





Considerable advances have been made in developing human papillomaviruses (HPV) prophylactic vaccines based on L1 virus-like particles (VLPs). However, there are limitations in the availability of these vaccines in developing countries, where most cases of cervical cancer occur. In the current study, the prime-boost immunization strategies were studied using a DNA vaccine carrying HPV-16 L1 gene (pcDNA/L1) and insect cell baculovirus-derived HPV-16 L1 VLP. The humoral immunity was evaluated by measuring the specific IgG levels, and the T-cell immune response was assessed by measuring different cytokines such as IFN-γ, TNF-α and IL-10. Results showed that although immunization with pcDNA/L1 alone could induce strong cellular immune responses, higher immunogenicity especially antibody response was achieved in pcDNA/L1 priming-VLP boosting regimen. Therefore, we suggest that prime-boost regimen can be considered as an efficient prophylactic and therapeutic vaccine.
INTRODUCTION
DNA-based immunization has been considered as one of the interesting approaches for vaccination. DNA vaccines may afford a series of advantages over traditional vaccines including safety, greater stability, technically simple design, time-saving and less expensive production process, ease of storage and transport. Moreover, these vaccines are capable of eliciting both antigen-specific humoral and cellular immune responses (Kutzler and Weiner 2008). However, one of the problems associated with DNA vaccines is lower DNA-raised antibodies in comparison with that of protein-raised ones (Kim et al., 1998; Tang et al., 2007; Ferraro et al., 2011). The prime-boost strategy, which is the combination of DNA vaccine priming step followed by boosting with related protein or other vectors each expressing similar antigens, can induce high levels of specific humoral immunity and in some cases can confer protection against infectious agents (Ramshaw and Ramsay 2000; Eo et al., 2001). This procedure has produced promising results against HIV and Leishmania in animal models (Ramshaw and Ramsay 2000).
Persistent infection with specific human papillomaviruses (HPV) has been found closely associated with the development of cervical cancers (Nobbenhuis et al., 1999; Kjaer et al., 2002; Dalstein et al., 2003). Of all the HPV types, HPV-16 is the most prevalent and represents the primary causative agent in more than 50% of all cervical cancer cases (Smith et al., 2007). Prophylactic vaccination is an important strategy for potentially reducing the impact of HPV in incidence and mortality rates of this cancer as they can prevent the primary infection by eliciting neutralizing antibodies against the incoming virus (Franco and Harper 2005).
L1, the major structural protein of the HPV capsid, is considered as a key component of prophylactic HPV vaccines because as expressed in eukaryotic cells, L1 can self-assemble into virus-like particles (VLPs) which have a highly ordered structure similar to native virions and display conformation-specific neutralization epitopes (Kirnbauer et al., 1992). VLPs are very efficient in inducing immune responses and currently are used in prophylactic quadrivalent (Gardasil®, Merck) and bivalent (Cervarix®, GSK) HPV vaccines (Hutchinson and Klein 2008). Both vaccines have been shown to prevent HPV infection because they can elicit strong systemic immune responses, involving both neutralizing antibodies and cell-mediated immunity (Moscicki 2008).
Despite the remarkable success of these vaccines against HPV infection, because of their considerable manufacturing costs for expression in and purification from eukaryotic cells (insect cells or yeast), the need to multiple intramuscular doses and cold chain transportation, they will likely remain unaffordable for developing countries where, in fact, 83% of all cervical cancer cases occur. For these reasons, an effective regimen should be adopted to produce prophylactic cervical cancer vaccines.
As an alternative approach, diminution of multiple doses administrated VLP and the use of DNA priming-protein (VLP) boosting strategy can improve the immune responses over those induced by DNA or protein vaccine alone and also can represent a cost-effective approach.
In this study, HPV-16 L1 gene was used to construct a DNA vaccine and also to prepare recombinant VLP using a baculovirus expression system in Sf9 insect cells. Purified VLP was administrated as a booster for enhancing the humoral immune responses induced by L1 DNA vaccine. The immunogenicity of different vaccine formulations was evaluated by assessment of specific antibody titers and also T-cell responses in C57BL/6 mice.
MATERIALS AND METHODS
Experimental animals
Female C57BL/6 mice (6–8 weeks old) were purchased from Pasteur Institute of Iran (Tehran, Iran), housed under filter top conditions with water and food and then were used for experimental purposes with approval of the Animal Ethics Committee of the Ministry of Health and Medical Education (Tehran, Iran).
Plasmid DNA construction
The pcDNA3/L1 was generated as previously described. Briefly, the HPV-16 L1 full gene was amplified from paraffin-embedded tissues, cloned to pTZ57R/T vector (Fermentase, Lithuania) and confirmed by sequencing (Teimoori et al., 2008). The 1595 bp L1 ORF was subcloned into the pcDNA3 (Invitrogen, Burlington, Canada) under the human cytomegalovirus immediate early promoter. The recombinant plasmid, pcDNA3/L1, was multiplied in a DH5α strain of Escherichia coli, and clones with the correct orientation of insert were prepared on a large scale. The pcDNA3/L1 was purified using a modification of alkaline lysis maxi-preparation (Sambrook and Russell 2001) and high-purity plasmid isolation using silica oxide procedures (Grimm and Voss-Neudecker 2003).
HPV-16 L1 VLP production and purification
Recombinant baculovirus encoding HPV-16 L1 gene was generated using the Bac-to-Bac system as previously described in detail (Abdoli et al., 2013). Sf9 cells were grown at 27°C in Grace's medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 50 U ml−1 penicillin, 50 μg ml−1 streptomycin and infected at a multiplicity of infection of 10 with the recombinant baculoviruses. After 72 h incubation, the infected cells were pelleted by centrifugation, washed once with ice-cold phosphate buffered saline (PBS) and stored at −70°C. Cell pellets were resuspended in extraction buffer (5 mM MgCl2, 5 mM CaCl2, 1 M NaCl, 0.01% Triton X-100, 20 mM Hepes pH 7.4 and 1 mM PMSF), subsequently lysed by three times sonication for 25 s on ice at 60 W and cleared by centrifugation at 10 000 rpm, 4°C for 30 min. The supernatant was taken and pellet was resuspended in extraction buffer, sonicated for 25 s on ice at 60 W and centrifuged again. Combined supernatants were layered onto a 40% sucrose (w/v in extraction buffer) cushion and centrifuged in a Hitachi swinging bucket rotors P40ST rotor (4°C, 3.5 h, 38 000 rpm). Pellets were resuspended in extraction buffer and cesium chloride (CsCl) was added to a final concentration of 0.4 g ml−1. Following ultracentrifugation in the same rotor (14°C, 24 h, 38 000 rpm), CsCl gradient fractions were harvested and the densities were calculated from the refractive index (20°C), as determined by an Abbe-3L refractometer (CARL/ZEISS 1404, Germany). The L1-containing fractions were determined by SDS-PAGE followed by staining with Coomassie blue and western blot analysis. Desired fractions were pooled and dialyzed against 20 mM Hepes (pH 7.4) and 0.5 M NaCl and stored at 4°C. The concentration of L1 protein was determined by using Bradford assay. The quality of VLPs was confirmed by electron microscopy and hemagglutination assay (HA).
Electron microscopy
The integrity of the purified VLPs was monitored by their morphology in the electron microscope. VLPs were absorbed to carbon-coated grids and stained with 1% uranyl acetate. Grids were analysed using a Hitachi transmission electron microscope (TEM, model HU 12-A) at final magnifications of 90 000×.
Hemagglutination assay
HA was performed for assessing the quality of purified VLPs since improperly folded viral L1 protein does not contribute to the HA (Roden et al., 1995). C57BL/6 mice blood was collected in a heparinized tube and the red blood cells (RBCs) were separated by centrifugation at 1000 rpm and 4°C for 5 min, washed twice with PBS and suspended in PBS at a concentration of 1% (v/v). Different amounts of purified VLPs (150, 300 and 600 ng ml−1) were titrated. Serial two-fold dilutions of VLPs in 50 μl of PBS in a round-bottomed 96-well plate were mixed with an equal volume of 1% suspension of RBCs in PBS (V/V) and incubated at 4°C for 3 h.
Vaccine design and immunization of mice
Based on the following program, five groups each containing five C57BL/6 mice were injected subcutaneously three times at 14-day intervals. Group l (V) received three doses of 5 μg purified VLPs, Group 2 (LV) received one dose of 100 μg pcDNA3/L1, boosted with two doses of 5 μg purified VLPs and Group 3 (L) received three doses of 100 μg pcDNA3/L1. Groups 4 and 5 were injected with three doses of the pcDNA3 (E) and PBS (P), respectively, as negative controls. To evaluate specific antibody responses, two weeks after the final immunization, blood was collected from the animals by puncture of the superficial temporal vein, allowed to clot, centrifuged and then serum samples were taken and kept at −20°C until use. Then, all mice were sacrificed and the spleens were isolated for ex vivo cytokine production.
Assay for immune responses
Detection of anti-VLP antibodies by ELISA
Sera were assayed for IgG-specific HPV-16 L1 VLP by endpoint dilution enzyme-linked immunosorbent assays (ELISAs). A 96-well flat-bottomed plate (Nunc, Denmark) was coated with VLP solution (2 μg ml−1) in PBS (Dulbecco's PBS without Ca2+ or Mg2+; PAA Laboratories GmbH, Australia) (pH 7.2) by overnight incubation at 4°C. The plates were washed three times with PBS containing 0.05% Tween 20 (PBST) and blocked by adding 100 μl of blocking solution (PBS-T containing 1% bovine serum albumin) to each well at 37°C for 1 h. After washing step, serial two-fold dilutions of sera in PBS were added to the plates and incubated on the shaker at 37°C for 2 h. To determine non-specific binding, the same dilutions of the sera were tested on uncoated wells. Following washing, horseradish peroxidase-conjugated goat anti-mouse IgG antibody (RaziBiotec AP8036) at 1/5000 dilution was added. After 1.5 h of incubation on the shaker at 37°C and subsequent washing, 100 μl TMB (3,3′,5,5′-tetramethyl benzidine substrate solution, Sigma T4444) as substrate was added to the wells and the plates were incubated at room temperature (RT) for 30 min. Finally, stop solution (2 N H2SO4, 50 μl) was added to the wells and absorbance was measured at 450 nm using an ELISA plate reader. Endpoint titers were defined as the reciprocal of the highest serum dilution giving an absorbance value greater than the average absorbance of negative control mice plus three times the standard deviation (SD) and were expressed as the group means ± SD for five mice.
Additionally in the other test, the total levels of mouse IgG in serum were determined by using commercial mouse IgG ELISA quantitation kit (Bethyl, INC, Montgomery) according to the manufacturer's instruction. The lower limit for detection of mouse IgG was 7.8 ng ml−1. Values were presented as ng IgG ml−1 (mean ± SD, n = 5).
Hemagglutination inhibition assay
To verify the specificity and neutralizing ability of the serum antibodies, hemagglutination inhibition (HI) assay was performed. Serum treatment for excluding non-specific hemagglutination activity and non-specific inhibitors was carried out according to the method described by Mannen et al., ( 1984) with some modification. Briefly, sera were heated at 56°C for 30 min and then diluted 1:2 with borate saline (pH 9) and an equal volume of 50 mg ml−1 Aerosil 380 (Evonik Industries) in borate saline. The mixture was incubated at RT for 30 min with occasional shaking, centrifuged at 1400 g and 4°C for 10 min and the supernatant was used for HI. Serial dilutions of the treated sera were incubated with VLPs (150 ng, 4 HA unite) at RT for 2 h, followed by mixing with 1% suspension of mouse RBC (v/v in PBS) in a round-bottomed 96-well plate. Inhibition of agglutination and appearance of spots that indicated VLP-neutralizing activity was read after 3 h incubation at 4°C. HI titers are expressed as the reciprocal serum dilution that caused complete inhibition of VLP-mediated RBC agglutination (Roden et al., 1995).
Cytokine ELISA assay
Spleens from immunized mice were removed two weeks after the final immunization, and single cell suspensions were prepared by gentle homogenization. RBCs were lysed by incubation in RBC lysis buffer (20 mM Tris, 160 mM NH4Cl, pH 7.4) for 5 min at RT, and the resulting splenocytes were resuspended in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamin, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. The cells at a density of 2×106 cells ml−1 were seeded in a flat-bottomed 24-well plate (Nunc, Denmark) and incubated with suitable mitogen (the 10 μg ml−1 Concanavalin A (Con A, Sigma C7275), 10 μg ml−1E. coli LPS (Sigma, L2630) or 10 μM H-2Db L1165–173 CTL epitope) and then incubated at 37°C with 5% CO2. Culture supernatants were collected after 72 h and frozen at −70°C until the samples were analysed to detect the presence of different cytokines. Levels of IFN-γ, IL-10 and TNF-α secretion were determined by using commercial DuoSet ELISA cytokine assay kits (R&D system, Minneapolis, MN) according to the manufacturer's instruction. The lower limit for detection of the cytokine was 32 pg ml−1. Values were presented as pg cytokine ml−1 (mean ± SD, n = 5).
T-cell proliferation assay
Statistical analysis
Analysis of antibody titers and different cytokine data was performed using one-way ANOVA followed by Turkey's post-test. Values are expressed as the mean ± SD. *P < 0.05 were considered statistically significant. All statistical analyses were performed using the GraphPad Prism 6.01 software (La Jolla, CA, USA).
RESULTS
Production of HPV-16 L1 VLPs
Baculovirus expression systems were employed to produce VLPs in Sf9 insect cells. Analysis of the purified VLPs preparation on 12% Coomassie blue-stained polyacrylamide gel revealed a single 55-kDa protein band that also reacted with the anti-HPV-16L1 monoclonal antibody in western blotting (Fig. 1A and B). When analysed by transmission electron microscopy, spherical particles with a diameter of about 55 nm were the indicative for L1 VLPs (Fig. 1C). The purified VLPs agglutinated mouse erythrocytes and increased amount of VLPs resulted in rising HA titer as shown in Fig. 1D.
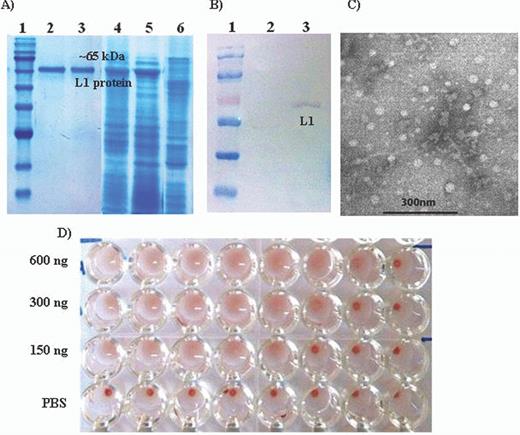
Analysis of the purified VLPs. HPV-16 L1 VLPs were expressed in and purified from recombinant baculovirus-infected Sf9 cells. (A) Samples were separated on 12% SDS-PAGE: 1, molecular size markers; 2 and 3, purified L1 VLP protein; 4 and 5, unpurified L1 VLP protein (Sf9 cells infected with HPV-16 L1 recombinant baculovirus) and 6, uninfected Sf9 cells. (B) Western blot: proteins on SDS-PAGE gel were transferred onto nitrocellulose membrane and L1 proteins were detected using the anti-HPV-16 L1 monoclonal antibody: 1, molecular size markers; 2, uninfected Sf9 cells; 3, Sf9 cells infected with HPV-16 L1 recombinant baculovirus. (C) Electron micrographs of negatively stained VLPs under TEM. Purified VLPs were negatively stained with 1% uranyl acetate and observed under TEM (Hitachi TEM, model HU 12-A) at 90 000 × magnifications. VLPs were approximately 55 nm in diameter. (D) HA: purified VLPs agglutinate mouse erythrocytes. Different amounts of purified VLPs were serially diluted two-fold in 50 μl PBS and mixed with an equal volume of the 1% mouse RBC suspension in PBS per well of a 96-well plate. The samples were incubated for 3 h at 4°C and photographed.
Analysis of serum antibody responses
Development of serum anti-VLP IgG antibody titers was monitored two weeks after the last immunization using endpoint dilution ELISA. As shown in Fig. 2A, DNA priming-VLP boosting regimen induced significantly (P < 0.01) higher VLP-specific IgG titer as compared with pcDNA3/L1 or VLP vaccine alone. The least antibody titer was obtained in the pcDNA3/L1 group as expected. There was no difference in serum total IgG amount between different groups (Fig. 2B) which indicate that both the VLPs and DNA priming-VLP boosting have been induced a specific B-cell response to L1 protein.
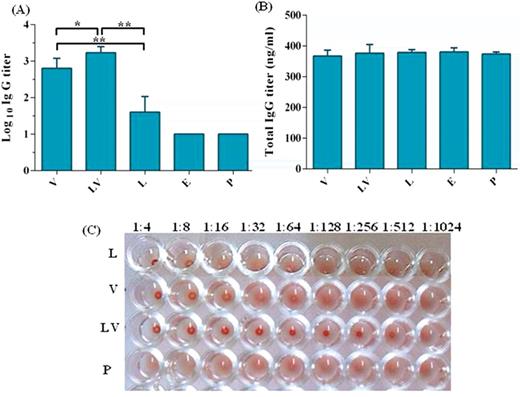
Induction of specific IgG antibodies by different vaccine preparations. Sera were collected from individual immunized mouse at two weeks after the last immunization. Presence of IgG antibodies in sera was detected by ELISA. (A) Specific HPV-16 L1 VLP IgG antibody titers and (B) total IgG antibody at dilution 1/100. Data are means ± SD, n = 5. The levels of statistical significance for differences between test groups were determined by using one-way ANOVA followed by Turkey's post-test. Statistical significance was indicated with *P < 0.001 and **P < 0.0001. (C) Inhibition of HPV-16 L1 VLPs induced hemagglutination. Sera from the same groups of mice were serially diluted and mixed with the VLPs before reacted with mouse erythrocytes. Data are expressed as the reciprocal dilution that completely inhibited agglutination of mouse RBCs by L1 VLPs. Group V received three doses of 5 μg VLPs, Group LV received one dose of 100 μg pcDNA3/L1 and boosted with two doses of 5 μg VLPs, Group L received three doses of 100 μg pcDNA3/L1, Group E received three doses of the pcDNA3 and Group P received three doses of PBS.
Specific antibody titer was further confirmed by VLP-mediated hemagglutination which is considered a surrogate method for virus neutralization. To exclude non-specific hemagglutination activity in HI assay, sera were precleared by heating for 30 min at 56°C followed by treatment with Aerosil 380. HI titer in VLP (V), prime-boost (LV) and pcDNA3/L1 (L) groups were 512, 64 and 8, respectively (Fig. 2C). These results demonstrate that antibodies in sera from prime-boost group were more elevated similar to ELISA data and also they have the highest neutralizing capacity.
Cytokines assay
Spleen cells of immunized mice were cultured in the presence of different mitogens ex vivo, and different cytokine profiles such as IFN-γ, TNF-α and IL-10 secreted by T cells were assessed by ELISA kit. Results showed that the IFN-γ level was significantly (P < 0.01) increased in the pcDNA3/L1 vaccinated mice as compared with other groups (Fig. 3A), but IL-10 assay did not show any statistically significant differences among tested groups (Fig. 3B).
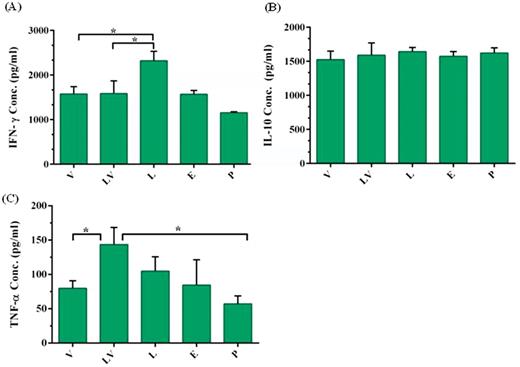
Measurement of cytokine levels secreted by splenocytes of immunized mice. Splenocytes were obtained from immunized mice 14 days after last vaccination with pcDNA3 plasmid (E), pcDNA3/L1 plasmid (L), VLP (V) pcDNA3/L1 priming and VLP boosting (LV) and PBS (P) via subcutaneously injection. Splenocytes were in vitro re-stimulated with LPS for measurement of IL-10 and TNF-α, and with Con A for measurement of IFN-γ. Cytokines released into culture media were determined after 72 h induction at 37°C by commercial ELISA kit. Data are mean ± SD, n = 5. The levels of statistical significance for differences between test groups were determined using ANOVA followed by Turkey's post-test. *indicates statistical significance (P < 0.01).
Among examined vaccine preparation, DNA priming-protein boosting (LV) could significantly induce the highest level of TNF-α production as compared with other vaccines (P < 0.01) (Fig. 3C).
Determination of the T-cell proliferation
The lymphocyte proliferation in response to the specific antigen re-stimulation was measured using MTT assay and SI was calculated. Results showed that incubation for 72 h led to increasing SI in V and LV groups as compared to 36 h. Moreover, SI was increased in splenocytes of LV group after 72 h, although the observed difference was not significant (Fig. 4).
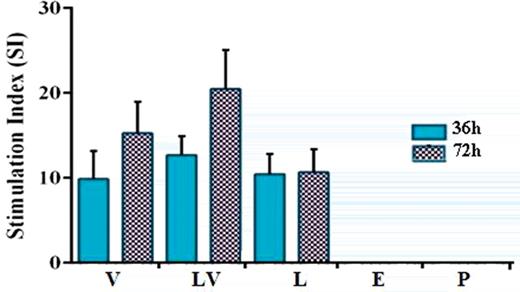
Lymphocyte proliferation shown as the stimulation index (SI) for vaccinated mice. Splenocytes from immunized mice were obtained and re-stimulated with specific antigen. After 36 and 72 h incubation, cell proliferation assay was performed using MTT solution. Formazan crystals were dissolved in dimethyl sulfoxide by vigorous pipetting. The optical densities were measured at 492 nm. The SI was calculated as mentioned in the section ‘Materials and Methods’.
DISCUSSION
Different vaccine models have been investigated to generate effective immune responses and protection against HPV infections. Nowadays, immunizing agents such as VLPs, recombinant fusion proteins or peptides, live recombinant bacteria, recombinant viruses and DNA vaccines are being evaluated for vaccination purposes (Kuck et al., 2006). DNA-based immunization has been proved to be a safe, effective and promising approach to induce host immune responses (Schreckenberger et al., 2000). However, due to relatively poor immunogenicity of DNA vaccines, numerous studies are performed to test different strategies aimed at enhancing and directing the potency of DNA vaccines. These strategies include selecting of appropriate site of immunization (Watts and Kennedy 1999; Jazayeri et al., 2009), improving of plasmid uptake using efficient vaccine delivery system (Touze and Coursaget 1998; Shi et al., 2001; Malboeuf et al., 2007; Lee et al., 2010), increasing expression of encoded antigen on a per-cell basis through employment of suitable expression vector and codon optimization (Leder et al., 2001; Cheung et al., 2004), improving formulation with proper adjuvants (Sanchez et al., 2004; Wang et al., 2006) and choosing prime-boost strategy (Doria-Rose and Haigwood 2003; Kutzler and Weiner 2008; Ferraro et al., 2011).
One of the potent strategies for increasing DNA vaccine efficacy involved DNA priming-protein boosting. In preclinical animal models, this approach has increased the amount of neutralizing antibodies and also boosted DNA-primed CTL responses (Kutzler and Weiner 2008). Using a DNA priming-protein boosting strategy has shown that although immunization with pDNA encoding desired gene or comparable protein alone could induce humoral immunity, a marked stimulation of humoral immune response occurred when DNA priming was followed by encoded antigen boosting (Soleimanjahi et al., 2006; Fotouhi et al., 2008).
Besides, Kowalczyk et al., developed a DNA vaccine and a recombinant adenovirus (Ad5), both expressing the L1 protein of HPV-16. The vaccines used in a prime-boost regimen, with the DNA given intramuscularly for priming, were followed by an intranasal booster immunization with the recombinant virus. They showed that using this strategy, a long-lived antibody response to conformation-dependent epitopes of L1 was induced both in sera and in vaginal secretions (Kowalczyk et al., 2001).
The effectiveness of prime-boosted regimen in enhancement of DNA-induced immune responses has been shown for the varieties of pDNA-encoded antigen and different animal species. In the current study, effect of this strategy was evaluated for pcDNA-encoding HPV-16L1 gene aimed at reducing crucial customary three VLP dosages. In order to achieve this aim, pcDNA3/L1 and self-assembled HPV-16L1 VLP were purified and applied as a subunit vaccine.
Our results showed that HI activity in mice vaccinated with prime-boosted regimen is higher than that in other groups and also compared to pcDNA3/L1 or VLP alone, combined pcDNA3/L1 and VLP as prime-boosted regimen provided a statistically significant enhancement in eliciting the specific antibody responses thereby functioning as an improved strategy for DNA vaccination.
Analysis of cytokine profiles revealed that although IFN-γ was significantly increased in the pcDNA3/L1 vaccinated mice as compared with other groups as expected, mice given VLP alone or prime-boost regimen induced as much as IFN-γ secretion. IL-10 as a critical cytokine for the maintenance of immune homeostasis was almost similar among tested groups.
Also, among different vaccine preparations, DNA priming-protein boosting (LV) could significantly induce the highest level of TNF-α production as compared with other groups. TNF-α has been implicated as an essential pro-inflammatory cytokine not only in the context of a link between innate and adaptive immunity, but also for optimal antibody responses (Ritter et al., 2003). HPV prophylactic vaccines should be capable of inducing neutralizing antibodies to prevent the initial infection of its target epithelial cells. So, increased antibody response and TNF-α secretion using a DNA priming-protein boosting strategy could play an important role in protecting individual against HPV infection.
The mechanisms by which prime-boosted regimen enhance immune response seem to be efficient antigen presenting through class I and class II MHC molecules, employing both arms of immune system and subsequently strengthen the humoral and cellular immune responses.
Weak antibody responses were detected upon pcDNA3/L1 administration without neutralizing activity. Thus, an L1 DNA vaccine alone was not a successful prophylactic vaccine, but it could serve as a good priming agent in a DNA priming-protein boosting regimen. DNA priming can drive strong cellular responses and boosting with VLP can elicit strong humoral responses, so this regimen takes advantage of both humoral and cellular immune responses. Therefore, presented data demonstrated that this regimen could induce immune responses higher than those obtained with DNA–DNA or VLP–VLP vaccination and provided a cost-effective approach in developing countries.
This work was supported by the Institute of Biochemistry and Biophysics, University of Tehran, Research Deputy of Tarbiat Modares University, Faculty of Medical Sciences and the Pasteur Institute of Iran. The authors thank Dr Ahmadian for kindly providing assistance with TEM.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}