





Anthrax and botulism are dangerous infectious diseases that can be fatal unless detected and treated quickly. Fatalities from these diseases are primarily due to endopeptidase toxins secreted by the pathogens. Rapid and sensitive detection of the presence of active toxins is the key element for protection from natural outbreaks of anthrax and botulism, as well as from the threat of bioterrorism. We describe an ultrasensitive polymerase chain reaction (PCR)-based assay for detecting proteolytic activity of anthrax and botulinum toxins using composite probes consisting of covalent peptide–DNA conjugate for the detection of anthrax, and noncovalent protein-aptamer assembly to assay botulinum toxin activity. Probes immobilized on the solid-phase support are cleaved by toxins to release DNA, which is detected by real-time PCR. Both assays can detect subpicogram quantities of active toxins isolated from composite matrices. Special procedures were developed to isolate intact toxins from the matrices under mild conditions. The assay is rapid, uses proven technologies, and can be modified to detect other proteolytic and biopolymer-degrading enzymes.
INTRODUCTION
Anthrax and botulinum toxins are responsible for fatality in their respective diseases: anthrax and botulism. Extreme toxicity and the way that it is exerted is common to these molecules, resulting in the proteolytic cleavage of host intracellular proteins (Rossetto et al. 2000). Small-molecule inhibitory drugs capable of blocking the intracellular activity of anthrax and botulinum proteases are yet to be developed. No treatment is available for the rapid reversal of their toxic effects. Both toxins are Tier One select agents, and pose a bioterrorism threat (Arnon et al. 2001; Inglesby et al. 2002; Cheng, Land and Stanker 2012; Balali-Mood, Moshiri and Etemad 2013; Adalja, Toner and Inglesby 2015). Therefore, it is very important to develop simple, robust and sensitive assays for unambiguous detection of the presence of these toxins.
The median lethal dose (LD50) for botulinum toxin in humans is 1 ng kg−1 or less when injected in the bloodstream (Arnon et al. 2001), while the dosage for lethal intoxication by the oral route is much higher due to toxin inactivation in the digestive system. It has been suggested that, because of toxin degradation and serum clearance, the sensitivity for diagnostic evaluation must be in the low to sub-pg/ml range (Cheng, Land and Stanker 2012).
In the case of systemic anthrax, the concentration of anthrax toxin in the blood shows builds up extremely rapidly after a relatively long (12–24 h) asymptomatic lag phase (Boyer et al. 2009). After the blood toxin concentration peaks at tens or even hundreds of ng/ml, the risk of reaching a ‘point of no return’ condition, after which all treatments may fail, increases dramatically (Inglesby et al. 2002; Cheng, Land and Stanker 2012). Therefore, the availability of an assay with subpicomolar detection of anthrax endopeptidase (lethal factor, LF) is critically important for timely initiation of therapy. Information about techniques for sensitive detection of anthrax and botulinum toxin has been summarized in several recent comprehensive reviews (Barr, Boyer and Quinn 2010; Capek & Dickerson 2010; Cheng, Land and Stanker 2012; Tevell Åberg, Björnstad and Hedeland 2013; Kim et al. 2015). Despite significant progress in the development of detection techniques, none have been converted into clinical diagnostic tests to date, and, for botulinum toxin, the mouse bioassay remains the main clinical detection tool (Capek & Dickerson 2010).
Analysis of the literature suggests that the assay method currently most accurate at detecting active toxin, via the detection of an inactivated protease (e.g. by immunological methods), can lead to a false-positive result (see e.g. Cheng, Land and Stanker 2012). Also, the majority of immunological detection methods do not achieve subpicogram levels of detection sensitivity. In contrast, a method that consistently shows high (pictogram or even attogram) sensitivity levels for botulinum neurotoxins (BoNTs) is immuno-polymerase chain reaction (IPCR), which involves PCR-based amplification of a signal obtained by recognition of a toxin by ligand-based probes such as antibodies (reviewed in Capek & Dickerson 2010) or components of natural toxin receptors (Mason et al. 2006; Kwon et al. 2014). This assay type has already been developed for other toxins such as ricin, including sensitive detection in complex matrices (He et al. 2010). However, to date, there are no reports describing the application of immuno-PCR or related techniques for the detection of anthrax toxin. Likewise, none of the PCR-based assays for toxin detection reported to date have addressed the issue of discrimination between catalytically competent and inactive toxins.
We have developed a technique termed proteolytic PCR, in which PCR is used to report the presence of catalytic activity of botulinum toxin type A (BoNT/A) and LF. To achieve this, we designed reporter probes in the form of DNA–protein complexes which, being attached to a solid phase, release DNA into solution upon cleavage by the target protease. We also developed methods for isolation of LF and BoNT/A from complex matrices under mild conditions, which exploit the natural properties of toxin components. Proteolytic PCR could be used for the development of highly sensitive clinical assays detecting the presence of subpicogram quantities of anthrax and botulinum toxins.
MATERIALS AND METHODS
Bacterial strains and media
Bacillus anthracis STI-1 is a toxigenic anthrax strain encoding fully active lethal toxin (LeTx), but lacking the poly-γ-D-glutamic acid capsule due to the absence of pXO2 plasmid (Okinaka et al. 2014). Bacillus anthracis STI-1 is avirulent in humans and is used for preparation of live anthrax vaccine in Russia (Stepanov et al. 1989). Bacteria were cultured in R-medium in accordance with the protocol described by Leppla (1988).
Clostridium botulinum type A strain ATCC19397 is the strain that produces fully active BoNT/A. For BoNT/A production, C. botulinum bacteria were grown in TGY media (30 g l−1 trypticase; 5 g l−1 glucose; 20 g l−1 yeast extract, 0.5 g l−1 cysteine hydrochloride; pH 7.5) in anaerobic conditions for 4 days at 37°C.
Both microorganisms were obtained from the State Collection of Pathogenic Microorganisms, Russia. Bacterial growth media and components were from BD Difco (USA).
Escherichia coli BL21(DE3) and CodonPlus (DE3)-RILP (Agilent Technologies, USA) was used for expression of all the recombinant proteins cited in this study.
Enzymes, chemicals and supplies
Unless indicated otherwise, all chemicals were purchased from Sigma-Aldrich (USA), and were graded as Molecular Biology reagents. SYPRO Red dye, DNA-modifying enzymes, neutravidin- and glutathione-coated magnetic particles were purchased from Thermo Pierce (USA). Standard oligonucleotides were from Evrogen (Russia), whereas modified oligos were from Syntol (Russia). Click chemistry reagents and SUMO protease were from Life Technologies (USA). Chromatography columns and media were from GE Life Sciences (USA) except for thiol-resistant immobilized-metal affinity chromatography (IMAC) beads, which were supplied by Roche Life Science (USA).
Toxins, toxin components, toxin substrates and monoclonal antibodies
The STI-1 strain was cultured as described previously (Leppla, 1988; Belova et al.2004). The overnight culture was centrifuged twice at 10 000 × g for 40 min to obtain supernatant that contained secreted anthrax toxin components. The supernatant was sterile filtered (syringe-mounted 0.22 μM low-protein binding membrane) for further processing. Sterile supernatant was spiked into serum samples in quantities based on the prior knowledge of the LF concentration in cultivation experiments. In parallel, aliquots of sterile supernatant were analyzed for actual LF concentration by ELISA, as described previously (Belova et al. 2004, 2008). Experiments involving recombinant and STI-1 derived LF and assays with recombinant LF used at concentrations determined by STI-1 secreted LF were repeated in triplicate.
Construction of plasmids expressing recombinant anthrax LF and protective antigen (PA), and relevant expression/purification protocols were described earlier (Belova et al. 2008; Zakharova et al. 2009). Briefly, LF and PA were produced by BL21(DE3) E. coli strains and contained corresponding coding sequences with upstream His6 tags, obtained by placing genes for mature LF and PA amplified from B. anthracis STI-1 strain in the pET22(b)+-derived plasmid pET-MASMT (Miller, Elliott and Collier 1999; Kolesnikov et al. 2007). Construction of PA with a C-terminal His6 tag was accomplished by PCR-guided addition of sequence coding for GGGSHHHHHHStop to the 3′-terminus of the PA construct, and removal of auxiliary sequences at its 5′-terminus (Belova et al. 2008). Sequences for cloning between Nde I and Xho I sites into pET22(b)+ plasmid were also attached by PCR. Purification of soluble LF and PA variants was performed by successive IMAC and size exclusion chromatography steps. Nicking of PA83 by trypsin to obtain PA63, and assembly and purification of PA63 heptamers was performed as described previously (Belova et al. 2004). Purified PA63 heptamers were frozen in 25 mM HEPES supplemented with 40% v/v glycerol and stored at −80°C until use.
The sequence coding for nontoxic non-hemagglutinin (NTNHA-A) was amplified from genomic DNA of the ATCC19397 strain and cloned into pET-MASMT between Bgl II and Xho I sites, introduced by PCR primers along with a stop codon preceding the Xho I site. NTNHA-A, containing a N-terminal His6 tag, was produced in E. coli BL21 CodonPlus (DE3)-RILP strain, and purified by IMAC and size-exclusion chromatography.
A BoNT/A-specific monoclonal antibody was prepared by immunization of mice with a C-terminal fragment of heavy chain of BoNT/A (HC50), and subsequent hybridoma construction and selection. The 2D5 clone was used for further work.
Anti-NTNHA-A monoclonal antibody was prepared by immunization of mice with full-length recombinant protein, and subsequent hybridoma construction and clone selection. Isolated clones were checked for the ability to immunoprecipitate high molecular weight species that were immunoreactive to the 2D5 mAb and displayed BoNT/A endopeptidase activity. Clones 3F2 and 1H7 were shown to be positive in both tests, and clone 1H7 was used to capture BoNT/A-NTNHA-A complexes during this study.
For antibody biotinylation, carbohydrates of 1H7 mAb were oxidized with sodium periodate, and treated with biotin hydrazide as detailed earlier (Wolfe & Hage 1995). Biotinylation of the mAb was checked by western blotting with NeutrAvidin horseradish peroxidase conjugate (Thermo Pierce, USA).
Mouse bioassay
Mouse assays were performed as described earlier (Sugiyama 1980). All laboratory animal testing was regulated under the Institutional Animal Care and Use Committee and performed by certified personnel at the institutional animal facility. Mice were injected intraperitoneally with serial toxin dilutions in gelatin-phosphate buffer (GPB) and observed for botulism symptoms or death for the standard 96-h period. After 96 h, all symptomatic mice were euthanized by narcosis. Control mice were injected either with GPB alone or with toxin mixed with polyclonal horse neutralizing antibody against BoNT/A (Microgen, Russia).
DNA–peptide and DNA–protein probes
Peptide consisting of an optimized LF substrate (Turk et al. 2004; Boyer et al. 2007), containing a biotin moiety separated from the LF substrate by a Gly spacer and terminated by a maleimide-reactive cysteine residue (Bio-GGGGGSPARRKKVYPYPMENFPPSTPSPTSC), was custom synthesized by ATG Service Gene (Russia).
Synthesis of peptide–DNA conjugate was performed using copper-free click chemistry. First, 5 mg of peptide was dissolved in 200 μl of 50% (v/v) DMSO in deionized water. Click-it maleimide DIBO alkyne (1 mg) was dissolved in 100 μl of dry DMSO. An approximately 5-fold molar excess of thiol-reactive alkyne (10 μM) was added to the peptide solution. The reaction was allowed to proceed for 6 h at +4°C, and the peptide–alkyne conjugate was subsequently purified using a Superdex Peptide 10/300 GL column (GE Healthcare Life Sciences, USA) equilibrated with HEPES buffer (25 mM HEPES, 300 mM NaCl) to remove unreacted alkyne molecules. Purified alkyne–peptide conjugate (10 mg ml−1 in 50% DMSO) was mixed with a 2-fold molar excess of PCR template. The template, designated Probe N3, was a 71-nt oligonucleotide modified at its 5′-terminus by azide (N3) and a 18-angstrom spacer, (5′-(N3)(SP_18 Å)CTGGGTTCTTGACAAGCTCAAAGCCGAGCGTGAGCGTGGTATCACCATTGATATCGCTCTCTGGAAGTTCG-3′). The reaction was allowed to proceed overnight, and the conjugate was separated from unreacted oligos by RP-HPLC. The resulting conjugate was checked by denaturing urea polyacrylamide gel electrophoresis for completion of the click reaction. The gel was stained with ethidium bromide and SYPRO RED dyes to visualize the DNA and peptide portions of the conjugate (Fig. 1).

DNA–peptide conjugate assayed by PAAG electrophoresis with CYPRO Ruby Protein Gel Stain (1–3) and ethidium bromide (4–7) staining. 1, 3, 4, 6 – peptide-DNA conjugate, 2, 5 – oligonucleotide, 7–50 bp DNA ladder (NEB).
The protein substrate for PCR-based detection of BoNT/A activity consisted of (in N- to C-terminal order) a His6 tag, in vivo biotinylated peptide (Beckett, Kovaleva and Schatz 1999), SUMO peptide, full-length SNAP 25 protein, and receptor-binding subunit of Shiga-like toxin from E. coli O157:H7 (Fig. 2). The construct, codon optimized for high-level expression in E. coli, was in-house synthesized using overlapping PCRs with a series of oligonucleotides, and cloned into the pET22(b)+ plasmid between Nde I and Xho I sites. The resulting fusion protein was amplified in E. coli BL21 (DE3) co-expressing untagged BirA biotin ligase from the low-copy ColE1-compatible pACYC184 plasmid under the control of the Lac promoter. Soluble fusion protein was purified by means of IMAC and size-exclusion chromatography, and checked for biotinylation by western blotting with a NeutrAvidin horseradish peroxidase conjugate (data not shown).

Structure of BoNT/A recombinant fusion substrate.
Toxin capture and release assays
All specimens were pretreated with Complete EDTA-free protease inhibitor (Roche Life Sciences, USA) according to the manufacturer's instructions.
To capture BoNT/A, NTNHA-A was added to the specimen to 1 μM. The pH of the solution was adjusted to 7.2 if needed. After 30 min incubation, the pH of the solution was adjusted to 6.2 by addition of a precalculated amount of MES buffer, the NTNHA-A-specific 1H7 mAb was added to a final concentration of 2 μM, and the mixture was incubated for 2 h. After incubation, 2 mg ml−1 of streptavidin-coated magnetic beads (Thermo Pierce, USA, typical binding capacity is 70 μg of biotinylated IgG per mg) were added to the mixture, and incubation continued for an additional 30 min. Beads were washed using an automated magnetic washer (Bio-Rad, USA) in 20 mM HEPES pH 6.8, 50 mM NaCl and 25 μl of elution buffer (20 mM Tris-HCl pH 8.5, 20 mM NaCl) were added per 1 mg of washed beads. Dissociated BoNT/A was collected and used for cleavage assay. All procedures were performed at room temperature (RT).
To capture LF, 2 μM of His6-tagged PA63 heptamers (molar concentration is given per monomeric PA63) were added to the diluted human plasma with LF spiked prior to dilution. After 1 h of incubation at RT, 2 mg of Talon magnetic beads (Clontech, USA) were added per 1 ml of solution (estimated binding capacity is 40 μg mg−1 for average sized His6-tagged protein) and incubated for 30 min. Further beads were captured and washed by automated magnetic washer (20 mM HEPES pH 6.8, 50 mM NaCl). After washing, beads were incubated for 15 min in elution buffer (25 μl mg−1 of beads, 20 mM MES, pH 5.6, 50 mM NaCl). The pH of the separated liquid phase was immediately brought to neutral by addition of 1/10 v/v of 300 mM HEPES.
Cleavage assays
BoNT/A cleavage assay was performed in cleavage/activation buffer (50 mM HEPES pH 7.0, 50 mM NaCl, 10 μM ZnCl2, 5 mM DTT, added to the eluted sample as a 10× concentrate) for 1 h at 37°C, with 2 mg of streptavidin-coated magnetic beads pre-saturated with the biotinylated fusion protein (200 μg in 50 μl), and Stx2b aptamer (50 μg in 25 μl) added sequentially to beads. The entire volume of BoNT/A eluate (approximately 50 μl, see above) was mixed with beads to perform a single assay.
Streptavidin beads were saturated with peptide-oligo LF substrate (100 μg of conjugate per mg of beads), washed thoroughly by automated washer, and mixed with eluted and neutralized LF:PA63 solution. LF cleavage assay was performed in optimized LF cleavage buffer. DTT and metal ion solutions were added to the neutralized LF solution to a final concentration of 0.1 mM DTT, 20 μM CaCl2, and 20 μM ZnCl2. Cleavage of a single LF sample was performed with 2 mg of probe-bound beads at 30°C for 30 min.
In both cases, 5 μl of the solutions containing the cleaved DNA–protein complexes were used in the subsequent PCR reaction.
Real-time PCRs
Real-time PCR was performed in a Mini Opticon thermal cycler (Bio-Rad, USA) using TaqMan probes. For amplification of the Stx2b aptamer (BoNT/A assay), forward primer StxF, reverse primer StxR and TaqMan probe TqStx were used. PCR in the LF assay was done with the Probe N3 template, forward primer N3_F3, reverse primer N3_R3, and TaqMan probe TaqM_178-119 (Fig. 3). For the BoNT/A assay, amplification was carried out as follows: denaturation step (95°C 5 min); followed by 40 cycles of 95°C for 15 s, 60°C for 10 s and 72°C for 15 s. For the LF assay: denaturation step (95°C 5 min); followed by 40 cycles of 95°C for 15 s, 58°C for 10 s and 72°C for 15 s.

Sequences of primers and TaqMan probes used for real-time PCR.
RESULTS
Assay design
The primary assay components were: (i) the probe consisting of protein or peptide substrate, specifically recognized and cleaved by the protease component of the toxin; and (ii) the reporter DNA molecule, which is an oligonucleotide designed to be amplified by PCR with high efficiency. The proteinaceous portion of the probe is attached to the solid phase via a specially designed anchor moiety, whereas the DNA component is linked to the anchor moiety via covalent or noncovalent bonding and is exposed to solution. The protease substrate is installed between the anchor moiety and DNA linkage site, and is cleaved if exposed to the cognate protease. Cleavage results in release of the DNA into solution. After separation of intact and cleaved probes, the latter can be amplified by PCR. Assay schemes for LF and BoNT/A are shown in Fig. 4. The assay's sensitivity benefits from two-step signal amplification: the first step is due to cleavage of multiple probe molecules by a single protease molecule, while the second amplification step is provided by PCR.

Assay schemes for detection of LF and BoNT/A by proteolytic PCR.
For LF, an artificial peptide substrate cleaved with high efficiency had previously been identified (Turk et al. 2004; Boyer et al. 2007). This substrate was extended by five Gly residues upstream of the extended LF recognition sequence (Boyer et al. 2007), biotinylated at the N-terminus and modified by addition of a Gly–Pro stretch terminated by Cys at the C-terminus. A terminal thiol group was conjugated with maleimide DIBO alkyne, a component of the copper-free Click chemistry pair. Another pair member, the azide moiety, was introduced at the 5′-terminus of a 71-nt oligonucleotide, selected for efficient PCR amplification. As shown in Fig. 5, the click reaction efficiently couples a single peptide to a single oligonucleotide.
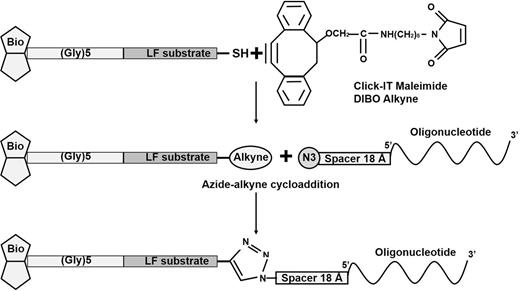
Conjugation of substrate peptide with oligonucleotide by click chemistry (azide-alkyne cycloaddition).
As it is known that peptide fragments of BoNT/A-encompassing cleavage sites are less efficient substrates compared to larger portions of SNARE family protein targets of botulinum toxin (Chen & Barbieri 2006), full-length SNAP-25 protein was chosen for the BoNT/A substrate probe. Attachment of single thiol- or primary amine reactive molecules such as maleimide or N-hydroxysuccinimide ester at a given site is not feasible for a protein that contains multiple Cys and Lys/Arg residues. On the other hand, fragments of full-length protein can fold poorly, hindering the production of recombinant soluble probe molecules.
As preparation of site-specific conjugates of DNA and long polypeptides containing more than one reactive group is difficult, noncovalent high-affinity binding of the DNA fragment to the probe was chosen for the BoNT/A assay. In the probe construct, SNAP-25 was genetically linked to the small subunit of Shiga toxin type II, to which a high-affinity DNA aptamer had previously been selected (Kozyr et al. 2014). To facilitate the production of a soluble fusion protein, and provide an option for testing the probe's performance, SUMO peptide was placed upstream of the SNAP25-Stx2b assembly. Two tags, His6 and in vivo biotinylated peptide, were placed at the N-terminus of the fusion probe to facilitate its purification and leakage-free attachment to the neutravidin-modified solid phase via the biotin moiety (Fig. 2). Cleavage of SNAP-25 by BoNT/A liberates Stx2b with bound aptamer in solution (Fig. 4).
The second important consideration for assay design was how to achieve high specificity. A protease assay capable of sensing subpicogram quantities of enzyme should be based on highly specific separation of the target protease from any contaminating enzymes. For LF, we developed isolation from test solutions by utilizing the intrinsic properties of the second LeTx component, PA, to bind LF without inhibiting its activity and to form heptamers and active toxin in vitro (Panchal et al. 2005). A chimeric PA heptamer was prepared from two types of PA: the first lacked the His6 tag in the heptamer-forming PA63 fragment, and the second had His6 attached to the C-terminus of PA63. The presence of hexahistidine at the C-terminus of PA63 does not alter its properties; likewise, PA63 readily forms heteroheptamers (Jennings-Antipov, Song and Collier 2011). Heptamerization of a PA63/PA63(His6) mixture results in the formation of a complex capable of binding one LF molecule per PA63 molecule with high specificity (Ren et al. 2004). The resulting PA:LF complex can be captured by an IMAC solid support and released with high selectivity into solution by lowering the pH value in a way similar to that used for natural LeTx trafficking inside the cell (Collier 2009). The released complex is used to cleave immobilized LF substrate (Turk et al. 2004, Boyer et al. 2007) conjugated with DNA, thus liberating the PCR template into solution.
As in the case of the LF assay, natural properties of BoNT/A were used to achieve highly selective isolation of botulinum toxin protease from complex mixtures. Native botulinum neurotoxins enter the organism as high molecular weight complexes consisting of NTNHA and hemagglutinins. These complexes protect the toxin from inactivation by proteases and low pH. Importantly, at low pH, NTNHA is tightly associated with BoNT (Kd = 30 nM), whereas at pH higher than neutral, NTNHA rapidly dissociates from the toxin (Maksymowych et al. 1999; Gu et al. 2012). Therefore, NTNHA can be used as efficient bait to isolate native intact BoNT from the sample of interest using a specific mAb attached to magnetic particles. To ensure that all the toxin molecules are saturated by bound NTNHA-A, a 10-fold molar excess of the recombinant protein with respect to BoNT/A (approx. 2 μM) was added to the mixture. After capture and extensive washing at low pH (5.5), the mAb–toxin complex was subjected to a pH shift (8.5), which dissociated BoNT/A from mAb-bound NTNHA-A, thus specifically releasing bound toxin to the solution and leaving impurities that would potentially co-purify with the toxin bound to the solid support. The pH value of the solution containing the released BoNT was brought to neutral (7.2), and the toxin activated by the addition of a reducing agent (Cai, Sarkar and Singh 1999).
Model experiments
Model studies were carried out with purified toxins and were set up to explore the lowest detection limits for the new assay. In addition, matrix interference with assay sensitivity was investigated.
Detection of LF in buffer solution
For anthrax toxin, purified recombinant LF in complex with PA63 heptamers was used. Immobilized peptide–DNA conjugates were treated by serial dilutions (1 ng ml−1 to 100 ag ml−1 in 10-fold increments) of LF:PA complex in HEPES buffer. The complex was precaptured by His6 magnetic beads and eluted by pH shift as described above. Cleaved DNA was amplified using real-time PCR detection, and Ct values generated by control reactions and toxin dilutions were calculated (Fig. 6A). Negative control contained no DNA–peptide conjugate. Additional controls for reaction specificity contained: (i) LF without PA63 heptamer; (ii) complex of PA63 with catalytically inactive LF mutant (Zakharova et al. 2009); (iii) trypsin which was capable of cleaving the LF peptide substrate used in this study (Zakharova et al. 2009), but was inactivated by an inhibitor cocktail and was not enriched by the LF capture and elution procedure.
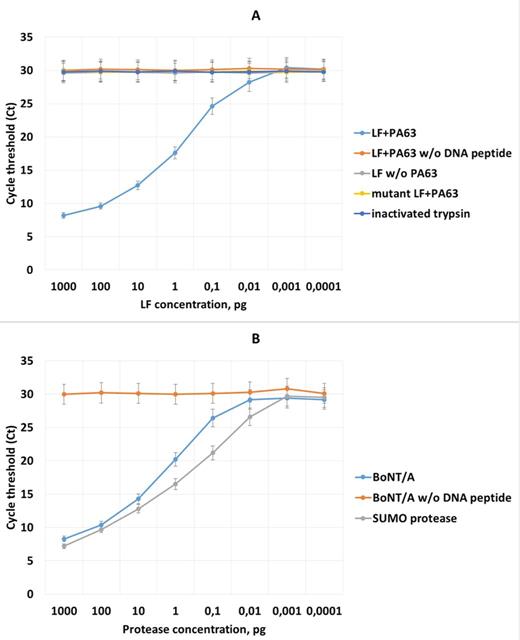
Real-time PCR detection of anthrax lethal toxin (LeTx) and botulinum neurotoxin type A (BoNT/A) in buffer solution by proteolytic substrate assay. Substrates were serially diluted in 10-fold increments (1 ng ml−1 to 100 ag ml−1, horizontal axis). (A) Detection of LeTx. Controls: negative (without DNA–peptide substrate); LF mock captured in the absence of the PA63 heptamer; complex the of PA63 heptamer with mutant LF; trypsin, added to the sample prior to dilution and addition of the protease inhibitor cocktail. (B) Detection of BoNT/A. Negative control contained no DNA-coupled recombinant fusion substrate, positive control represented treatment of substrate by serial dilutions of SUMO protease.
The new PCR-based assay for sensing LF protease activity was able to detect as little as 10 fg of LF spiked to HEPES, which is 50–100 times below the threshold suggested for LF detection. All the controls yielded signals below the indicated LOD, although values of signals obtained from different controls showed a certain degree of variation (Fig. 6A).
Detection of anthrax LF in human serum
Human blood and serum samples are the primary specimens for detection of LF in the clinical setting. Clinical specimens often provide additional challenges for detection, as they are heterogeneous, contain PCR inhibitors (Konet et al. 2000; He et al. 2010) and proteases (Kalb et al. 2006), and their contents can sequester and mask target molecules. To decrease the influence of such inhibitors, a serum sample with spiked LF was diluted five times before starting the assay. Dilution of the sample after spiking with the target toxin would not affect the assay sensitivity provided that the volume of the assayed sample is increased by the factor of the dilution. Accordingly, the amount of PA63 heptamer, as well as the beads added to the diluted sample, was increased by a factor of 5 to ensure efficient capture of LF molecules present in solution. As in the previous experiment, the amount of spiked LF varied from 1 ng to 100 ag per ml. The detection limit for LF spiked into the human serum was 50 fg (Table 1), which corresponded to the subpicomolar assay sensitivity necessary for timely detection of LF in clinical samples. Specimen interference was indeed observed in the serum sample, but the assay sensitivity was still sufficient to detect LF a few hours postinfection (Boyer et al. 2009).
Results of proteolytic PCR limit of detection (LOD) determination for anthrax toxin and botulinum neurotoxin. Data present average Ct values determined by real-time PCR.
| Sample | 5 pg | 2 pg | 1 pg | 0,5 pg | 200 fg | 100 fg | 50 fg | 10 fg |
|---|---|---|---|---|---|---|---|---|
| LaTx in human serum | 14,3 | 16,2 | 19,4 | 23,2 | 25,7 | 27,6 | 28,9 | 29,1 |
| BoNTA in human serum | 20,3 | 22,2 | 25,4 | 27,8 | 29,6 | 29,5 | 29,4 | 29,3 |
| BoNTA in carrot juice | 22,3 | 24,2 | 26,4 | 28,0 | 27,9 | 28,1 | 27,6 | 27,5 |
| Sample | 5 pg | 2 pg | 1 pg | 0,5 pg | 200 fg | 100 fg | 50 fg | 10 fg |
|---|---|---|---|---|---|---|---|---|
| LaTx in human serum | 14,3 | 16,2 | 19,4 | 23,2 | 25,7 | 27,6 | 28,9 | 29,1 |
| BoNTA in human serum | 20,3 | 22,2 | 25,4 | 27,8 | 29,6 | 29,5 | 29,4 | 29,3 |
| BoNTA in carrot juice | 22,3 | 24,2 | 26,4 | 28,0 | 27,9 | 28,1 | 27,6 | 27,5 |
Results of proteolytic PCR limit of detection (LOD) determination for anthrax toxin and botulinum neurotoxin. Data present average Ct values determined by real-time PCR.
| Sample | 5 pg | 2 pg | 1 pg | 0,5 pg | 200 fg | 100 fg | 50 fg | 10 fg |
|---|---|---|---|---|---|---|---|---|
| LaTx in human serum | 14,3 | 16,2 | 19,4 | 23,2 | 25,7 | 27,6 | 28,9 | 29,1 |
| BoNTA in human serum | 20,3 | 22,2 | 25,4 | 27,8 | 29,6 | 29,5 | 29,4 | 29,3 |
| BoNTA in carrot juice | 22,3 | 24,2 | 26,4 | 28,0 | 27,9 | 28,1 | 27,6 | 27,5 |
| Sample | 5 pg | 2 pg | 1 pg | 0,5 pg | 200 fg | 100 fg | 50 fg | 10 fg |
|---|---|---|---|---|---|---|---|---|
| LaTx in human serum | 14,3 | 16,2 | 19,4 | 23,2 | 25,7 | 27,6 | 28,9 | 29,1 |
| BoNTA in human serum | 20,3 | 22,2 | 25,4 | 27,8 | 29,6 | 29,5 | 29,4 | 29,3 |
| BoNTA in carrot juice | 22,3 | 24,2 | 26,4 | 28,0 | 27,9 | 28,1 | 27,6 | 27,5 |
No significant background signal was generated in control experiments, indicating an absence of nonspecific protease cleavage, and high selectivity of the elution procedure.
To obtain more information on LF detection in serum samples, we compared the aforementioned data with the data collected from samples spiked with sterile supernatant obtained after culturing the STI-1 anthrax vaccine strain which is defective in the synthesis of capsular antigen but secretes fully active anthrax toxin. In addition, we directly analyzed the effect of sample dilution on the sensitivity and reproducibility of the LF assay. To do this, a series of dilutions of sterilized culture supernatant from B. anthracis STI-1 strain was spiked into the serum sample; the sample was diluted and assayed for LF activity, as described above. In parallel, same dilutions of recombinant LF were tested without serum dilution after LF spiking.
The results are shown in Fig. 7. No significant difference in the assay sensitivity values was found for recombinant and STI-1-produced LF. The assay sensitivity in the undiluted sample appeared to be in the same range as that for the diluted sample. However, the accuracy of the obtained data was low compared to that of the diluted sample because the fluctuation of the Ct values for each concentration point was significantly larger than that of the diluted serum.
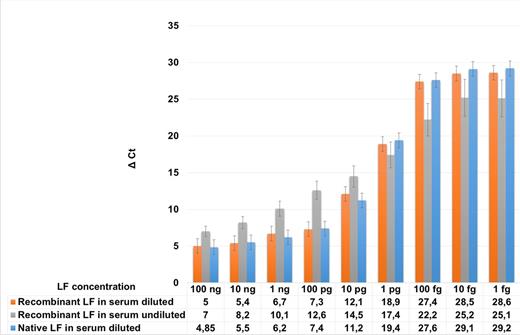
Influence of serum sample dilution on detection efficiency of native and recombinant LF. Recombinant LF, anthrax lethal factor produced in E. coli and purified according to the protocol described in Materials and Methods. Native LF, sterile supernatant of B. anthracis STI-1 strain secreting functional anthrax toxin.
Detection of BoNT/A in buffer solution
The mouse assay has been reported to have 10 pg LD50 for BoNT/A (Capek & Dickerson 2010; Cheng, Land and Stanker 2012). However, this value varies depending on the preparation method, and values ranging from 30 (Cheng, Land and Stanker 2012) to less than 5 pg can be found in the literature (Wilder-Kofie et al. 2011). This is partially because a positive result of the mouse bioassay can be observed at a range of sublethal concentrations: some animals can die, whereas others survive but show clear intoxication symptoms. In addition, BoNT preparations are heterogeneous and contain various quantities of nontoxic associated proteins (Maksymowych et al. 1999; Gu et al. 2012), making precise determination of the toxin concentration by conventional protein quantitation methods challenging (Bradford or A280 assays). In addition, as mentioned above, a number of factors affecting the activity of BoNT protease, such as pH, stoichiometry of associated proteins, and the concentration of reducing agents such as DTT in the reaction mixture (Cai, Sarkar and Singh 1999). Various commercial preparations of BoNT/A have yielded a discrepancy between the indicated amount of toxin, and data obtained in the mouse bioassay. Therefore, to test the assay's performance, we used in-house purified BoNT/A and determined its LD50 via mouse bioassay. The amount of toxin tested in the mouse bioassay was 1, 5, 10, 15 and 20 pg per animal. It was found that the LD50 for the BoNT/A preparation was 10 pg ml−1. This value was further used in all experiments.
Freshly purified BoNT/A was serially diluted (1 ng ml−1 to 100 ag ml−1 in 10-fold increments). The results of the PCR-based proteolytic BoNT/A activity detection are shown in Fig. 6B. The LOD determined for this assay was 100 fg. This value is 10-fold higher than the LOD value for LF, and we set up an experiment to check whether the increased LOD was because of the properties of the reporter or BoNT/A protease. To this end, immobilized substrate with bound aptamer was cleaved with SUMO protease under experimental conditions identical to those used for BoNT/A. SUMO protease is a cysteine endopeptidase activated by reducing agents, which cleaves SUMO peptide with exquisite specificity under a broad range of conditions (Panavas, Sanders and Butt 2009). The detection limit for the SUMO cleavage assay was 10 fg, indicating that the reduced assay performance for BoNT/A was likely due to the catalytic efficiency of the endopeptidase. In contrast, noncovalent DNA attachment to the BoNT/A probe was not likely to be the cause of the reduced sensitivity of the BoNT assay.
Detection of BoNT/A in human serum and carrot juice
Basing on the LOD assay data in buffer solution, serial dilutions of BoNT/A were spiked into human plasma and vegetable (carrot) juice. These specimens potentially represent the most widespread matrices to be tested for BoNT/A presence: clinical samples in wound or infant botulism, ingested or inhaled toxin, and samples obtained from potentially contaminated food. Both serum and juice were diluted five times with HEPES buffer before addition of recombinant NTNHA-A and beads. The proteolytic PCR assay successfully detected 0.5 and 1 pg of BoNT/A in serum and carrot juice, respectively (Table 1). Control experiments conducted with diluted samples devoid of BoNT/A yielded signals with value below LOD.
DISCUSSION
Botulinum and anthrax toxins are causes of life-threatening conditions that can be fatal unless quickly detected and treated. Extreme toxicity of BoNT, the rapid onset of symptoms and a lack of available treatment to inhibit the intracellular toxin and reverse paralysis necessitate BoNT detection at the earliest opportunity. It has been noted that the subpicogram detection of the toxin in clinical samples is needed for efficient treatment of botulism (Cheng, Land and Stanker 2012). The same is true for anthrax LeTx, given its ultrafast accumulation after a lag-phase observed early in infection (Boyer et al. 2009). Although anthrax LeTx is not as toxic as BoNT, its slow accumulation early in infection rapidly peaks after about a day, leading to a ‘no-return’ state after which attempts at treatment fail (Inglesby et al. 2002; Barr, Boyer and Quinn 2010). As for BoNT, no treatment is available to inhibit LeTx inside the host cell. Both toxins can arise in the environment as the result of a bioterrorist attack, resulting in a need to be able to detect them outside the clinical setting.
Sensitivity of the mouse bioassay for botulinum toxin is approximately 5–20 pg (Sugiyama 1980; Capek & Dickerson 2010; Cheng, Land and Stanker 2012). Despite the fact that it is the mainstream assay for BoNT detection, it is laborious, time-consuming, and requires 4 days for BoNT type determination, which is unacceptably long. Another sensitive method for detecting both BoNT and LeTx active toxins is mass spectrometry (MS), which detects cleavage products after treatment of a peptide substrate with the target toxic protease. However, the assay sensitivity for clinical or environmental samples does not reach the subpicogram limit for either toxin type (Kalb et al. 2006; Boyer et al. 2011). In addition, MS requires expensive equipment and dedicated personnel for experimental setup and data analysis. Therefore, alternative assay formats are under development to increase detection sensitivity.
Recent advances in so-called ‘immuno’ or, more generally, affinity-dependent PCR have made possible the detection of subpicogram or even attogram quantities of microbial targets (Mehta et al. 2014). Recent technological advances helped to efficiently overcome the background issues that affected the sensitivity of earlier developed immuno-PCR assays (Niemeyer, Adler and Wacker 2007). Several studies have reported the detection of subpicogram quantities of BoNT (Mason et al. 2006; Capek & Dickerson 2010; Cheng, Land and Stanker 2012) and other toxins by various modifications of affinity-dependent PCR, including some works focused on detection in complex matrices such as plasma, feces, vegetable juices or milk (He et al. 2010; Mehta et al. 2014). Interestingly, to the best of our knowledge, there are no publications describing the detection of anthrax toxin components by immuno-PCR or related technologies.
Despite the extremely low detection limits obtained in some works, immuno-PCR assays are not tailored to detect the endopeptidase activity of a toxin. Hence, despite the extreme assay sensitivity, application of affinity-based PCR cannot provide information about the actual amount of dangerous (active) substance, and can result in false-positive detection of inactive (e.g. cooked, denatured or partially degraded) toxin.
Here, we attempted to combine the high sensitivity of PCR assay with the ability of such an assay to detect endopeptidase activity of BoNT/A and LeTx. To do this, we prepared composite substrates containing DNA and protein parts linked to each other. The protein or peptide part that contains the cleavage site for the endopeptidase component of the toxin can be attached to a solid phase via a streptavidin–biotin bond. The DNA part is attached to the protein and, upon cleavage, is released into solution. Released DNA can then be further separated from the noncleaved probe and amplified by PCR. For LF, an efficiently cleaved short peptide substrate was developed (Turk et al. 2004; Boyer et al. 2007) and used to prepare a site-specific covalent peptide–oligonucleotide conjugate. Click chemistry appeared to be a very efficient method of peptide–oligonucleotide coupling, resulting in almost 100% conversion of the alkyne–peptide to DNA conjugate. For BoNT/A, no short peptides have been described that are cleaved with comparably high efficiency, so an alternative strategy was chosen for the design of a cleavage probe. A fusion protein was designed comprising full-length SNAP-25, a small subunit of a Shiga-like toxin (Stx2b; Strockbine et al. 1986) binding high-affinity DNA aptamer (Kozyr et al. 2014), and auxiliary polypeptides such as SUMO and in vivo biotinylated peptide.
This strategy, when applied to a model reaction, resulted in highly sensitive detection of toxins, with LOD for LF as low as 10 fg, whereas for BoNT the LOD was 100 fg. Decreased LOD for BoNT/A compared to that of LF was likely due to lower cleavage efficiency of the BONT endopeptidase, as judged by control cleavage of the same reporter substrate by SUMO protease.
Apart from assay sensitivity, cleavage specificity of the reporter substrate is extremely important, especially if we consider detection of toxin endopeptidase activity in complex matrices such as food, blood samples or vegetables. We developed efficient procedures for the rapid isolation and gentle elution of LF and BoNT/A from complex mixtures. In both cases, isolation techniques relied upon natural properties of holotoxin complexes generating a quasinatural environment during the toxin isolation procedure, which efficiently preserved the peptidase activity of the target molecules. The developed procedures helped to achieve low background signals, which were comparable to the backgrounds observed in the experiments with highly purified toxin. In BoNT detection, direct toxin capture is possible only with a panel of serotype-specific monoclonal antibodies, because polyclonal antibody preparations (pAbs) were shown to inhibit the toxin's proteolytic activity (Kalb et al. 2010). Use of NTNHA-specific pAbs would not inhibit BoNT protease, thus potentially eliminating the need of using expensive mAb panels and the cumbersome engineering of poly-specific anti-BoNT mAbs (Kalb et al. 2010) in the attempt to optimize the toxin capture procedure. Use of an efficient commercially available protease inhibitor cocktail which does not inhibit activity of both toxins, further reduced the risk of false-positive signals.
We believe that further optimization of this detection technique is possible to increase the detection efficiency. For example, selection of highly efficient artificial peptide substrates for BoNTs, or optimization of the divalent metal ion environment for LF (Lo et al. 2014), may further increase the assay sensitivity. Additional analysis of the assay performance in samples rich in lytic enzymes, such as stool samples, which are important for BoNT detection, is needed to design substrates appropriate for such an assay format and to design sample preparation procedures to eliminate the possibility of false-negative or false-positive results caused by background enzymatic activity.
In summary, we have developed a new method of PCR-based detection of activity of LF and BoNT/A endopeptidases. The proteolytic PCR assay is highly sensitive and permits detection of subpicogram quantities of anthrax and botulinum toxins in complex matrices. In addition, it is based on techniques already used for the detection of these toxins in clinical and environmental samples. The new PCR-based assay can be developed as a diagnostic tool to detect extremely low amounts of active toxins. In addition, the assay could be applied to the detection of other clinically important proteases, such as other bacterial toxins and cancer-associated enzymes. In general, the assay could be adapted to detect the activity of virtually any enzyme, the actions of which result in the cleavage of a polymer or oligomer followed by the physical separation of cleaved parts.
The authors express their gratitude to SRCAMB staff researcher Olga Krasavtseva and research technicians Valentina Lebedeva and Valentina Gusarova for technical support during the experiments.
FUNDING
The work was supported by Russian Science Foundation [grant number 14-15-00630].
Conflict of interest. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}