





Clostridium difficile infection (CDI) is the most common cause of antibiotic-associated diarrhea and colitis in developed countries. The disease is mainly mediated via two major exotoxins TcdA and TcdB secreted by the bacterium. We have previously developed a novel, potently neutralizing, tetravalent and bispecific heavy-chain-only single domain (VHH) antibody to both TcdA and TcdB (designated as ABA) that reverses fulminant CDI in mice. Since ABA has a short serum half-life, in this study a replication-deficient recombinant adenovirus expressing ABA was generated and the long-lasting expression of functional ABA was demonstrated in vitro and in vivo. Mice transduced with one dose of the adenovirus displayed high levels of serum ABA for more than1 month and were fully protected against systemic toxin challenges. More importantly, the ABA delivered by the adenovirus protected mice from both primary and recurrent CDI. Thus, replication-deficient adenoviral vector may be used to deliver neutralizing antibodies against the toxins in order to prevent CDI and recurrence.
INTRODUCTION
Clostridium difficile is the most common cause of nosocomial antibiotic-associated diarrhea and is the etiological agent of pseudomembranous colitis (Cloud and Kelly 2007). Clostridium difficile infection (CDI) is primarily caused by two large exotoxins: TcdA and TcdB. It is estimated that over 500 000 cases of CDI occur annually in the USA and that CDI causes over 29 000 deaths (Lessa et al.2015). The incidence of CDI-associated mortality in patients is increasing rapidly due to the emergence of hypervirulent and antibiotic-resistant strains (Kelly and LaMont 2008). Systemic complications are the major cause of death in C. difficile-associated disease (Siemann, Koch-Dorfler and Rabenhorst 2000).
Primary treatment for CDI involves the use of the antibiotics metronidazole and/or vancomycin. However, neither antibiotic is entirely effective as high rates of recurrence occur after initial successful treatment with these antibiotics (Huang et al.2009). The incidence of recurrence is estimated at 20%–35% after which there is an even greater probability (as high as 50%) of additional recurrences (Zar et al.2007; Johnson 2009). The newly FDA-approved drug fidaxomicin shows an improved efficacy over vancomycin at lowering the relapse rate (Louie et al.2011). Several experimental therapies are currently under development, including novel antibiotics (Garey et al.2011; Louie et al.2011; Butler et al.2012), probiotic and fecal transplant therapies (Gough, Shaikh and Manges 2011; Brandt et al.2012), vaccines (Ward et al.1999; Kotloff et al.2001; Gardiner et al.2009; Seregin et al.2012; Wang et al.2012) and antibody-based therapies (Kelly et al.1996; Lowy et al.2010; Marozsan et al.2012; Roberts et al.2012).
Camelidae species, which includes camels, llamas and alpacas, produce both conventional IgG antibodies containing a heavy chain and light chain (IgG1) and unconventional IgGs (IgG2 and IgG3) that consist of a single heavy chain (HCAb) (Wesolowski et al.2009). HCAbs bind antigens only through the variable domain of the heavy chain, thus allowing easy cloning of the DNA encoding this binding domain, which is known as a single domain antibody or variable heavy-chain-only antibody (VHH) (Wesolowski et al.2009). Several groups have used VHHs for treating toxin-mediated diseases (Adams et al.2009; Tremblay et al.2013). Recently, we have developed a novel, potently neutralizing, tetravalent and bispecific heavy-chain-only single domain (VHH) antibody to both TcdA and TcdB (designated as ABA) (Yang et al.2014). A systemic infusion with ABA prevented mice from fulminant CDI (Yang et al.2014). In this study, we generated a replication-deficient recombinant adenovirus expressing ABA and demonstrated the expression of functional ABA in vitro and in vivo. Mice transduced with one dose of the adenovirus displayed high levels of serum ABA and were fully protected against oral challenge of C. difficile spores. Thus, replication-deficient adenoviral vector may be used to express neutralizing antibodies against the toxins in order to prevent CDI.
MATERIALS AND METHODS
Construction of recombinant adenoviral vector expressing ABA tetramer (rAd-ABA)
ABA tetramer was constructed as described previously (Yang et al.2014). pAdEasy-1, part of the AdEasy Adenoviral Vector System (Agilent Technologies, Santa Clara, CA), served as the adenoviral vector to express ABA as described previously (Liu et al.2004). Briefly, a DNA fragment consisting of the CMV promoter, His-tag, ABA tetramer and E-tag was amplified from pSEC91 by PCR with primers terminated with an upstream KpnI site and a downstream XbaI site, and subcloned into KpnI/XbaI digested pShuttle vector to form the psABA construct. MyFi DNA polymerase (Bioline USA Inc, Taunton, MA) was used to maintain a high fidelity, which was confirmed by sequencing the insert. The PmeI site in the 3′ untranslated region of the ABA sequence was removed by PCR-based site-directed mutation as described previously (Liu et al.2004). Recombinant adenoviral DNA was generated by cotransforming BJ5183-Ad competent cells with PmeI-linearized psABA plus pAdEasy-1. Correct recombinant DNA was selected by restriction digestion and transfected into AD-293 cells to produce recombinant virions, which were then purified by double CsCl density-gradient ultracentrifugation (Jager et al.2009). The band of intact rAd-ABA virions was collected and desalted via PD-10 column (GE Healthcare Life Sciences, Pittsburgh, PA). Virion concentration was evaluated by A260 absorbance and also tested functionally by plaque formation by transfecting cultured human embryonic kidney cell line HEK293 cells (ATCC, Manassas, VA), which are then immobilized by agar. The titer was calculated as plaque-forming units (PFU) per ml. The rAd-ABA virion preparation was saved in 1× PBS 10% glycerol at –80°C until use. The control GFP plasmid construction and rAd-GFP preparation were carried out the same way except that the ABA gene was replaced with the GFP gene in the constructs.
rAd-ABA infection of cultured cell lines and purification of ABA from culture supernatants
rAd-ABA virions were used to infect Africa green monkey kidney epithelial cell line Vero cells (ATCC, Manassas, VA). Vero cells were seeded at density of 2.0 × 106 cells in a T75 flask 1 day before rAd-ABA virion infection. When cells reached 70% confluency, the culture media was aspirated. Then 1.5 ml of cell culture medium including concentrated rAd-ABA virions were applied to Vero cells, which were then cultured in a 37°C incubator for 1 h. The flask was rocked every 10–15 min for better infection. After that, 10 ml of growth culture media was added to virus-infected cells, which were cultured until dead. Culture supernatant was harvested, centrifuged and further filtered. His-tagged ABA were purified via Ni-affinity chromatography. The purity of the ABA was visualized by SDS-PAGE and binding to the two toxins was examined by dot blotting. To perform dot blotting, serially diluted TcdA and TcdB proteins were spotted on a nitrocellulose paper. The paper was then blocked with non-fat milk before adding ABA at a final concentration of 1 μg ml−1. After washing, horseradish-peroxidase (HRP)-conjugated anti-E-tag antibody (Bethyl Laboratories, Inc. Montgomery, TX) was used as a secondary antibody for detection.
ELISA assays
Microplates were coated with 0.5 μg ml−1 of recombinant TcdA or 0.5 μg ml−1 of TcdB (Yang et al.2008) overnight at 4°C and incubated with the purified ABA or with the sera from rAd-ABA-infected mice. After washing, HRP-conjugated anti-E-tag antibody was added to plates, followed by standard ELISA detection.
In vitro neutralizing assays
mRG1 cells that express human CD64 (He et al.2009) were cultured in DMEM medium with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U ml−1 penicillin-G and 40 μg ml−1 streptomycin sulfate. Subconfluent mRG1 cells (2.0 × 104 cells per well) were seeded in 96-well plates for 24 h before the addition of toxin and VHH antibodies. Serially diluted TcdA or TcdB was mixed with 1 μg ml−1 ABA for 1 h at 37°C. Then 100 μl of the mixture was applied to 90% confluent mRG1 cells. In wells with TcdA, A1H3 was added to enhance TcdA cytotoxicity (He et al.2009; Yu et al.2015). After culturing for 24 h, cell rounding was observed under microscopy.
Infection of Mice with rAd-ABA and measurement of ABA levels in the blood
Six-week-old female C57BL/6 mice (Envigo, Frederick, MD) were maintained in a pathogen-free ABSL2 facility. All mice used in the experiments were housed in groups of five per cage under the same conditions. Food, water, bedding and cages were autoclaved. All procedures involving mice were conducted under protocols approved by the Institutional Animal Care and Use Committee. Mice (five per group) were administered rAd-ABA virions at 109, 1010 or 1011 PFU per mouse by intravenous (i.v.) injection. Serum samples were collected from the infected mice and the amounts of ABA in blood were measured by ELISA.
Systemic challenge with Clostridium difficile toxins
Mice (five per group) were administered rAd-ABA or rAd-GFP virions (1010 PFU) by i.v. injection 4 days before intraperitoneal challenge with a mixture of TcdA and TcdB (25 or 250 ng of each toxin per mouse). Mice were monitored hourly for signs of illness including hunched posture, ruffled coat and rapid breathing. Animals that became moribund were sacrificed.
Mouse primary and recurrent CDI
C57BL/6 mice were orally administered 105C. difficile spores from the UK1 (BI/NAP1/027) strain after antibiotic treatment as previously described (Wang et al.2012). Mice (10 per group) were administered rAd-ABA or rAd-GFP virions (1010 PFU) by i.v. injection 4 days before spore challenge. In a mouse recurrent/reinfection model, all surviving mice from rAd-GFP groups (10 mice from three repeated experiments) and 11 age-matched mice from rAd-ABA groups were treated with antibiotic cocktail before rechallenge with 105 UK1 C. difficile spores 2 weeks after the initial challenge as described previously (Sun et al.2011). Mouse weights were measured daily and the development of disease symptoms was monitored twice a day. Animals that became moribund or lost more than 15% of their body weight within 24 h or 20% of their body weight during the course of the infection were sacrificed.
Statistical analysis
Data were analyzed by the Kaplan–Meier survival analysis with the log-rank test of significance, analysis of variance (ANOVA) and by one-way ANOVA followed by Bonferroni post-tests using the Prism statistical software program. Results are expressed as mean ± standard error of mean.
RESULTS
Construction of rAd-ABA and virion production
We subcloned the ABA expression cassette into the shuttle vector of the AdEasy system and successfully constructed recombinant adenovirus expressing the ABA protein under control of the human CMV promoter. After double CsCl banding by ultracentrifugation and removing extra salt via PD10 column, we were able to obtain rAd-ABA at a titer of 4 × 1012 PFU ml−1. The capability of virion infection was confirmed on cultured HEK293 cells before application in transduction of Vero cells and in living animal.
Cells infected with rAd-ABA secrete functional ABA that neutralizes both TcdA and TcdB
Supernatants from Vero cells infected with rAd-ABA were collected and ABA was purified via Ni-chromatography. The purified ABA was the dominant band on a SDS-PAGE with a molecular weight around 66 kDa (Fig. 1a). Dot blotting showed that ABA recognized decreasing amounts of both TcdA and TcdB in a dose-dependent manner (Fig. 1b). The binding of ABA to the toxins was further measured by ELISA and the serially diluted ABA purified from rAd-ABA-infected cells exhibited dose-dependent binding to TcdA and TcdB (Figs 2a and b). Moreover, ABA neutralized the cytopathic effects of both TcdA and TcdB in a dose-dependent manner (Fig. 2c). TcdA and TcdB caused cell rounding of mRG1 cells in a dose-dependent manner and the dose curves were shifted left to substantially higher toxin concentrations in the presence of ABA (Fig. 2c).

ABA purification and function. ABA was purified from culture supernatants of Vero cells infected with rAd-ABA. (a) The purified ABA exhibited a single dominant band at 66 kDa on a SDS-PAGE gel. (b) Dot blotting showed binding of ABA to TcdA and TcdB. Serially diluted TcdA and TcdB proteins were spotted on nitrocellulose paper. The paper was then blocked with milk before adding ABA at 1 μg ml−1 final concentration. The binding of ABA to both TcdA and TcdB was determined by an HRP-conjugated anti-E-tag antibody.
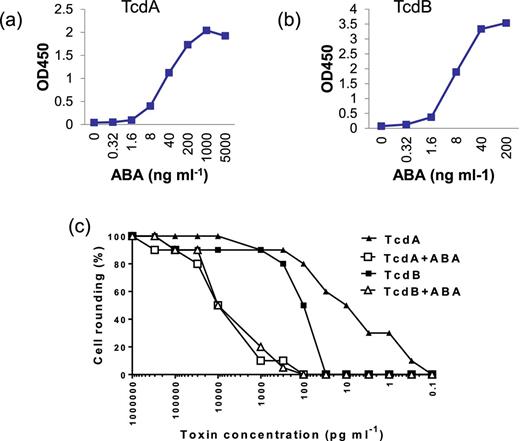
ABA binding and neutralizing activities. Serially diluted ABA (ng ml−1) was added to microplate wells coated with TcdA (a) or TcdB (b). The binding of ABA to the toxins was determined by an HRP-conjugated anti-E-tag antibody. (c) Serially diluted TcdA or TcdB mixed with or without 1 μg ml−1 of ABA was added to mRG1 monolayers in a 96-well plate. The cell rounding was examined under a phase-contrast microscope after 24 h incubation.
Mouse infected with rAd-ABA express high serum levels of ABA
Next we evaluated the secretion of ABA in mice after their transduction with rAd-ABA. Groups of mice were i.v. injected with 1011, 1010 or 109 rAd-ABA virions. The non-replicable recombinant adenoviral infection did not cause any observable symptoms in mice even at the highest dose. The virally infected cells rapidly secreted ABA as indicated by serum levels of ABA 2 days after inoculation. Mice injected with 1011 virions rapidly reached saturating amounts of ABA in circulation within 2–4 days after inoculation, whereas the injection of 1010 virions led to a lower serum level of ABA 2 days after inoculation but reached similar levels as mice injected with higher dose 4 days after inoculation (Fig. 3a). Furthermore, mice injected with of 109 virions exhibited even lower amounts of serum ABA within the first week, but the ABA levels gradually reached similar levels as those mice infected with higher viral doses 2 weeks after inoculation (Fig. 3a). A repeated pharmacokinetic study of mice transduced with 1010 rAd-ABA virions showed that the high serum levels of ABA lasted approximately 35 days and then gradually declined during the second month of the study (Fig. 3b). Therefore, we chose 1010 virions as the infectious dose for the rest of the experiments.
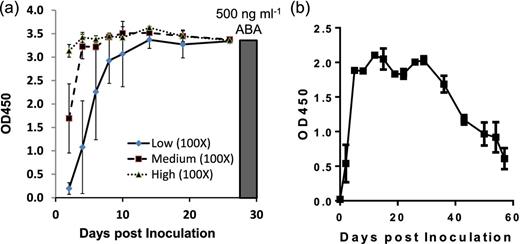
Serum ABA levels in mice transduced with rAd-ABA. (a) Mice were i.v. injected with 109 (low), 1010 (medium) or 1011 (high) PFU of rAd-ABA. Serum samples were collected over 25 days and serum levels of ABA was measured by ELISA. The data show the mean OD values of serum samples (100× dilution) from three mice. The black bar shows the OD value of purified ABA (500 ng ml–1). (b) Mice were i.v. injected with 1010 PFU of rAd-ABA. Serum samples were collected over 60 days and serum levels of ABA was measured by ELISA.
rAd-ABA protects mice from systemic toxin challenge
Since mice infected with rAd-ABA secreted high levels of ABA in circulation, we assessed whether rAd-ABA transduced mice are protected from systemic challenge of lethal Clostridium difficile toxins. Four days after the injection of rAd-ABA, mice were challenged with a mixture of TcdA and TcdB (25 ng of each toxin per mouse). Mice were completely protected against a lethal challenge of mixed TcdA and TcdB when they were pre-treated with rAd-ABA, whereas mice injected with either PBS or the same dose of rAd-GFP died within 24 h of toxin challenge (P = 0.003) (Fig. 4). Moreover, the rAd-ABA-infected mice were significantly protected against a challenge of 10 times the lethal dose of toxin (250 ng of each toxin per mouse) as compared with PBS-treated or rAd-GFP-infected mice (P = 0.01) (Fig. 4).
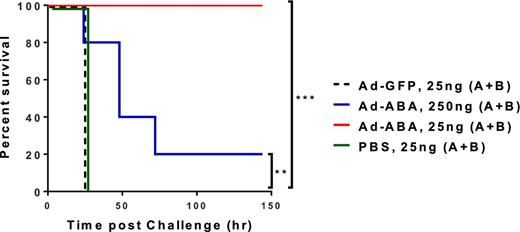
rAd-ABA protection of mice against systemic toxin challenges. PBS, rAd-GFP (1010 PFU) or rAd-ABA (1010 PFU) was i.v. administered 4 days before mice were challenged with a mix of TcdA and TcdB (25 or 250 ng of each toxin per mouse) and mouse survival was monitored. Overall mouse survival was analyzed by Kaplan-Meier survival curves. **P = 0.01 and ***P = 0.003.
rAd-ABA protects mice from primary and recurrent CDI
Finally, we evaluated the efficacy of rAd-ABA to prevent severe disease symptoms in mice challenged with C. difficile spores (Fig. 5a). Mice were injected with a single dose of rAd-ABA, or rAd-GFP as a control, during antibiotic treatment. Four days later, mice were challenged with C. difficile spores from the NAP1/027 strain. Treatment of mice with rAd-ABA significantly prevented the severe weight loss associated with CDI as compared with rAd-GFP treatment (Fig. 5b). More importantly, mice treated with rAd-ABA were significantly protected from mortality induced by C. difficile challenge compared with the rAd-GFP control (Fig. 5c). None of the mice in rAd-ABA groups died, whereas 70% of the mice succumbed in the rAd-GFP control group (Fig. 5c). Since rAd-ABA infection led to high serum levels of ABA for over a month (Fig. 3), we hypothesized that those mice were also protected against CDI recurrence. The surviving mice from both groups were rechallenged with the same dose of UK1 spores. rAd-GFP-treated mice had significant weight loss and 30% of the mice succumbed from the second episode of CDI, whereas the rAd-ABA-treated group had less weight loss and all mice survived (Figs 5d and e).
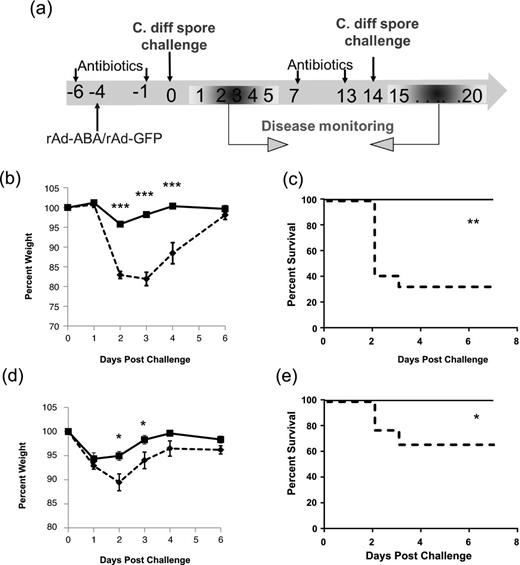
rAd-ABA protection of mice against primary and recurrent CDI. (a) rAd-GFP (1010 PFU, dashed lines) or rAd-ABA (1010 PFU, solid lines) was i.v. administered during antibiotic treatment. Four days later, mice were challenged with 105 PFU of C. difficile UK1 spores. Mouse weight loss (b) and survival (c) were monitored and are plotted. All surviving mice from rAd-GFP groups (9 mice) and 11 surviving mice from rAd-ABA groups were treated with another episode of antibiotic cocktail and then rechallenged with 105 PFU of C. difficile UK1 spores 14 days after the initial challenge (a). Mouse weight loss (d) and survival (e) were monitored and are plotted. Error bars show standard errors of mean (SEM). *P < 0.05, **P < 0.01 and ***P < 0.001.
DISCUSSION
The bispecific heavy-chain-only single domain (VHH) antibody ABA neutralizes TcdA and TcdB simultaneously and protects mice from fulminant CDI (Yang et al.2014). However, ABA has a short serum half-life that limits its potential in CDI prevention. In this study, ABA was designed to be delivered by a replication-deficient adenovirus and the viral infection of mice led to a high level of serum ABA for more than1 month. Mice infected with rAd-ABA were protected from systemic toxin challenge and also Clostridium difficile spore challenge in a prophylactic CDI model. Therefore, this study proved the principle that adenovirus can be utilized for the delivery of therapeutic antibodies for the prevention of CDI and is effective against both primary and recurrent disease.
Live vectors have been used for antibody delivery for a variety of therapeutic applications and offer an alternative to injectable preparations of purified antibodies. The most widely used viral vectors are adenovirus and adeno-associated virus (AAV) for delivering antibodies against both cancers and infectious diseases in in vivo experimental settings (Jiang et al.2006; Fang et al.2007; De et al.2008; Skaricic et al.2008; Sofer-Podesta et al.2009). Recently, adenovirus has been used to deliver VHHs against toxins from infectious diseases and illnesses, such as botulism and Escherichia coli infection (Mukherjee et al.2014; Sheoran et al.2015). The virus-based live vectors enable the achievement of sustained antitoxin protection against disease targets and virulence factors thus enhancing therapeutic efficacy. In previous studies where adenoviral vector encoding an anti-anthrax single-chain variable fragment (scFv) was i.v. administered to mice, scFv was detected in serum over a 2-week period conferring antitoxin protection for 14 days before toxin challenge (Kasuya et al.2005). Another study using intrapleural administration of an AAV vector encoding the anti-anthrax antibody protected mice against toxin challenge from 2 weeks up to a 6-month period (De et al.2008), demonstrating delayed but sustained efficacy of AAV-delivered antitoxins. In this study, a single intravenous dose of rAd-ABA achieved more than 30 days of high serum levels of ABA in mice.
An ideal antitoxin therapeutic should provide protection with an easily administered single application that is rapid acting and long lasting, providing a high level of protection for the duration of a toxin exposure threat. In this study, we combined two technologies for the design of the therapeutic: a novel bispecific ABA antibody that potently and broadly neutralizes both TcdA and TcdB (Yang et al.2014) and a replication-deficient adenoviral delivery system that provides high, long-lasting levels of ABA production. The level of ABA in mouse sera rapidly reached therapeutic doses within a few days (Fig. 3) and mice were either completely or significantly protected against lethal or supralethal (10x) doses in systemic toxin challenges, respectively (Fig. 4).
These rapid and long-lasting transgene expression kinetics obtained using adenovirus renders this vector particularly applicable as an antitoxin delivery vehicle for CDI treatment and prevention. The options for treating CDI patients are limited and the recurrence rate is high (20%–35% of patients). Standard therapy with antibiotic treatments is suboptimal and has a disruptive effect on gut microflora leading to multiple relapses (Barbut et al.2000; Zar et al.2007; Gerding, Muto and Owens 2008). Numerous independent studies have demonstrated that neutralizing antibodies against the toxins confer protection against CDI (Fernie et al.1983; Kim, Iaconis and Rolfe 1987; Lyerly et al.1991; Katchar et al.2007; Leav et al.2010; Lowy et al.2010; Wang et al.2012). Merck's phase III clinical trials showed that bezlotoxumab (anti-TcdB) exhibited a 40% reduction of relapse rate compared with placebo. At present, no prevention/treatment approaches are available that are effective against both primary and recurrent CDI. In this study, we found a single dose of rAd-ABA-protected mice from both primary and recurrent CDI. The mechanism of protection is likely mediated by long-lasting high levels of ABA in rAd-ABA-treated mice. In fact, surviving mice maintained a high level of ABA throughout entire experiments (data not shown), which is consistent with the mouse pharmacokinetic study (Fig. 3). Thus, our study provided a potential new therapeutic strategy for preventing high-risk patients from developing primary and recurrent CDI.
FUNDING
Thisworkwas supported by the National Institute of Allergy and Infectious Diseases and National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health [R01AI088748, R01DK084509 and U19 AI109776]. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of interests
HF has been a paid speaker/consultant for the following companies: Merck, Vaxinnate, Genesis Biotechnology Group. HF is a co-founder of FZata Inc. These companies had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}