tumors. Some studies have reported EBV coinfection with L. infantum during VL. This coinfection causes hemophagocytic lymphohistiocytosis (HLH). Although there are reports suggesting the reactivation of EBV, there is not much clarity regarding its mechanism and cause. It can occur through the proliferation of EBV-infected lymphocytes due to immunomodulation or the independent occurrence of these events (Tascini et al. 2018). Tryparedoxin is a toxin produced by L. infantum. This toxin has the potential to stimulate the B cell activation, which, in turn, may result in the reactivation of EBV.
EBV reactivation by fungi
Aspergillus flavus
Aspergillus flavus is an opportunistic fungus that is commonly associated with clinical manifestations, such as chronic granulomatous sinusitis, allergic fungal sinusitis, keratitis, cutaneous aspergillosis, wound infections, and osteomyelitis after trauma (Hedayati et al. 2007, Rudramurthy et al. 2019). Aspergillus flavus produces aflatoxin B1 (AFB1), which is a mycotoxin that is harmful to animals and humans. It is also a highly toxic and potent carcinogen. In a study by Accardi et al. (2015), hen primary human B cells were precultured in the presence of AFB1 and infected with EBV, AFB1 boosted the number of immortalized B cells. When AFB2 was considered, it was reported that, unlike AFB1, AFB2 does not promote EBV infection. Acyclovir, which is an antiviral drug that inhibits viral DNA polymerase, suppressed the AFB1-dependent EBV genome amplification, which could imply that EBV DNA replication is responsible for increased infection efficiency observed in AFB1-treated Burkitt’s lymphoma cells (Loukes). When cells were treated with AFB1 for 48 h and then infected with EBV, the methylation levels in the CpG sites, including the meZREs, in the promoters of the BZLF1 target genes, BRLF1 and BBLF4, increased, which is required for the activation of the EBV lytic cascade. Furthermore, AFB1 reactivated EBV in latently infected cells (Accardi et al. 2015).
In the same study, the effect of AFB1 on EBV infection was assessed using an animal model. Mice that were treated with AFB1/EBV had a higher EBV DNA load in their blood compared to mice that were not treated with AFB1. They also had higher EBNA2-positive B counts, EBER RNA levels, and BZLF1 mRNA levels in the spleen. The presence of these two factors indicated the transformation of B cells into the spleen. Hence, AFB1 stimulates EBV reactivation and it also promotes EBV-induced B cell transformation (Accardi et al. 2015).
AFB1 and at lower efficiency, AFB2 reactivates latently infected cells by activating the P13K pathway, which results in BZLF1 expression. AFB1 could possibly bind to receptors on the surface of lymphoid cell lines, such as receptor tyrosine kinase (RTK) or G-protein-coupled receptors (GPCR). Upon binding to receptor tyrosine kinases (RTK), RTKs autophosphorylate their intracellular domains upon stimulation. This autophosphorylation activates the P13K Pathway, which, in turn, activates a number of downstream proteins, such as AKT, which further activates the mammalian target of rapamycin (mTOR) protein and the transcription factor AP-1. As a result, EBV was reactivated. Additionally, signaling pathways such as p38 MAPK and c-Jun are also crucial for AFB1-mediated reactivation of the EBV lytic cycle, as inhibitors of these two pathways impaired AFB1’s ability to induce BZLF1 expression (Accardi et al. 2015).
Conclusion
EBV reactivation is one of the pivotal steps in developing any EBV-associated complications such as cancer. Many types of biological agents, including bacteria, viruses, fungi, and protozoa have been reported to reactivate EBV. In one way, some of these biological entities can either directly interact with EBV-infected cells to drive the EBV reactivation as seen in the case of Pl. falciparum-driven BCR stimulation and EBV reactivation (Fig. 4). In another way, the biological agents can modulate nearby microenvironments or produce molecules that may stimulate the cell for EBV lytic cycle progression as seen in the case of Helicobacter pylori, periodontal pathogens, Aspergillus flavus, etc. Most of the viruses can drive the process from inside the cells to activate lytic gene promotors as observed in the case of HSV-1 and HHV6. We also additionally have shown here the host proteins interacting with SARS-CoV-2 protein and having a potential role in EBV reactivation. This information would be important for gaining additional insight into SARS-CoV-2-mediated EBV reactivation by further research. There is indeed an increased need to avail the details of biological agents causing EBV reactivation as the main cause of observed complications may stay hidden. The original culprit of EBV-associated complications could actually be some other biological agents as observed in case of the development of Burkitt’s lymphoma post malaria-mediated EBV reactivation. Future research will help in understanding the interplay of pathogens playing role in EBV reactivation. It may aid in developing therapeutic interventions to minimize the complications associated with such scenarios.
Author contributions
Omkar Indari (Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft), Subhrojyoti Ghosh (Formal analysis, Investigation, Methodology), Adhiraj Singh Bal (Investigation, Methodology), Ajay James (Investigation, Methodology), Mehek Garg (Investigation, Methodology), Amit Mishra (Visualization, Writing – review & editing), Krishanpal Karmodiya (Visualization, Writing – review & editing), and Hem Chandra Jha (Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing).
Conflict of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}