





Nontypeable Haemophilus influenzae (NTHi) is an important cause of lower respiratory tract infections, resulting in exacerbations of chronic obstructive pulmonary disease (COPD). Despite its pathogenic potential, little is known regarding the role of intracellular NTHi in pathogenesis of pulmonary infection. Kinetics of NTHi internalization was studied using gentamicin protection assays. NTHi strains isolated from COPD patients efficiently adhere to and invade type II alveolar (A549) cells. During early stages, that is, 6 h postinfection, we noted a substantial increase in NTHi invasion with no evidence of intracellular replication. Electron microscopy revealed that the majority of internalized NTHi resided within membrane bound acidic endocytic vacuoles. However, at later stages, that is, 8 h postinfection, significant reduction in viable intracellular NTHi was observed and vacuoles were found to be empty with NTHi escape into the cytosol. By 12 h, cytopathic changes of cells were evident with massive vacuolization of cytoplasm, intense chromatin condensation, and intact plasma membrane. Furthermore, analysis of apoptotic markers confirmed that infected A549 cells underwent apoptosis at later stages. In addition, inhibition of internalization of NTHi by cytochalasin D prevented apoptosis of cells. Collectively, these findings suggest that internalization of NTHi and its escape from vacuolar compartments triggers cytotoxicity of alveolar cells via apoptosis during the infection process.
INTRODUCTION
Nontypeable Haemophilus influenzae (NTHi) is a Gram-negative, unencapsulated human pulmonary pathogen. NTHi enters the body during respiration and establishes either an asymptomatic colonization in the nasopharynx or a candid infectious process within the host respiratory mucosa. NTHi is mainly implicated in the majority of chronic infections of lower respiratory tract such as exacerbations of chronic obstructive pulmonary disease (COPD) and cystic fibrosis (Murphy et al., 2004; Sethi and Murphy 2008). NTHi is also associated with respiratory tract infections such as otitis media (OM), conjunctivitis, sinusitis, and community-acquired pneumonia (Murphy 2003). The success of NTHi as a colonizer and a pathogen is due to enormous strain-to-strain variability and reliance on diverse mechanisms of attachment and invasion. NTHi has emerged as a potential pathogen in post-H. influenzae serotype b (Hib) conjugate vaccine era (Ulanova and Tsang 2009) and furthermore, emergence of antibiotic resistance strains of NTHi and intricacy in developing an efficacious vaccine support further efforts to understand the host response mechanisms involved in NTHi infections.
NTHi has long been considered as an extracellular pathogen based on its binding to mucin and adherence to epithelial cells (St Geme and Falkow 1990; Read et al., 1991). Several in vitro and ex vivo studies on NTHi-host cell interactions, however, now establish that NTHi has both an extracellular and intracellular niche in the human respiratory epithelial cells (St Geme and Falkow 1990; van Schilfgaarde et al., 1995; Ketterer et al., 1999), endothelial cells (Virji et al., 1992), macrophages (Craig et al., 2001; Marti-Lliteras et al., 2009), and adenoidal epithelial cells (Forsgren et al., 1994; Nistico et al., 2011).
During the last two decades, intracellular NTHi has been decisively linked to symptomatic infection in adults with COPD. Several studies have demonstrated extensive presence of NTHi in lung tissue (submucosa of the bronchi, the bronchioles, the interstitium, and the alveolar epithelium) in 25–80% subjects who had exacerbations of COPD (Moller et al., 1998; Bandi et al., 2001; Dromann et al., 2010).
Until recently, much of the NTHi research related to pathogenesis has been focused on studying the invasion and trafficking mechanisms. Most of the reports have clearly shown that NTHi invades into host cells and is located inside vacuolar compartments, which may provide a protective reservoir for persistence (St Geme 2002; Morey et al., 2011). But these studies failed to speculate the precise role of intracellular NTHi inside host cells.
The alveolar epithelium functions as the first line of protection against potential respiratory pathogens and is comprised of two morphologically different alveolar epithelial cell (AEC) types – type I and type II. Type II AECs are cuboidal cells which cover 3–5% of the alveolar surface area and have traditionally been considered as labeled defenders of the alveolus for their immuno-modulatory functions. The role of Type II cells as nonprofessional antigen-presenting cells has been more recently proposed. In contrast, type I AEC covering 96% of the alveolar surface area, mostly play a role for gas exchange but do not participate in the active cellular immune response of the lung (Fehenbach 2001; Mason 2006). Our hypothesis is that being in the interface, type II AEC may play an important role in the pathogenesis of NTHi infection in the lower airways. Here, we have used A549 human Type II AEC line, defined as a model of human AECII cells (Lieber et al., 1976).
Thus, a broader understanding is needed regarding the potential role of NTHi internalization into major tissue target of airways, that is, alveolar epithelium and its survival and virulence strategies involved in the pathogenesis of NTHi infection. Keeping all these concerns in mind, the aim of this study was to focus on the intracellular life of NTHi and fate of host epithelial cells during the infection process. In sum, to the best of our knowledge, this study demonstrated for the first time that internalization of NTHi mediates cytotoxic effect on type II AECs via apoptosis, a crucial stage in the pathogenesis of persistent NTHi infections.
MATERIALS AND METHODS
Bacterial strains
NTHi strain isolated from broncho alveolar lavage (BAL) of a patient suffering from COPD for more than 10 years was used in this study. An ATCC standard strain of NTHi 49247 was also used. The serotyping was performed by (1) slide agglutination using polyvalent or serotype-specific antisera against H. influenzae capsular antigens (a–f) (Difco, US) and (2) molecular capsular typing of bexA gene by PCR.
Bacterial growth conditions
NTHi was grown overnight on chocolate agar (CA) with 300 μg mL−1 bacitracin (Hi-Media) at 37°C with 5% CO2 and stored at −80 °C in chocolate broth with 20% glycerol. For growth in broth, bacteria were suspended in BHI broth (brain heart infusion, Hi-Media) supplemented with hemin (10μg mL−1) and NAD (3.5μg mL−1) (Sigma). For all assays, NTHi was streaked from fresh frozen stocks onto CA.
Cell culture and culture conditions
Type II AEC line (A549) was obtained from the American Type Culture Collection (ATCC) derived from human lung carcinoma. A549 cells were grown and maintained in Ham's F-12 Medium (Sigma) containing 10% (v/v) heat-inactivated fetal calf serum (FCS, Sigma), 0.15% (v/v) sodium-bicarbonate (Hi-Media), 5 mM l-glutamine (ICN, US), streptomycin and penicillin (100μg mL−1, Hi-Media) at 37°C, and 5% CO2. The confluent stock cultures were trypsinized (0.1% Trypsin, Sigma) and cells were seeded at a density of 105 cells per well in fresh tissue culture plates (Griener Bio-One) and incubated for 24 h. Cell viability was determined by trypan blue dye exclusion. The culture medium was replaced by medium without antibiotics before carrying out infection experiments.
Adherence assay
We adapted the assay as previously described (St Geme and Falkow 1990). Cells were grown in culture plates until 80% confluence. NTHi strain was grown to mid-log phase at OD600 nm of 0.5 in BHI broth, washed in phosphate-buffered saline (PBS, pH-7.4), and resuspended in medium containing 10% FCS. A549 cells were infected with NTHi to achieve multiplicity of infection (MOI) 10. During all the experiments, cells were infected over various time points as indicated in the results section. At selected time intervals, cells were washed with PBS to eliminate nonspecific bacterial attachment, treated with 0.05% trypsin for 5 min at 37°C and serial dilutions of lysate were spread plated on CA plates. Colony-forming units (CFU) recovered per well were counted to yield the number of adherent bacteria.
Invasion assay
Assays were performed as described previously (Isberg and Falkow 1985). The cells were infected as outlined above for 2 h, washed thrice with PBS and fresh medium containing gentamicin (100μg mL−1, Hi-Media) was added for additional 2 h to kill the extracellular bacteria (internalized bacteria are protected from its effect). Preliminary experiments indicated that 100μg mL−1 of gentamicin was sufficient to kill 100% of initial inoculum added and did not cause any cytotoxic effect (data not shown). After washing, 0.5% Triton X-100/EDTA was added to permeabilize cells and serial dilutions were plated and results are expressed as CFU per well.
Light microscopy
Confluent monolayer was grown on coverslips in 12-well tissue culture plates and infections were performed as described above. When indicated, cells were washed with PBS thrice, fixed with methanol for 1 min, and stained with 2.5% Giemsa stain for 10 min. Stained cells were washed thrice with PBS, and coverslips were mounted on glass slides. The morphological changes were recorded using a light microscope (Leitz, Biomed) equipped with a camera (Leica, MPS 32).
Transmission electron microscope (TEM)
Nearly confluent A549 cells seeded in culture flasks were infected as outlined above. The cells were harvested, fixed with 1% glutaraldehyde, washed with Sorensen's buffer (pH 7.4), postfixed with 1% OsO4, washed with Millong's buffer (pH 7.4), and dehydrated with a series of graded ethanol solutions. The samples were then embedded in Spurr's low viscosity Epoxy resin to cut into ultrathin sections and counterstained with 0.5% uranyl acetate and 1% lead citrate. Electron microscopic examination was conducted using a 906 Zeiss TEM.
Detection of autophagic/acidic vacuole formation
The volume of the cellular acidic compartment was visualized by acridine orange (AO) staining, as previously described (Arthur et al., 2007). Briefly, when indicated, cells were washed with PBS and incubated with medium containing 1 mg mL−1AO (Sigma) for 15 min. Finally, cells were washed with PBS and immediately fluorescent micrographs were taken using an inverted fluorescent microscope (40× objective, Olympus CKX41, Japan) equipped with a filter system (excitation filter: 460–490 C and barrier filter: 520 IF).
A fluorescent compound, monodansylcadaverine (Sigma), has been used to confirm the abundance of autophagic vacuoles in cells (Biederbick et al., 1995). Ten millimolar stock solution of monodansylcadaverine was prepared fresh in 1 : 1 DMSO/Ethanol prior to its use. After infection for indicated time, cells were incubated with 0.05 mM monodansylcadaverine in PBS at 37°C for 10 min. Subsequently, cells were washed with cold PBS and immediately examined by fluorescence microscopy (oil immersion) using an inverted microscope (Olympus BX51, Japan) equipped with a UV filter system (excitation filter: 360–370 nm and barrier filter: 420–460 nm).
Assessment of autophagic vacuoles with monodansylcadaverine
Following infection, the cells were incubated with 0.05 mM monodansylcadaverine at 37°C for 10 min as previously described (Munafo′ and Colombo 2001). After incubation, cells were washed four times with PBS and collected in 10 mM Tris-HCl, pH 8 containing 0.1% Triton X-100. Intracellular monodansylcadaverine was determined using a Cary Eclipse fluorometer (excitation wavelength 380 nm and emission filter 525 nm). To normalize the measurements to the number of cells present in each well, a solution of ethidium bromide was added to a final concentration of 0.2 mM and the DNA fluorescence was measured (excitation wavelength 530 nm and emission filter 590 nm). The monodansylcadaverine incorporated was expressed as specific activity (arbitrary units).
Propidium iodide (PI) uptake assay
PI is a fluorescent dye that intercalates into the DNA strands and the extent of PI incorporation in cells can be monitored by flow cytometry as described previously (Nicoletti et al., 1991). Briefly, following infection with NTHi for desired time intervals, cells were harvested by trypsin (0.01%) and fixed with 1% paraformaldehyde in PBS at 4 °C. The cells were washed with 70% ethanol and then permeabilized in hypotonic buffer (0.1% sodium citrate and 0.1% Triton X-100 in PBS) containing RNAase H (10μg mL−1, Roche) for 15 min. Then after washing, PI (10μg mL−1, ICN, US) was added and kept for 20 min at 4 °C in dark. Analysis was carried out on a flow cytometer (FACS Calibur equipped with 15 MW, 488 nm air cooled argon laser, Becton Dickinson BD) using the cell quest software.
Assessment of apoptosis by flow cytometry
An early marker of apoptosis is the loss of symmetry of phospholipid membrane, resulting in increased level of phosphatidylserine in outer leaflet which was assessed using annexin V-FITC by FACS as previously described (Vermes et al., 1995). As the translocation of phosphatidylserine to the external cell surface is not unique to apoptosis but also occurs during necrosis, a combination of annexin V-FITC and PI can be used to distinguish between apoptotic and necrotic cells. This assay was carried out using annexin V-FITC apoptosis detection kit I (BD, US) according to manufacturer's instructions. Briefly, 100μL of Ca2+-based binding buffer and 5μL of annexin V-FITC was added to infected cells for indicated time intervals. The samples were incubated at RT for 30 min, mixed with 400μL of binding buffer, and 0.5μg of PI prior to analysis. The cells were analyzed by flow cytometer (FACS Calibur, using filter of 488 nm for FITC and > 600 nm for PI detection), and results were expressed as mean fluorescence intensity of labeled cells in the Quadrants. Each region shows percentage of live (lower left, LL), PI stained, that is, necrotic (upper left, UL), annexin V stained, that is, early apoptotic (lower right, LR), and dual stained, that is, late apoptotic (upper right, UR) cells.
Differentiation of living, apoptotic, and necrotic cells
Ethidium bromide (EtBr) and AO are fluorescent intercalating DNA dyes that allow differentiation of living, apoptotic, and necrotic cells (Grossman et al., 1998). Following infection at indicated time intervals, cells were harvested, resuspended in 20μL of PBS and 2μL of a combined dye of EtBr, and AO (100μg mL−1 each) was added to the suspension. Five microliter of the stained cell suspension was rapidly transferred to a glass slide for immediate analysis using an ultraviolet fluorescent microscope (40×, Olympus, CKX41, Japan) equipped with a filter system (excitation filter: 480–550 C, barrier filter: 590 C). Cells were scored into four categories: C1: live with green, noncondensed nuclei; C2: early apoptotic with green nuclei and signs of nuclear condensation; C3: late apoptotic with orange/red nuclei and nuclear bead formation; and C4: necrotic with large red, noncondensed nuclei. Two-hundred cells per sample were counted in multiple randomly selected fields and scored. The apoptotic index was calculated as the sum of early and late apoptotic cells (C2 + C3)/total number of cells scored × 100.
DNA fragmentation analysis
Infections were performed as described above. When indicated, cells were harvested and lysed with hypotonic buffer (10 mM Tris pH 7.4), 1 mM EDTA, 0.2% Triton X-100) on ice for 15 min. The lysates were treated with 0.5% sodiumdodecyl sulfate (ICN, US) and 300μg mL−1 proteinase K (Qiagen) for 1 h. After complete lysis, RNAase (10 mg mL−1, Roche) was added and incubated for 5 min. DNA was extracted with an equal volume of phenol–chloroform–isoamyl alcohol (25 : 24 : 1 [v/v/v]). DNA in upper aqueous phase was precipitated overnight at −20 °C in cold 100% ethanol, and pellet was washed with cold 70% ethanol. The pellet was air dried, dissolved in Tris-EDTA (TE), separated by 1.8% agarose gel electrophoresis, stained with EtBr, and visualized under Chemilmager™ 4400 (Applied Biosystem). DNA fragments were sized by comparison with a 100 bp DNA ladder (Bangalore Genei).
Inhibition of internalization
For invasion blocking experiment, cells were preincubated for 1 h at 37°C with appropriate concentration of cytochalasin D (0.25, 0.5 and 1μg mL−1) in culture medium prior to infection. The cells were incubated with the inhibitor during 2-h invasion period. Afterward, cells were washed thrice with PBS and incubated for another 2 h in fresh medium containing gentamicin (100μg mL−1). Subsequently, cells were washed and internalized bacteria were counted as described above. Infected cells without exposure to inhibitor were taken as control. Invasion in the presence of cytochalasin D was expressed as relative invasion defined as percentage invasion compared with bacteria in experimental medium alone which was arbitrarily set as 100%. The level of apoptosis in cytochalasin D-treated and untreated cells were checked using annexin V-FITC apoptosis detection kit as described above.
Statistical analysis
For statistical analysis, each experiment was carried out at least in triplicate, and the results were expressed as mean ± SD. The significance of the differences was analyzed using one-way analysis of variance followed by post hoc comparisons or 2-tailed Student's t-test. P ≤ 0.05 represents a significant difference while P ≤ 0.001 was considered highly significant.
RESULTS
Adherence and invasion efficiency of NTHi strains
Preliminary experiments were carried out to examine the ability of NTHi strains isolated from patients suffering from chronic respiratory diseases (COPD and OM) and nasopharyngeal swabs of colonizers to adhere and invade type II alveolar (A549) cells. Among nine strains tested, NTHi strains isolated from BAL of COPD patients showed the maximum ability to adhere and invade A549 cells (Supporting Information, Table S1). Standard strain of NTHi (ATCC 49247) showed much less adherence and invasion efficiency in A549 cells as compared to the clinical strains of NTHi. However, COPD strain showing the maximum adherence (3.23 ± 0.20) and invasion efficiency (1.15 ± 0.08) was used as a representative strain in this study for all the experiments. Also, nonviable (bacteria pretreated with gentamicin) and metabolically inactive bacteria (bacteria incubated at 4 °C) showed marked inhibition of adherence and invasion in A549 cells (data not shown).
Morphological changes of A549 cells
The morphological changes in infected A549 cells were analyzed by light microscopy at 2-h intervals. The intervals were restricted up to 8 h as the monolayer began to lose its integrity beyond this time period. The number of adherent bacteria increased gradually from 2 to 6 h (Fig. 1a). By 8 h, there was a decline in their number with evidence of epithelial cell damage such as detachment from substratum, clumping and rounding of cells, and mottled appearance of cell membrane suggesting cellular injury (Fig. 1a). Similar observations were also noticed with other clinical COPD strains initially isolated in this study (data not shown).
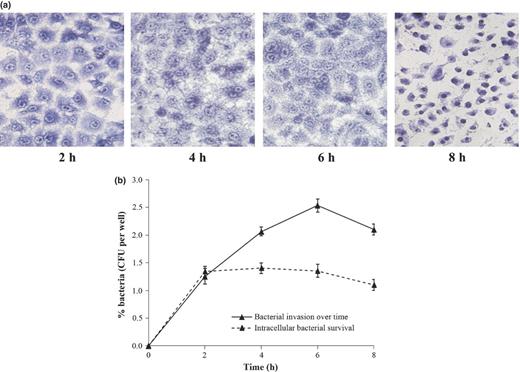
Infection of A549 cells with NTHi strain at MOI of 1 : 10. (a) A549 cells infected for indicated time intervals (2, 4, 6, and 8 h) were fixed and stained with Giemsa. Images were obtained from light microscopy analysis (40×) of three independent experiments. (b) Percent invaded bacteria were enumerated at selected time points (after treatment with gentamicin for additional 2 h) by plating of serial dilutions of cell lysate on CA as shown by ‘solid line’. Intracellular survival rate was determined at desired time points, keeping time of infection constant for 2 h while gentamicin treatment was extended up to 8 h as shown by ‘dashed line’. All results were expressed as CFU recovered bacteria per mL (mean ± SEM from three separate experiments).
Intracellular survival assay
We studied the time course of invasion efficiency to determine the effect of infection time over a period of 8 h. Interestingly, we observed that the number of intracellular bacteria recovered showed a significant increase from 2 to 6 h. However, there was a significant decrease in numbers of intracellular bacteria from 2.533 ± 0.112 at 6 h to 2.10 ± 0.111 at 8 h (P ≤ 0.01) (Fig. 1b).
Next, we tested the ability of the NTHi strain to replicate and survive intracellularly over time in A549 cells. In this experiment, the time of infection remains constant for 2 h, after which fresh medium containing gentamicin was added and invasion was determined at 2, 4, 6, and 8 h. No net increase in intracellular CFU between 2 and 6 h in presence of gentamicin was observed, showing that no intracellular replication takes place. However, similar to the above findings, intracellular bacteria were found to be significantly decreased from 1.356 ± 0.115 to 1.103 ± 0.108 at 8 h (P ≤ 0.05) (Fig. 1b). This result was further confirmed by TEM.
Cytopathic changes induced in A549 cells
Qualitative evidence of the intracellular behavior was explored using TEM. The uninfected cells exhibited normal morphological appearance typical of type II AECs with numerous organelles (Fig. S1a) and surfactant bodies (Fig. S1b). Time–kinetics experiment indicated that 15-min postinfection, bacteria were located near the structures resembling coated pits and were bound to thin pili-like extensions of host cell membrane containing microvilli (Fig. 2a). Few internalized bacteria were seen at the tip of the cell by 1 h (Fig. 2b). After 2 h, the uptake process was found to be accompanied by the formation of the membrane bound vacuoles enclosing few bacteria (Fig. 2c). After 4 h, it was observed that the number of membrane bound vacuoles increased with many internalized bacteria (Fig. 2d). Interestingly, after 8 h, vacuoles were found to be empty and degraded intracellular bacteria free in the cytoplasm (Fig. 2e). Furthermore, for longer infection time, that is, after 12 h, the morphological features characteristics of apoptotic cells were manifested clearly, with intense chromatin condensation and intact plasma membrane (Fig. 2f). Beyond 24 h of infection, damaged cellular morphology with reduced size and lack of surface projections and profound fusion of endocytic vacuoles with expulsion of bacteria was observed (Fig. 2g).
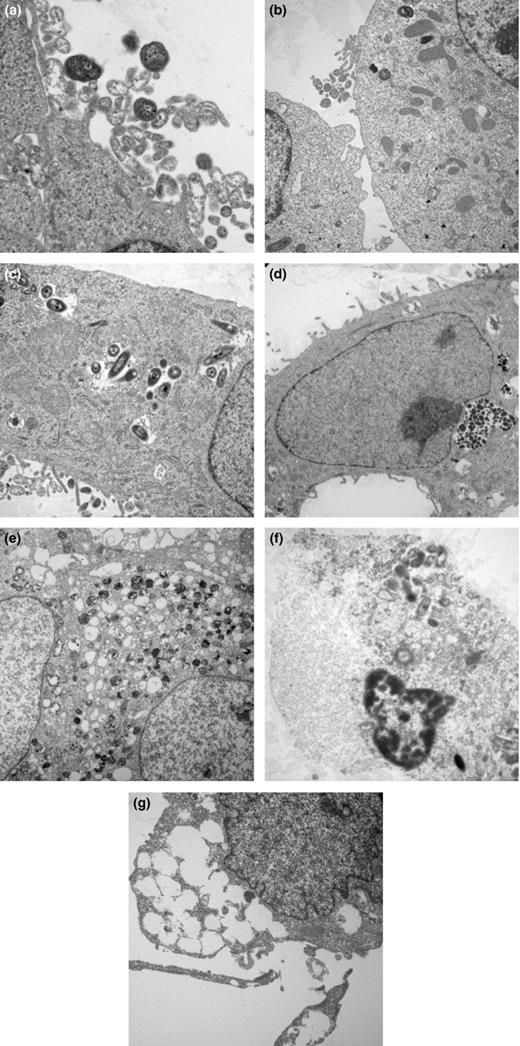
Cytopathic changes induced in A549 cells during interaction with NTHi strain. A549 cells were infected with NTHi strain at MOI of 1 : 10, fixed at various time points after infection and examined by transmission electron microscopy (a) 15 min (×6450), (b) 45 min (×3000), (c) 2 h (×3900), (d) 4 h (×3000), (e) 8 h (×1800), (f) 12 h (×2325), and (g) 24 h (×3900). Images shown are representative of two independent experiments.
Detailed examination of TEM micrographs has revealed the extensive formation of membrane bound vacuoles and degradation of intracellular bacteria. Therefore, we further examined the possibility if these vacuoles were derived from autophagosomes. To confirm, AO was used to visualize the volume of cellular acidic compartments by fluorescent microscope. Uninfected cells showed very few orange-stained vesicles. However, kinetic study showed visible increase in number of vesicles from 2 to 6 h postinfection. However, after 8 h, cells showed weakly stained vesicles as monolayer started to lose its integrity from substratum with more pronounced effect after 12 h of infection (Fig. 3a).
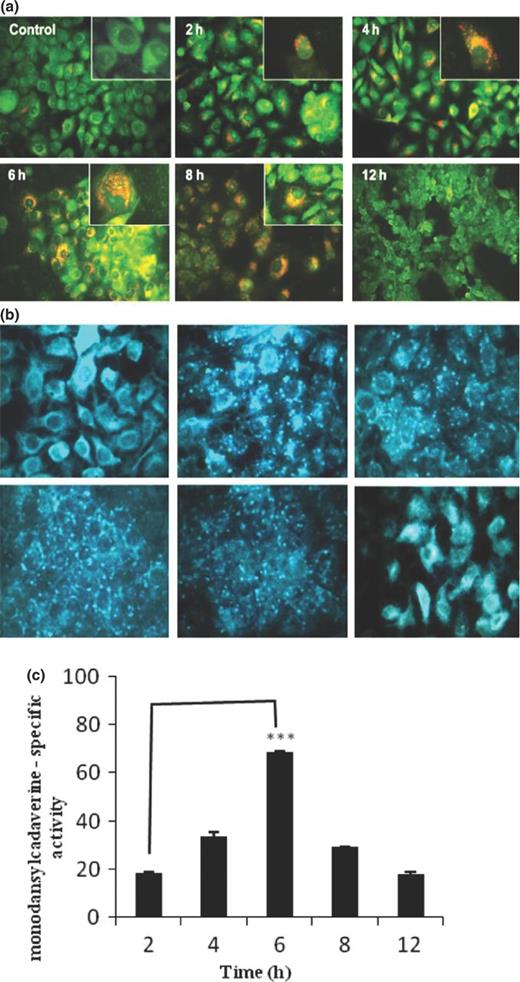
Evaluation of autophagic vacuole formation in A549 cells infected with NTHi strain. After infection for indicated times (2, 4, 6, 8, and 12 h), A549 cells were washed with PBS and incubated with fresh medium containing (a) AO (1 mg mL−1) for 15 min and (b) 0.05 mM monodansylcadaverine in PBS for 10 min, washed with cold PBS and immediately examined by fluorescence microscopy. Uninfected cells were taken as control. Representative images were obtained from two independent experiments. (Inset: Enlarged microscopic view, 100×). (c) After incubation with 0.05 mM monodansylcadaverine at 37°C for 10 min, cells were washed with PBS and collected in 10 mM Tris-HCl, containing 0.1% Triton X-100. The amount of incorporated monodansylcadaverine was quantitated using Cary Eclipse fluorometer. The data represent the mean ± SEM of three independent experiments. ***P ≤ 0.001 indicates statistically significant difference between time points of infection.
Assessment of autophagic vacuole formation in A549 cells
To further substantiate our finding, we examined the labeling of cytoplasmic acidic vacuoles by monodansylcadaverine staining. We observed that monodansylcadaverine concentrated in spherical structures distributed in cytoplasm of infected cells in contrast to the weak, diffuse, overall cytoplasmic staining in uninfected cells. Interestingly, NTHi strain showed a marked increase in number of monodansylcadaverine-stained vesicles from 2 to 6 h postinfection (Fig. 3b). Similar morphological changes were observed 8 h postinfection, as noticed in previous experiments. To quantify the number of autophagic vacuoles, we analyzed the amount of monodansylcadaverine incorporation in infected cells. As shown in Fig. 3c, the amount of monodansylcadaverine accumulation in autophagic vacuoles significantly increases from 18.45 ± 0.514% at 2 h to 68.407 ± 0.725% at 6 h postinfection, with a remarkable decrease afterward.
Cytopathic effects by TEM studies have shown that infected Type II AECs (A549) revealed morphological changes consistent with apoptosis. Further experiments were carried out to determine whether apoptosis was a cause of cytotoxicity in these cells.
PI uptake assay for assessment of dead cells
The cell death at various time periods (8, 12, 18, and 24 h) was assessed by PI uptake assay. As shown in Fig. 4a, percentage of dead cells significantly increases from 12.09 ± 0.97% at 8 h to 52.16 ± 2.05% at 24 h postinfection.
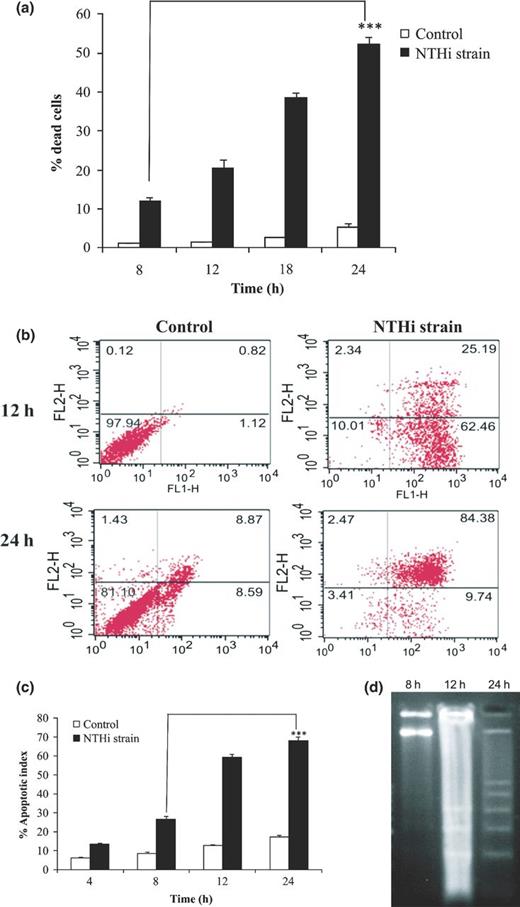
Assessment of cell death in A549 cells infected with NTHi strain. A549 monolayers were infected with NTHi strain at MOI of 1 : 10 for desired time intervals. (a) Cells were harvested at indicated time points (8, 12, 18, and 24 h) after infection, fixed with 1% paraformaldehyde, and permeabilized in hypotonic buffer for 15 min. Finally, cells were stained with PI for 20 min at 4 °C in dark and analyzed by flow cytometry. Uninfected cells cultured for indicated time points served as control. Data shown are mean ± SEM of three independent experiments. (b) A549 cells were harvested after 12 and 24 h of infection and exposure of membrane phosphatidylserine was analyzed using dual staining (annexin V-FITC and PI) by flow cytometry. The dot plots represent fluorescent intensities of annexin V-FITC (FL-1H) and PI (FL-2H)-labeled cells on respective axis. The respective cell populations were delineated to eliminate background signals originating from cell debris. To assess background fluorescent signals from the tested cell populations, nonstained samples were included. Uninfected A549 cells cultured for indicated time points served as control. Dot plots shown were from one of three independent experiments. (c) Infected A549 cells were harvested at indicated time interval (4, 8, 12, and 24 h), stained with combined dye of EtBr and AO (100μg mL−1 each), and immediately analyzed by fluorescent microscopy. The results expressed as the mean percentages of apoptotic cells per 500 cells enumerated from three separate experiments. Uninfected cells cultured for indicated time points were taken as control. (d) DNA was isolated from A549 cells after 8, 12, and 24 h postinfection and analyzed for internucleosomal DNA fragmentation by 1.8% agarose gel electrophoresis. Lane M represents 100 bp DNA ladder. ***P ≤ 0.001 indicates statistically significant difference between time points of infection.
Quantification of apoptotic cells
To further confirm the nature of cell death, we assessed the extent of apoptosis using annexin V-FITC apoptosis kit. Both the uninfected and infected cells after 12 and 24 h postinfection showed low levels (< 5%) of PI staining (necrotic cells). A considerable increase in percentage apoptotic cells (early apoptotic, 62%) at 12 h was observed as compared to uninfected cells (1%). However, 24 h postinfection, a significantly higher percentage of apoptotic cells (early + late apoptotic, 84%) was observed as compared to uninfected cells (9%) (Fig. 4b). This result indicated that during transition from 12 to 24 h, infected cells had entered from early apoptosis to late apoptosis.
To substantiate the above findings, AO/EtBr staining of infected cells at various time periods (4, 8, 12, and 24 h) was performed. Figure S2 depicts the four different cell populations – normal, early apoptotic, late apoptotic, and necrotic. In agreement with the previous results, the number of apoptotic cells (early + late) increased significantly with increase in duration of infection, that is, 26.5 ± 1.76% at 8 h to 68.25 ± 2.12% at 24 h postinfection (Fig. 4c).
DNA fragmentation analysis
Formation of internucleosomal DNA ladder is a hallmark of apoptosis. No DNA laddering was observed in cells after 8 h of infection; however, laddering was observed at 12 h, which was more pronounced at 24 h postinfection (Fig. 4d).
Effect of cytochalasin D on apoptosis of A549 cells
To determine whether bacterial internalization was required for NTHi-induced apoptosis of epithelial cells, uninfected A549 cells were pretreated with cytochalasin D (a specific inhibitor of F-actin polymerization). This agent blocked internalization of NTHi strain in cells by c. 92–95% (P < 0.001) at a concentration of 1μg mL−1 as compared to untreated control (Table S2). Further, we checked the level of apoptosis in these cytochalasin D-pretreated cells by FACS. The level of apoptosis was reduced markedly (c. fivefold) in infected cells treated with cytochalasin D as compared to untreated cells (Fig. 5). This showed that NTHi internalization is indispensable for host cell death.
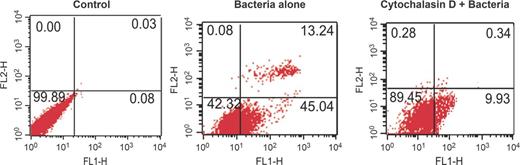
Evaluation of apoptosis of A549 cells preincubated with cytochalasin D prior to NTHi infection. For invasion blocking experiment, A549 cells were either nontreated or pretreated with cytochalasin D inhibitor (1μg mL−1) for 1 h at 37°C prior to infection with NTHi strain for 2 h. The level of apoptosis in cytochalasin D-pretreated and nontreated cells was checked using annexin V-FITC apoptosis detection kit as described above. The uninfected cells without exposure to inhibitor were taken as negative control. The dot plots represent fluorescent intensities of annexin V-FITC (FL-1H) and PI (FL-2H) on respective axes. Dot plots were obtained from FACS analysis of one experiment, representative of three independent experiments.
DISCUSSION
NTHi is an astute bacterial pathogen most commonly associated with asymptomatic carriage, but under predisposing conditions develops strategies to cause persistent infections of the respiratory tract. Relative to the interest in attachment and internalization of NTHi, this study highlights the succeeding steps in the pathogenesis including the intracellular life of NTHi and fate of infected epithelial cells.
In this study, among several NTHi strains tested, COPD strains shown the maximum ability to adhere and invade AECs. Several studies have suggested the existence of enormous strain-to-strain heterogeneity in terms of NTHi invasion abilities (Ketterer et al., 1999; Swords et al., 2000; Hotomi et al., 2010). A few studies have also reported that COPD strains adhered to host cells in greater numbers as compared to colonizers (van Schilfgaarde et al., 2000; Chin et al., 2005).
Kinetic experiment by viable plate count assay and light microscopy demonstrated reduced adherence and invasion by 8 h with clear features of cellular injury of epithelial cells, suggesting damage of necessary or complimentary epithelial receptors required for efficient NTHi binding. Intracellular survival assay showed stable numbers of intracellular NTHi until 6 h postgentamicin, providing evidence that once internalized, NTHi did not replicate within epithelial cells. However, a sharp decrease by 8 h demonstrated degradation/killing of intracellular NTHi. Morey et al. (2011) have also suggested nonproliferative intracellular stage of NTHi maintained for 6 h, with a decrease afterward. We were able to enumerate the viable number of intracellular NTHi until 8 h as monolayer begins to lose its integrity at later time points of infection. Recently, Clementi et al. (2014) reported killing of intracellular NTHi in lysosomes 24 h postinfection. Differences observed in terms of time points of infection between our data and previous reports could be due to variation in experimental conditions, procedures, host cell types, NTHi strains, and MOI used.
TEM studies carried out to document the cytopathic effects shed light on important observations of host pathogen interplay. It is important to know that at early time points of infection, most of the NTHi reside inside membrane bound vacuoles. Previous studies have also demonstrated that intracellular NTHi resides in vacuoles of epithelial cells, providing temporary or long-term niche for protection against immune clearance mechanisms (Clementi and Murphy 2011). Morey et al. (2011) also provided experimental evidence showing intracellular survival of NTHi inside subcellular compartments named as NTHi-containing vacuoles (NTHi-CV). Collectively, our data provide evidence that type II alveolar cells act as temporary reservoirs of NTHi during infection.
Furthermore, the transition period from 8 to 12 h was characterized by critical features pointing toward NTHi-mediated cytotoxicity of infected host cells via apoptosis. The characteristic apoptotic events were confirmed using various apoptotic markers such as expression of phosphatidylserine and DNA fragmentation. A number of studies have addressed the question of whether infection of AECs interferes with apoptosis, which play a crucial role in the pathogenesis of bacterial infection (Zychlinsky and Sansonetti 1997). Several studies have demonstrated that infection by wide range of pathogens such as Streptococcus pneumoniae (Marriott and Dockrell 2006), Legionella pneumophila (Gao and Abu Kwaik 1999), and Mycobacterium tuberculosis (Danelishvili et al., 2003) has been associated with apoptosis of human epithelial cells. Recent discoveries have also revealed that for pathogens that have evolved an intracellular habitat, escaping the confines of an infected host cell is a crucial stage in microbial pathogenesis for dissemination and avoiding an infectious dead end (Hybiske and Stephens 2008). An array of escape mechanisms can be exploited by diverse pathogens such as lysis of host cell (Hybiske and Stephens 2007), protrusion into and engulfment by neighboring cells (Yoshida et al., 2006), exocytic fusion (Ma et al., 2006), extrusion into membrane bound compartments (Hybiske and Stephens 2007), and induction of either proinflammatory or apoptotic cell death (Fink and Cookson 2005). Our data, for the first time suggest that induction of apoptosis could be an important strategy employed by NTHi to egress/escape from intracellular destination in host cells.
Autophagy is an emerging pathway of intracellular host cell defense machinery that bacteria must confront upon cell invasion. Several pathogens can manipulate the autophagy pathway as a strategy to establish persistent infection (Orvedahl and Levine 2009). The intracellular lifestyle of NTHi including persistence of NTHi inside acidic vacuoles, following its degradation and escape into cytoplasm has pointed toward the role of autophagy in this study. Also, the appearance of bacteria in cytoplasm coincides with features of host cell death. A growing number of intracellular pathogens, such as Listeria monocytogenes, Francisella tularensis, and Shigella flexneri, group A Streptococcus escape out of the phagocytic vacuoles into the cytosol of infected cells (Kumar and Valdivia 2009; Ray et al., 2009). Interestingly, AO and monodansylcadaverine staining have revealed the nature of acidic vacuolar compartments containing large numbers of NTHi to be derived from autophagosomes. Recent studies have suggested the role of lysosomal machinery (lysotracker, lamp-1, lamp-2, CD63, and Rab7) (Morey et al., 2011; Clementi et al., 2014) but no autophagic markers had been linked with NTHi vacuolar uptake. Future studies are required to broadly monitor the role of autophagy during NTHi infection using more rigorous markers.
It is unclear, however, whether host or bacterial cell factors are responsible for induction of apoptosis of epithelial cells. Cytoskeleton depolymerization using cytochalasin D inhibits NTHi invasion into epithelial cells (St Geme and Falkow 1990; Holmes and Bakaletz 1997; Ahren et al., 2001). Interestingly, inhibition of NTHi internalization by cytochalasin D prevented apoptosis of AECs, indicating that internalization of NTHi is decisive for induction of apoptosis of epithelial cells, similar to reported data for Staphylococcus aureus (Menzies and Kourteva 1998).
The ability of invasive NTHi to survive in an intracellular niche, that is, acidic vacuoles during beginning of infection, would facilitate NTHi to resist the antibiotic therapy and evade host immune response. In addition, at later stages of infection, the induction of apoptosis of infected cells could benefit NTHi by eliminating epithelial cells to allow deeper tissue penetration within the respiratory tract. Or perhaps NTHi escape from dead cells could initiate a new round of host cell infection, thus avoiding the infectious dead end; and prolonged disease process particularly in case of chronically colonized COPD patients with deficient immune system. This study also suggested that by directly penetrating the alveolar epithelial lining of an infected lung, NTHi might have the ability to gain access to the host's lymphatic and circulatory system. Moreover, the tissue damage of alveolar cells may have harmful effects on alveolar permeability leading to alveolar filling and worsening oxygenation. Our study will have an important implication in understanding the disease pathology of NTHi and may help to explain the clinical picture of severe exacerbations episodes characteristic of COPD patients. The intracellular lifestyle of NTHi and its ability to induce programmed cell death in AECs and thus orchestrate normal host cell processes for its own advantage suggests that NTHi is a more versatile and adaptive pathogen than previously believed.
The unique requirements for intracellular survival and vacuolar escape of NTHi during infection process need to be rigorously proven by experimentation. Future research in this direction will help us to better understand the underlying mechanism of induction of apoptosis and the anti-internalization strategy to prevent NTHi infections.
The first author M.G. acknowledges the financial assistance of Senior Research Fellowship from the Indian Council of Medical Research, New Delhi, India. We would also like to thank Dr C. S. Ryat and Ms Meena, Department of Histopathology, PGIMER, Chandigarh for electron microscopic examination.
Conflict of interest statement. None declared.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Transmission electron micrographs of uninfected A549 cells.
Fig. S2. Identification of different cell populations during apoptosis of A549 cells infected with NTHi strain by AO/EtBr staining.
Table S1. Adherence and invasion abilities of NTHi strains isolated from different sources.
Table S2. Blocking of actin filaments by Cytochalasin D inhibits internalization of NTHi strains in A549 cells relative to untreated control.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}