





DNA sensors are formidable immune guardians of the host. At least 14 cytoplasmic DNA sensors have been identified in recent years, each with specialized roles in driving inflammation and/or cell death. Of these, AIM2 is a sensor of dsDNA, and forms an inflammasome complex to activate the cysteine protease caspase-1, mediates the release of the proinflammatory cytokines IL-1β and IL-18, and induces pyroptosis. The inflammasome sensor NLRP3 can also respond to DNA in the forms of oxidized mitochondrial DNA and the DNA derivative RNA:DNA hybrids produced by bacteria, whereas the putative inflammasome sensor IFI16 responds to viral DNA in the nucleus. Although inflammasomes provoke inflammation for anti-microbial host defense, they must also maintain homeostasis with commensal microbiota. Here, we outline recent advances highlighting the complex relationship between DNA-sensing inflammasomes, bacterial host defense and the gut microbiota.
INTRODUCTION
Pattern-recognition receptors (PRRs) are a set of germ-line encoded sensors, which provide host defense against infectious agents and physiological perturbation (Takeuchi and Akira 2010). PRRs recognize conserved microbial motifs known as pathogen-associated molecular patterns (PAMPs), including lipopolysaccharides (LPS), lipoproteins, flagellin, microbial DNA, RNA, metabolites and toxins. They also detect danger-associated molecular patterns (DAMPs) released from damaged host cells, including ATP, alarmins and host-derived DNA. PRRs can be membrane-bound (Toll-like receptors (TLRs) and C-type lectin receptors), or cytoplasmic (nucleotide and oligomerization domain, leucine-rich repeats-containing proteins (NLRs), RIG-I-like receptors (RLRs) and AIM2-like receptors (ALRs)).
DNA is a source of PAMPs and DAMPs. During infection by cytosolic bacteria, damage to bacteria can lead to a release of bacterial DNA in the cytoplasm that activates cytoplasmic DNA sensors (Hornung and Latz 2010; Keating, Baran and Bowie 2011; Wu and Chen 2014; Xiao 2015). In healthy host cells, DNA is sequestered in the nucleus and mitochondria, precluding its ability to activate cytoplasmic DNA sensors. However, cellular injury might result in the liberation of nuclear or mitochondrial DNA and activates cytoplasmic DNA sensors (Muruve et al. 2008; Shimada et al. 2012).
Activation of cytoplasmic DNA sensors leads to transcriptional upregulation of type I interferons (IFNs), proinflammatory cytokines and other host defense genes, assembly of macromolecular signaling machineries, and induction of programmed cell death. In addition to the membrane-bound TLR9, at least 14 cytoplasmic DNA sensors have been described so far. Of these, the DNA sensor cGAS and its adaptor STING are essential components driving the type I IFN signaling pathway in response to cytoplasmic DNA (Ablasser et al. 2013; Sun et al. 2013; Wu et al. 2013). Several other DNA sensors have been suggested to contribute to transcriptional regulation of type I IFNs and/or pro-inflammatory cytokines, including ZBP1 (also known as DLM-1 or DAI) (Fu et al. 1999; Schwartz et al. 2001; Takaoka et al. 2007), RNA polymerase III (Ablasser et al. 2009; Chiu, Macmillan and Chen 2009), ADAR1 (Herbert et al. 1997, 1998; Schwartz et al. 1999a,b; Wang et al. 2008), IFI16 (Unterholzner et al. 2010), DHX9 (Kim et al. 2010b), DHX36 (Kim et al. 2010b), DDX41 (Zhang et al. 2011), DDX60 (Miyashita et al. 2011), LRRFIP1 (Yang et al. 2010), RAD50 (Roth et al. 2014) and SOX2 (Xia et al. 2015). The biological functions of these DNA sensors have been reviewed elsewhere and will not be the focus of this review article (Hornung and Latz 2010; Keating, Baran and Bowie 2011; Wu and Chen 2014; Xiao 2015).
Unlike DNA sensors which control the process of gene transcription, AIM2 recognizes DNA and assembles an inflammasome—a cytoplasmic protein complex which drives pyroptosis and proteolytic maturation of the pro-inflammatory cytokines IL-1β and IL-18. Other than AIM2, the sensors NLRP1, NLRP3, NLRC4 and pyrin are capable of forming an inflammasome complex (Man and Kanneganti 2016). Although AIM2 is a specific sensor of cytosolic DNA (Burckstummer et al. 2009; Fernandes-Alnemri et al. 2009; Hornung et al. 2009; Roberts et al. 2009), there is some evidence to suggest that NLRP3 can recognize DNA or DNA derivatives to drive inflammasome responses (Shimada et al. 2012; Kailasan Vanaja et al. 2014). Furthermore, the nuclear DNA sensor IFI16 has also been suggested to bind DNA and form an inflammasome complex (Kerur et al. 2011). Here, we outline the role of DNA-sensing inflammasomes in innate immunity, and focus primarily on their roles in the host defense against bacterial infection and the regulation of the gut microbiota.
AIM2 inflammasome
Earlier studies observed that delivery of DNA into macrophages by electroporation or transfection results in death of the cells (Stacey, Ross and Hume 1993; Van De Parre et al. 2005), a phenomenon we now called pyroptosis. In 2008, Muruve and colleagues reported that transfection of bacterial, viral, or mammalian DNA activates caspase-1 and induces secretion of IL-1β in mouse bone marrow-derived macrophages (BMDMs) (Muruve et al. 2008). The DNA-induced inflammasome responses are dependent on the inflammasome adaptor protein ASC (apoptosis-associated speck-like protein containing a carboxyl-terminal CARD), but not any of the established inflammasome sensors at the time—NLRP1, NLRP3 and NLRC4 (Muruve et al. 2008). These earlier studies suggest that a bona fide inflammasome sensor of DNA exist in the cytoplasm to activate the inflammasome.
In 2009, four groups found that the cytosolic sensor AIM2 binds dsDNA and assembles a functional inflammasome platform in the presence of ASC and caspase-1 (Burckstummer et al. 2009; Fernandes-Alnemri et al. 2009; Hornung et al. 2009; Roberts et al. 2009). Prior to the identification of AIM2 as an inflammasome sensor, the Monack laboratory demonstrated that the Gram-negative bacterium Francisella novicida also activates an unknown ASC-dependent inflammasome (Mariathasan et al. 2005). Further studies revealed that the then-unknown inflammasome sensor mediating the recognition of F. novicida was in fact AIM2 (Fernandes-Alnemri et al. 2010; Jones et al. 2010; Rathinam et al. 2010).
AIM2 has a HIN-200 domain at its carboxyl terminus, which binds directly to dsDNA of at least 80 base-pairs in length, irrespective of its sequence composition (Jin et al. 2012). The pyrin domain of AIM2 interacts with the pyrin domain of the pyrin-CARD-containing protein ASC. The CARD of ASC then binds the CARD of the cysteine protease procaspase-1, fulfilling the archetype of a functional inflammasome complex (Fig. 1). In the absence of DNA, the HIN-200 domain interacts with the pyrin domain of AIM2 to provide autoinhibition (Jin et al. 2012, 2013).
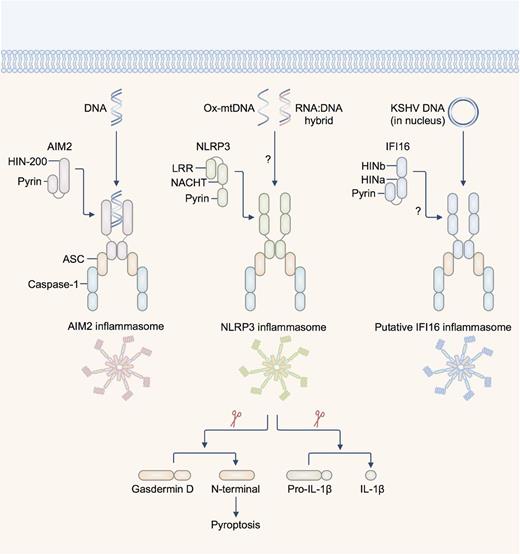
Inflammasome-activating DNA sensors. AIM2 is a dsDNA sensor which binds DNA of microbial or host origin. The HIN-200 domain binds DNA in a sequence-independent manner. NLRP3 has been suggested to recognize oxidized DNA released from the mitochondria (ox-mtDNA) and RNA:DNA hybrids released by bacteria. The pyrin domain of AIM2 and NLRP3 recruit via pyrin–pyrin interaction, the pyrin-CARD-containing protein ASC (apoptosis-associated speck-like protein containing a carboxyl-terminal CARD). The CARD of ASC interacts with the CARD of pro-caspase-1. The DNA sensor IFI16 has been shown to colocalize with the DNA of Kaposi's sarcoma-associated herpesvirus (KSHV) and with ASC in the nucleus of infected endothelial cells. This complex then redistributes to the cytoplasm where it interacts with caspase-1 to form a putative IFI16 inflammasome. There is also evidence to suggest that IFI16 is required to drive caspase-1-dependent pyroptosis in CD4+ T cells infected with HIV-1. Activation of the inflammasome leads to cleavage of the pro-pyroptotic substrate gasdermin D, initiating pyroptosis and rupture of the cell membrane. Active caspase-1 also cleaves pro-IL-1β and pro-IL-18, generating the bioactive form of the cytokines. Abbreviations: LRR—leucine-rich repeats; NACHT—NB-ARC or domain present in NAIP, CIITA, HET-E and TP1.
AIM2 contributes to the host defense against microbial pathogens from virtually all domains of life, including bacteria, DNA viruses, fungi and protozoa (Man, Karki and Kanneganti 2016). The mechanism governing activation of the AIM2 inflammasome is best characterized in the context of bacterial infection. Bacterial nucleic acids are concealed within the bacterial cell wall and cell membrane; DNA must be liberated to activate the AIM2 inflammasome. Indeed, the AIM2 inflammasome is activated only when the cytosolic pathogen F. novicida escapes the vacuole to the cytoplasm (Mariathasan et al. 2005; Belhocine and Monack 2012; Man et al.2015a; Meunier et al. 2015). Bacterial mutants of F. novicida which lack genes important for disruption of the vacuole cannot engage the AIM2 inflammasome, and mutants which readily undergo membrane lysis hyperactivate the AIM2 inflammasome (Mariathasan et al. 2005; Peng et al. 2011; Belhocine and Monack 2012; Sampson et al. 2014; Man et al. 2015a; Meunier et al. 2015). Therefore, the ability of F. novicida to access the cytoplasm and release its DNA is a prerequisite for activation of the AIM2 inflammasome.
Recent studies have provided a clue as to how DNA from F. novicida might be released in the cytoplasm to activate AIM2. Multiple studies have found that type I IFN signaling is essential for activation of the AIM2 inflammasome in response to infection by F. novicida, but not in response to infection by DNA viruses or transfection of dsDNA (Henry et al. 2007; Jones et al. 2010; Man et al. 2015a; Meunier et al. 2015). These findings already suggest that the regulatory mechanisms of the AIM2 inflammasome differ depending on the pathogen encountered by the cells or by the way DNA is introduced into the cytoplasm. Our lab and others connected the requirement for type I IFN signaling and liberation of DNA to the activation of the AIM2 inflammasome (Man et al. 2015a; Meunier et al. 2015). In this non-canonical pathway (analogous to the non-canonical NLRP3 inflammasome pathway requiring type I IFN signaling), the transcription factor IRF1 is induced in response to type I IFN signaling during F. novicida infection, whereby IRF1 is crucial for activation of the AIM2 inflammasome (Man et al.2015a). This pathway is specific to F. novicida infection because IRF1 is dispensable for activation of the AIM2 inflammasome induced by transfected dsDNA or infection by mouse cytomegalovirus (MCMV) (Man et al.2015a). Further investigation revealed that IRF1 is required to induce the expression of a family of IFN-inducible proteins called guanylate-binding proteins (GBPs) (Man et al. 2015a).
Our work and the work of Meunier and colleagues further demonstrated that GBP2 or GBP5 are recruited to the bacteria in the cytoplasm, where they mediate the exposure of DNA from F. novicida for sensing by the AIM2 inflammasome (Man et al. 2015a; Meunier et al. 2015). Precisely how GBPs mediate the release of bacterial DNA is still unclear. It is possible that GBPs alone or in concert with other proteins induce lysis of the bacteria in the cytoplasm.
The host sensors which initiate the production of type I IFNs in the non-canonical AIM2 inflammasome pathway have also been identified. The DNA sensor cGAS detects F. novicida DNA and induces type I IFN production and IRF1 expression (Man et al. 2015a; Storek et al. 2015). In addition, Storek and colleagues further identified a partial contribution from p204, a DNA sensor encoded by the Ifi204 gene in mouse; a functional ortholog of human IFI16 (Storek et al. 2015). Both cGAS and p204 signal through STING to drive robust type I IFN production, ultimately leading to activation of the AIM2 inflammasome (Jones et al. 2010; Man et al. 2015a; Meunier et al. 2015; Storek et al. 2015).
These findings culminate in a model, whereby the cGAS-p204-STING axis initially detects DNA released by F. novicida in the cytoplasm, triggering type I IFN signaling and expression of IRF1. IRF1 further induces the expression of GBPs; GBP2 and GBP5 attack F. novicida via a yet-undefined manner, causing bacteriolysis and additional DNA release to a level sufficient to activate the AIM2 inflammasome. In this model, how DNA released by F. novicida activates cGAS and p204, but not AIM2 at the same time have remained unclear. One possibility is that the threshold of DNA which can activate cGAS and p204 is lower than the threshold required to activate AIM2. Additional bacteriolysis orchestrated by IFN-inducible proteins seems to be required to fully engage the AIM2 inflammasome.
In addition to F. novicida, AIM2 contributes to the recognition of Listeria monocytogenes (Kim et al. 2010a; Sauer et al. 2010; Warren et al. 2010; Wu, Fernandes-Alnemri and Alnemri 2010), Mycobacterium species (Saiga et al. 2012, 2015; Yang et al. 2013; Wassermann et al. 2015), Streptococcus pneumoniae (Fang et al. 2011), Porphyromonas gingivalis (Park et al. 2014), Brucella abortus (Gomes et al. 2013), Chlamydia muridarum (Finethy et al. 2015), the DNA viruses MCMV and vaccinia virus (Rathinam et al. 2010; Man et al. 2015a), the fungal pathogen Aspergillus fumigatus (Karki et al. 2015) and the protozoan parasite Plasmodium (Kalantari et al. 2014). How DNA is liberated from these pathogens for sensing by AIM2 during infection has remained largely unclear. However, Sauer and colleagues have shown that lysis of the L. monocytogenes in the cytoplasm is essential for activation of the AIM2 inflammasome (Sauer et al. 2010). Finethy and colleagues showed that activation of the AIM2 and NLRP3 inflammasomes by C. muridarum partially requires GBPs encoded on chromosome 3, but not the ability of GBPs to recruit to the pathogen-containing vacuole (Finethy et al. 2015). This observation is interesting and suggests an alternative mechanism by which ligands are released in the cytoplasm, one that is presumably not dependent on GBP-mediated rupture of the bacterial or vacuolar membrane.
The importance of access to ligands for sensing by cytosolic DNA sensors is further supported by recent studies. Mycobacterium tuberculosis infection activates cGAS and the AIM2 and NLRP3 inflammasomes (Collins et al. 2015; Wassermann et al. 2015; Watson et al. 2015). In this case, sensing of DNA by cGAS requires M. tuberculosis to access the cytoplasm from the vacuole via its type VII secretion system ESX-1 (Collins et al. 2015; Wassermann et al. 2015; Watson et al. 2015). Unlike the case in F. novicida infection, type I IFN production is not necessary for the activation of inflammasomes in the THP-1 human cell line infected by certain strains of M. tuberculosis (Wassermann et al. 2015). This finding provides examples of how the interplay between DNA sensors is determined by the pathogen encountered by the host cell.
Interestingly, the ability of AIM2 to protect the host against infectious agents is not confined to its ability to sense microbial DNA. A recent study suggests that AIM2 mediates host defense against oral Salmonella enterica serovar Typhimurium infection (Hu et al. 2016), a pathogen which normally activates the NLRC4 and NLRP3 inflammasomes (Broz et al. 2010; Man et al. 2014). AIM2 contributes to the upregulation of tight junction proteins, increases barrier permeability and reduces invasion of S. Typhimurium at the intestinal epithelium (Hu et al. 2016). Further, another study found that host DNA released in the lung microenvironment during infection by the RNA virus Influenza A Virus is recognized by AIM2, which leads to a reduction in inflammation-associated lethality in mice (Schattgen et al. 2016). How AIM2 orchestrates its immunosuppressive effects and modifies barrier integrity remain to be determined. Nevertheless, the ability of AIM2 to respond to pathogens from all domains of life highlights its importance in pathogen host defense and innate immunity.
Nucleic acid-sensing potential of the NLRP3 and putative IFI16 inflammasomes
In addition to the AIM2 inflammasome, NLRP3 and the putative IFI16 inflammasomes have been implicated in cytosolic sensing of nucleic acids (Kanneganti et al. 2006; Unterholzner et al. 2010) (Fig. 1). NLRP3 is a global sensor of PAMPs and DAMPs. Activation of the NLRP3 inflammasome is more complex and requires two signals—a priming signal and an activation signal. In principle, the priming signal is provided by TLRs and other receptors which trigger activation of the transcription factor NF-kB, resulting in the upregulation of NLRP3 expression (Bauernfeind et al. 2009). The activation signal is provided by an NLRP3 activator in the forms of PAMPs and DAMPs not limited to ATP, bacterial RNA, pore-forming toxins and crystalline substances (Man and Kanneganti 2015). Activation of the NLRP3 inflammasome is governed by multiple factors, which include potassium efflux, calcium influx, mitochondrial reactive oxygen species, translocation of cardiolipin within the mitochondria and phagosomal destabilization (Rathinam, Vanaja and Fitzgerald 2012).
In the context of DNA sensing, the NLRP3 activators ATP and nigericin can induce the release of oxidized DNA from the mitochondria (Shimada et al. 2012). Oxidized mitochondrial DNA associates with NLRP3 and partially induces NLRP3-dependent IL-1β secretion independently of AIM2 (Shimada et al. 2012). This finding provides some evidence that oxidized mitochondrial DNA could be a potential activator of the NLRP3 inflammasome (Fig. 1). Further, accumulation of damaged mitochondria and ROS and excessive release of mitochondrial DNA in the cytoplasm owing to impaired autophagy or mitophagy (autophagy of mitochondria) can lead to hyperactivation of the NLRP3 inflammasome (Nakahira et al. 2011; Zhou et al. 2011; Lupfer et al. 2013; Zhong et al.2016). From the perspective of microbial pathogenesis, it is tempting to speculate that release of oxidized mitochondrial DNA as a consequence of generalized damaged caused by infection could trigger the NLRP3 inflammasome.
The ability of NLRP3 to mediate the recognition of RNA:DNA hybrids has also been proposed recently (Kailasan Vanaja et al. 2014) (Fig. 1). RNA:DNA hybrids are generated by DNA replication and transcription in bacteria and can be found in the cytoplasm of BMDMs infected with Enterohemorrhagic Escherichia coli. Transfection of synthetic RNA:DNA hybrids into BMDMs activates the NLRP3 inflammasome (Kailasan Vanaja et al. 2014). Further, an E. coli strain deficient in the RNase H enzyme cannot degrade the RNA primer during DNA replication, and therefore, accumulates long and stable RNA:DNA hybrids, triggers elevated level of NLRP3 inflammasome activation (Kailasan Vanaja et al. 2014). Of interest is that the E. coli strain deficient in the RNase H enzyme is cleared from macrophages at a similar rate as wild-type E. coli (Kailasan Vanaja et al. 2014), suggesting that increased level of inflammasome activity does not always correlate with increased bacterial clearance in macrophages. It will be worthwhile to investigate whether RNA:DNA hybrids produced by other bacteria can activate the NLRP3 inflammasome and how these hybrids secure entry to the cytoplasm.
Human IFI16 (mouse p204) is a dsDNA sensor which has the ability to induce transcription of the gene encoding IFN-β in response to infection by herpes simplex virus 1, a DNA virus that replicates in the nucleus (Unterholzner et al. 2010). IFI16 directly binds IFN-stimulating viral DNA in the cytosol, and drives the production of type I IFNs and cytokines via STING (Unterholzner et al. 2010). The identification of IFI16 as a DNA sensor prompted establishment of the AIM2-like receptor family (ALRs, or the pyrin and HIN domain-containing or PYHIN family) (Unterholzner et al. 2010; Brunette et al. 2012; Cridland et al. 2012).
In addition to the ability of IFI16 to induce transcriptional regulation of genes encoding type I IFNs, a study by Kerur and colleagues suggests that IFI16 has the ability to assemble an inflammasome in endothelial cells infected with the DNA virus Kaposi's sarcoma-associated herpesvirus (KSHV) (Kerur et al. 2011). The proposed mechanism is that the viral DNA genome that has invaded the nucleus is recognized by IFI16. IFI16 then associates with ASC and translocates to the cytoplasm where the IFI16-ASC complex recruits caspase-1 to drive inflammasome activation (Kerur et al. 2011) (Fig. 1). Further studies also suggest that IFI16 and caspase-1 are essential for mediating pyroptosis in HIV-1-infected CD4+ T cells (Doitsh et al. 2014; Monroe et al. 2014). In bacterial infection, p204, the mouse ortholog of IFI16 contributes to induction of type I IFN production in response to F. novicida infection, as discussed earlier (Storek et al. 2015). The IFI16 inflammasome speck has also been visualized in human macrophages infected with the gastrointestinal pathogen Campylobacter concisus (Kaakoush et al. 2015). Since bacteria are not known to enter the nucleus of the host cell, detection of bacterial DNA by IFI16 or p204 probably occurs in the cytoplasm. Further studies are required to investigate the importance of IFI16 or p204 in bacterial recognition. Furthermore, how IFI16 or p204 achieves functional synergism rather than redundancy with cGAS and AIM2 in the context of DNA sensing warrants further clarification. A recent study provided insights to this question and found that cGAS can interact and stabilize IFI16 to promote recognition of nuclear herpesviral DNA in fibroblasts (Orzalli et al. 2015).
DNA-sensing inflammasomes and the gut microbiota
Although innate immune sensors mediate recognition of pathogens and trigger immune responses, the host must also maintain homeostasis with commensal microbiota found in the gut, skin, oral cavity and respiratory and urogenital tracts. Genetic predispositions or a change in environment and diet contributes to aberrant activation of innate immune sensors that might lead to dysbiosis, a condition defined by a breakdown in the balance between ‘protective’ and ‘harmful’ bacteria (Man, Kaakoush and Mitchell 2011). Dysbiosis is one of the driving factors which predisposes an individual to IBD, cancer and other clinical manifestations.
Cytoplasmic DNA sensors have emerged as critical regulators of the gut microbiota, especially in the context of colitis and colorectal cancer (Zhu et al. 2014; Man et al.2015b; Wilson et al. 2015). Recently, our work and the work of Wilson and colleagues have shown that mice lacking AIM2 are prone to developing colorectal tumors in a model of colitis-associated cancer (Man et al.2015b; Wilson et al. 2015). In this context, the function of AIM2 is uniquely inflammasome-independent (Man et al. 2015b; Wilson et al. 2015), but requires the ability of AIM2 to induce inhibition of the DNA-dependent protein kinase (DNA-PK) complex (Wilson et al. 2015). AIM2 suppresses DNA-PK-dependent activation of AKT and c-Myc, providing AIM2 the license to ultimately control proliferation of intestinal stem cells in the face of damage and inflammation (Man et al. 2015b; Wilson et al. 2015).
In the gastrointestinal tract, untreated Aim2–/– mice harbor increased levels of Akkermansia muciniphila and Anaeroplasma, and decreased levels of Anaerostipes, Bifidobacterium, Flexispira, Prevotella and Paraprevotella species compared with WT mice (Man et al. 2015b). Differences in the microbiota profile between WT and Aim2–/– mice can be counterbalanced by cohousing WT and Aim2–/– mice (Man et al. 2015b). After treatment with the carcinogen AOM and inflammation-inducing agent DSS, Aim2–/– mice cohoused with WT mice have a reduced tumor burden compared with non-cohoused Aim2–/– mice, whereas cohoused WT mice have an increased tumor burden compared with non-cohoused WT mice (Man et al.2015b).
A further study confirmed that the susceptibility of Aim2–/– mice to DSS-induced colitis is associated with a dysregulated host response to the gut microbiota (Hu et al. 2015). This study shows that antibiotic treatment protects Aim2–/– mice from colitis, suggesting a role for microbiota in acute colitis. Fecal analysis identified a higher burden of E. coli and Enterobacteriaceae in Aim2–/– mice compared with WT mice, both prior to and after the onset of colitis (Hu et al. 2015). However, no significant differences in the abundance of Bacteroides, Lactobacillus, mouse intestinal Bacteroides, Eubacterium rectale, segmented filamentous bacteria, Staphylococcus, Streptococcus, Enterococcus, Clostridium and Porphyromonas have been observed between WT and Aim2–/– mice prior to and after the onset of colitis (Hu et al. 2015).
Although previous studies have demonstrated a role for AIM2 in colitis and colitis-associated cancer to be inflammasome-independent (Man et al.2015b; Wilson et al. 2015), the study by Hu and colleagues argued that AIM2 controls the development of colitis via an inflammasome-dependent mechanism (Hu et al. 2015). The exact reason for this discrepancy is unclear. However, the mouse lines used may account for this difference. The Aim2–/– mouse line used by Hu and colleagues (Hu et al. 2015) was generated using embryonic stem cells from the 129 background, whereas the two other independent lines of Aim2–/– mice used by Man et al. (2015b) and Wilson et al. (2015) were generated using embryonic stem cells from the C56BL/6 background. Importantly, the Ifi202 gene, which encodes p202, is in close proximity to the gene encoding AIM2 (Sester and Stacey 2015). Sester and Stacey recently reported that the Ifi202 gene from the 129 background is expressed, whereas the Ifi202 gene from C57BL/6 background is not expressed at an appreciable level (Sester and Stacey 2015). Owing to the close proximity between the Aim2–/– alleles generated in the 129 background and the 129 version of the Ifi202 gene, both genes were inherited as a haplotype due to a low probability of recombination, essentially introducing the Ifi202 gene from the 129 background into the Aim2–/– mouse gene trap line during backcrossing to C57BL/6 mice (Sester and Stacey 2015). The p202 protein contains two HIN domains, which can bind dsDNA (Roberts et al. 2009). The consequence of introducing the Ifi202 gene from the 129 background into the Aim2–/– mouse line used by Hu and colleagues (Hu et al. 2015) could have an impact on the interpretation of the mechanism driving the development of colitis and colitis-associated colorectal cancer.
In addition to AIM2, the relationship between NLRP3 and the gut microbiota has been highlighted in the context of both colitis and non-alcoholic steatohepatitis (NASH). NLRP3 is required for mediating protection against chemically-induced colitis in mice (Allen et al. 2010; Zaki et al. 2010a,b; Hirota et al. 2011). NLRP3 activates caspase-1 and mediates the release of IL-18, a crucial cytokine which ameliorates inflammation and reduces the risk of developing colorectal cancer (Allen et al. 2010; Dupaul-Chicoine et al. 2010; Zaki et al. 2010a,b). Hirota and colleagues further reported that WT and Nlrp3−/− littermate mice can be distinguished by the fecal microbiota profile they carry (Hirota et al. 2011). Nlrp3−/− mice harbor Enterobacteriaceae, Mycobacterium, Collinsella, Subdoligranulum, Clostridium and Ralstonia, which are not found in WT littermate animals (Hirota et al. 2011). Although these bacterial species could have a role in driving increased susceptibility of the Nlrp3−/− mice to DSS-induced colitis, how NLRP3 prevents colonization of these disease-associated bacteria in the gastrointestinal tract has remained unclear. It is possible that constitutive release of IL-18 driven by NLRP3 could play a role in maintaining a healthy microbiota profile, as seen in the case for NLRP6 (Elinav et al. 2011).
A role for the NLRP3 inflammasome in the modulation of the gut microbiota extends to clinical manifestations other than colitis. Mice deficient in NLRP3, ASC or IL-18 showed enhanced severity of NASH (Henao-Mejia et al. 2012). Interestingly, cohousing mice deficient in NLRP3, ASC or IL-18 with WT mice for 4 weeks increases the severity of NASH in the cohoused WT mice (Henao-Mejia et al. 2012). Treatment with ciprofloxacin and metronidazole reduces the severity of NASH in Asc−/− mice, and blocks increased NASH severity in WT mice carrying disease-associated microbiota from Asc−/− mice (Henao-Mejia et al. 2012). These studies highlight the importance of the NLRP3 inflammasome in the maintenance of a ‘disease-resistant’ gut microbiota. Overall, the influence of cytoplasmic DNA sensors on the gut microbiota is beginning to be realized and further studies are needed to elucidate their relationship with one another.
CONCLUSIONS
Pathogens often enter the cytoplasm of a host cell to replicate and avoid extracellular immunity. Cytoplasmic sensors provide intracellular immunosurveillance by sensing PAMPs released from the pathogen or DAMPs released by the host cell. Microbial or host DNA is normally concealed, and when released, are potent activators of the immune system. Of the compendium of DNA sensors identified, AIM2 is the unequivocal inflammasome-initiating sensor of dsDNA, capable of mediating proteolytic maturation of the key proinflammatory cytokines IL-1β and IL-18, as well as driving pyroptosis. In doing so, IL-1β and IL-18 are secreted to the surrounding milieu where they exert their biological activities. Pyroptosis ensues and physically dismantles the cell to prevent further unwanted replication of the pathogen intracellularly. The capacity of AIM2 to mediating sensing of pathogens from all domains life signifies its importance in host defense. What is unclear is how DNA from each class of pathogens is liberated and presented to the AIM2 inflammasome. This question will be a focal point in future investigations aimed at understanding the regulation of cytosolic DNA sensing. Elucidation of how exactly DNA is released by pathogens would provide insights into the molecular mechanism of DNA sensing in the cytoplasm.
The existence of other putative DNA sensors raised the question of how these can operate synergistically in the cytosolic compartment. From the perspective of inflammasome sensors, AIM2 and NLRP3 are highly expressed and active in macrophages and dendritic cells, with each sensor recognizing a unique subset of DNA derivatives. The putative IFI16 inflammasome has so far been described in endothelial cells infected with KSHV and CD4+ T cells infected with HIV-1. In addition, AIM2 and NLRP3 are cytoplasmic, whereas IFI16 is predominately nuclear. The cell-type-specific activity and compartmentalized distribution of these sensors may account for some of the reasons as to why these sensors are not functionally redundant.
Research into the gut microbiota in health and disease is accelerating at a rapid pace. The microbiota establishes a symbiotic relationship with the host immune system. Yet the importance of DNA-sensing inflammasomes in the regulation of gut microbiota is only beginning to be realized. How DNA sensors specifically regulate the composition of the microbiota is an important question that could yield insights into therapies to prevent or treat diseases associated with dysbiosis. Overall, our knowledge of the relationship between cytosolic DNA sensors and microbial pathogenesis is only at its infancy. Untangling the interplay between DNA sensors against the backdrop of infectious and chronic diseases could unmask therapeutic options for prevention and treatment of human diseases.
FUNDING
Work from our laboratory is supported by the US National Institutes of Health (AR056296, CA163507 and AI101935 to T.D.K.), the American Lebanese Syrian Associated Charities (to T.D.K.) and the R.G. Menzies Early Career Fellowship from the National Health and Medical Research Council of Australia (to S.M.M.).
Conflict of interest. None declared.
REFERENCES

{kind=link}