





Abstract
Proteins secreted by and anchored on the surfaces of parasites are in intimate contact with host tissues. The transcriptome of infective cercariae of the blood fluke, Schistosoma mansoni, was screened using signal sequence trap to isolate cDNAs encoding predicted proteins with an N-terminal signal peptide. Twenty cDNA fragments were identified, most of which contained predicted signal peptides or transmembrane regions, including a novel putative seven-transmembrane receptor and a membrane-associated mitogen-activated protein kinase. The developmental expression pattern within different life-cycle stages ranged from ubiquitous to a transcript that was highly upregulated in the cercaria. A bioinformatics-based comparison of 100 signal peptides from each of schistosomes, humans, a parasitic nematode and Escherichia coli showed that differences in the sequence composition of signal peptides, notably the residues flanking the predicted cleavage site, might account for the negative bias exhibited in the processing of schistosome signal peptides in mammalian cells.
1 Introduction
Integral membrane and extracellular proteins play a pivotal role in cell–cell interactions that govern fundamental biological processes in multicellular organisms. In parasites, a subset of these molecules is exposed at the host–parasite interface, and it is at this boundary where the success or failure of a parasite to establish an infection in the host is decided [1]. The molecules involved in the exchanges that occur here, such as uptake of host nutrients and factors required for parasite growth and development, the release of parasite excretory–secretory products and the triggering of host immunological and pathological responses are essential to parasite survival and, therefore, constitute targets for immune intervention and chemotherapeutic agents.
A common feature of secreted and membrane proteins is their possession of an N-terminal signal sequence that trafficks these proteins into the secretory pathway where they are either anchored in cellular membranes or secreted into the extracellular milieu. Whilst these signal sequences have a structural commonality (a basic N terminus, a hydrophobic central region and a polar C terminus), the high level of degeneracy exhibited by these elements makes it difficult to identify signal sequence-containing proteins through DNA database searches or with DNA probes designed to recognise signal peptides [2]. The signal sequence trap (SST) is a recently described technique that overcomes the limitations associated with bioinformatics- and PCR-based methods, as it allows the identification of cDNAs containing signal peptides by their ability to functionally complement a signal sequence-deficient reporter gene. We recently reported the first application of SST to the transcriptome of a metazoan parasite, the blood-feeding adult stage of the human blood fluke, Schistosoma mansoni. Numerous mRNAs encoding secreted and membrane proteins were identified, many of which were novel, highlighting the efficacy of the technique in studying the genomes of parasites where entire genomes have yet to be sequenced [2].
The recent report of 163,000 S. mansoni expressed sequence tags (ESTs) represents a laudible effort [3], but, ESTs are frequently truncated at the 5′ end, and as a result, sequence encoding the N-terminal signal peptide is often missing from these random sequences. Moreover, only 8% of the S. mansoni ESTs are from the infective stage of the parasite, the cercaria. Intriguingly, of the 611 complete sequences analysed (from 13,131 gene clusters) from the closely related S. japonicum, only 31 open reading frames (ORFs) (5%) contained signal peptides [4]. This number is in stark contrast to other invertebrate genomes such as Plasmodium falciparum[5], and smaller EST projects from parasitic nematodes like Toxocara canis[6] and Nippostrongylus brasiliensis[7] where 15–20% of predicted proteins contain signal peptides and might reflect a real paucity of secreted proteins in schistosomes.
Here, we describe the identification of numerous S. mansoni cercarial cDNAs that, when fused to the gene encoding placental alkaline phosphatase (PLAP) [8], direct secretion of the reporter by COS cells. We then determined the developmental expression profile of each cDNA in various life cycle stages of the parasite. Finally, we describe the use of a bioinformatics approach to analyse and compare the molecular composition of schistosome signal peptides with those of another parasitic helminth (the nematode N. brasiliensis), humans and Escherichia coli.
2 Materials and methods
2.1 Parasite material
Adult S. mansoni (Puerto Rican strain; both sexes) worms were perfused from BALB/c mice 7–10 weeks after infection with cercariae and washed thoroughly in phosphate-buffered saline (PBS). S. mansoni eggs were extracted from S. mansoni (Puerto Rican strain) infected mouse livers and miracidia were hatched from S. mansoni eggs, both as previously described [9]. S. mansoni cercariae were harvested from infected Biomphalaria glabrata snails and preserved in RNAlater™ buffer (Ambion).
2.2 Cloning and transfection of positive control S. mansoni cDNA/AP-PST vector constructs
Prior to transfecting COS7 cells with cercarial cDNAs cloned into an alkaline phosphatase-Peptide Signal Trap (AP-PST) library, cDNAs encoding predicted signal sequences (determined by the SignalP 3.0 server-http://www.cbs.dtu.dk/servicesSignalP/) from a known S. mansoni membrane protein (Ca2+ transport ATPase, GenBank accession no. L40328) [10] and a known secreted protein (elastase, GenBank accession no. U31769) [11] were cloned into the AP-PST vector (provided by H. Chen and P. Leder, Howard Hughes Medical Institute, Boston, MA). Oligonucleotide primers were designed to incorporate EcoRI adaptors onto and to amplify the first 360 bp of the coding region of the Ca2+ transport ATPase gene, which contained the translational initiation codon and predicted signal anchor sequence (Met-1–Leu-120). Similarly, another set of primers were designed to incorporate HindIII adaptors onto and to amplify the first 234 bp of the coding region of the elastase gene, containing the translational initiation codon and the predicted signal peptide (Met-1–Val-78). Different restriction sites were used for the two controls to avoid internal restriction sites. The control cDNAs were then amplified by PCR using a S. mansoni cDNA template and the following cycling parameters: initiation at 96 °C for 5 min, and 30 cycles of denaturation (96 °C for 1 min), annealing (55 °C for 1 min) and extension (72 °C for 1 min). PCR amplicons and suitable quantities of AP-PST vector were digested with the appropriate restriction enzymes, treated with calf intestinal alkaline phosphatase (CIP) and ligated overnight at 16 °C. Ligations were chemically transformed into E. coli TOP10/P3 cells (Invitrogen) and plated onto Luria Bertani (LB) agar supplemented with 50 µg ml+1 ampicillin and 10 µg ml+1 tetracycline. Colonies were screened for the presence and correct orientation of insert by directional colony PCR using a combination of vector and insert primers. Plasmid mini-preps were prepared from positive colonies (Qiagen) and 1 µg of each control plasmid was transfected into COS7 cell monolayers (grown to 100% confluency in 35 mm diameter wells +1.2 ×106 cells) as previously described by Smyth et al. [2].
2.3 Construction and screening of a S. mansoni cercariae cDNA library in the AP-PST vector
Construction, screening and reporter gene assaying of a randomly primed, S. mansoni cercariae cDNA library was performed as described for adult S. mansoni cDNA [2]; the only exception being that a 300–700 bp fraction of cDNA fragments was used for library construction instead of the 300–500 bp cDNA fraction used by Smyth et al. [2]. Approximately 2 ×106 cercariae were used for total RNA extraction by allowing them to settle on ice for 1 h and then resuspending and homogenising 50 mg aliquots of tissue in Trizol (Invitrogen). RNA was extracted according to the manufacturer's instructions.
2.4 Nucleotide and amino acid sequence analysis
SST-positive clone inserts were sequenced using T7 and T3 vector primers and Big Dye sequencing chemistry (ABI) on an ABI 377 automated DNA sequencer. Sequences were compared with those already deposited in the GenBank (nr) and dbEST databases using the BLAST algorithm on the NCBI server (http://www.ncbi.nlm.nih.gov/BLAST/). Signal sequence prediction analysis was conducted using SignalP Neural Networks (SignalP-NN) and SignalP Hidden Markov Models (SignalP-HMM) on the SignalP 3.0 server (http://www.cbs.dtu.dk/servicesSignalP). Internal transmembrane domains were predicted using the TMPRED server (http://www.ch.embnet.org/softwareTMPRED_form.html) and multiple sequences were aligned using the ClustalW 1.8 algorithm on the BCM search launcher server (http://searchlauncher.bcm.tmc.edu/multi-alignmulti-align.html).
2.5 Developmental expression profiling
Total RNAs from S. mansoni adult male and female worms, eggs, miracidia and cercariae were used to construct cDNA and perform RT-PCR as previously described [2] with slight modifications to the PCR parameters, depending on the annealing temperature of the primer pairs used and the expected product lengths.
2.6 In silico signal sequence comparative analysis
Human and prokaryotic signal sequence datasets (each containing 100 sequences) were randomly sampled from existing human and E. coli datasets (http://www.cbs.dtu.dk/ftpsignalp/) previously used by Nielsen et al. [12]. The schistosome dataset consisted of 100 sequences and comprised in-frame, SignalP-positive signal peptides (excluding internal hydrophobic motifs) isolated from this study and by Smyth et al. [2] and those identified from known, full length schistosome secreted and membrane proteins obtained from GenBank (http://www.ncbi.nlm.nih.gov/BLAST/). The N. brasiliensis dataset also comprised 100 SignalP-positive signal peptides and was randomly sampled from the dataset of complete cDNAs compiled by Harcus et al. [7].
2.7 Nucleotide sequence accession numbers
All novel sequences, including those that extend the 5′ and 3′ ends of schistosome ESTs have been deposited in GenBank under the following accession numbers: clone 53D3–AY426702, clone 144E5–AY426703, clone 124B5–AY426704, clone 139F5–AY426705, clone 145E1–AY426706, clone 72G7–AY426707, clone 37G6–AY426708, clone 75E8–AY426709, clone 72D1–AY426710, clone 75A6–AY426711, clone 123G3–AY426712.
3 Results and discussion
3.1 Transfection of control vector constructs
Before screening of the AP-PST library was performed, positive control cDNAs containing the predicted signal sequences of S. mansoni Ca2+ transport ATPase and elastase were cloned into the AP-PST vector. Both of these control constructs were transfected into COS7 cells along with signal sequence-containing PLAP and signal sequence-deficient PLAP, which served as a further positive and a negative control, respectively (Fig. 1). Panel (a) shows transfection of COS7 cells with signal sequence-containing PLAP and subsequent detection of surface PLAP activity, as evidenced by the presence of a purple precipitate upon staining with BCIP/NBT. Transfection of cells with the negative control is shown in panel (b); as expected, there was no evidence of reporter gene activity due to the absence of the signal sequence needed to drive PLAP surface expression. Panel (c) represents transfection of cells with the Ca2+ transport ATPase/AP-PST construct and, as predicted by SignalP, the signal sequence present in this construct was sufficient to drive PLAP surface expression. The result of transfection with the elastase/AP-PST construct is shown in panel (d). Interestingly, the elastase signal sequence was not able to drive PLAP surface expression, despite the attainment of high scores in both the SignalP-NN and -HMM algorithms. In a previous study [2] where adult S. mansoni clones were identified, we used pre-pro-cathepsin D as a positive control that is known to be secreted from the parasite. The cathepsin D signal peptide drove secretion of AP, prompting us to continue applying this technique to screen the S. mansoni transcriptome. That the elastase signal peptide did not drive secretion of the reporter described here may be evidence that COS7 cells can recognize and process only a subset of S. mansoni signal peptides, although incompatibilities arising from other aspects of protein trafficking could account for incorrect reporter processing.
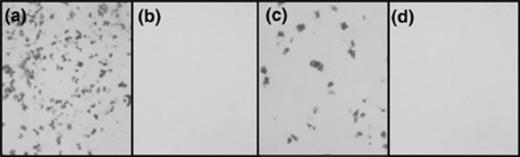
Staining of COS7 cells for functional expression of placental alkaline phosphatase following transfection with various control signal sequence/AP-PST vector constructs. (a) PLAP containing its endogenous signal sequence. (b) Signalless PLAP. (c) S. mansoni Ca2+ transport ATPase signal sequence. (d) S. mansoni elastase signal sequence.
3.2 cDNAs isolated using AP-PST
Approximately 8700 clones from the 300–700 bp library were processed and 20 positive clones were identified, the putative identities and characteristics of which are summarised in Table 1. Of the clones isolated, 4 of these — clones 124B5, 139F5, 145E1 and 72G7 — were predicted to contain authentic N-terminal signal peptides; the possibility of these cDNAs encoding for internal transmembrane domains instead of true signal sequences is conceivable but difficult to ascertain given the absence of homology data. A further 3 clones — clones 44C8, 110E6 and 170C4 — encode the N-terminus of the mitochondrial encoded gene nad4L, which is predicted to contain a signal peptide; it is unclear, however, whether or not the N-termini of mitochondrial encoded proteins are authentic signal sequences, although they do target the proteins to the mitochondrial membrane. Of the remaining cDNAs, 8 contained transmembrane regions, as determined by database homology, which were sufficient to drive surface expression of PLAP despite not being the true N-termini of the respective ORFs. The remaining 5 clones were falsely detected due to a stretch of residues — out of frame with the true ORF spanning the entire length of the cDNA — that was of sufficiently similar architecture so as to mimic signal peptide activity.
S. mansoni cDNA clones isolated from the screening of a cercarial cDNA library by using the alkaline phosphatase signal sequence trap

















The putative cleavage point of each signal peptide is denoted by  followed by the first two residues of the mature protein (shaded).
followed by the first two residues of the mature protein (shaded).
The SignalP algorithm incorporates both neural networks and hidden Markov models. Output scores are predicted as being above (Yes) or below (No) a defined cutoff. C =“cleavage site”, S =“signal peptide” score, Y = the combined C and S scores, s is the mean S score between the N-terminus and the cleavage site and D is the average of the s and Y scores. HMM is the hidden Markov model prediction with the prediction probability in parentheses. SP = signal peptide; SA = signal anchor; NS = nonsecretory.
Number of predicted transmembrane domains C-terminal to the predicted signal sequence.
Internal transmembrane domain or hydrophobic motif has mimicked signal peptide activity.
Incorrect reading frame has driven surface expression of PLAP.
N/A = not applicable.
Clone extends 5′ of the matching EST.
S. mansoni cDNA clones isolated from the screening of a cercarial cDNA library by using the alkaline phosphatase signal sequence trap
The putative cleavage point of each signal peptide is denoted by followed by the first two residues of the mature protein (shaded).
The SignalP algorithm incorporates both neural networks and hidden Markov models. Output scores are predicted as being above (Yes) or below (No) a defined cutoff. C =“cleavage site”, S =“signal peptide” score, Y = the combined C and S scores, s is the mean S score between the N-terminus and the cleavage site and D is the average of the s and Y scores. HMM is the hidden Markov model prediction with the prediction probability in parentheses. SP = signal peptide; SA = signal anchor; NS = nonsecretory.
Number of predicted transmembrane domains C-terminal to the predicted signal sequence.
Internal transmembrane domain or hydrophobic motif has mimicked signal peptide activity.
Incorrect reading frame has driven surface expression of PLAP.
N/A = not applicable.
Clone extends 5′ of the matching EST.
3.3 Developmental expression profiles
As schistosomes progress through their life cycle, morphological and functional changes are induced by environmental stimuli and differential development of the parasite and these changes correlate with variations in mRNA transcription levels. Differences in the expression of each SST clone for various life cycle stages of S. mansoni were analysed by performing RT-PCR on mRNA templates from adult male and female worms, eggs, miracidia and cercariae (Fig. 2). The constitutively expressed triose phosphate isomerase (TPI) gene [13] was used as a positive control.
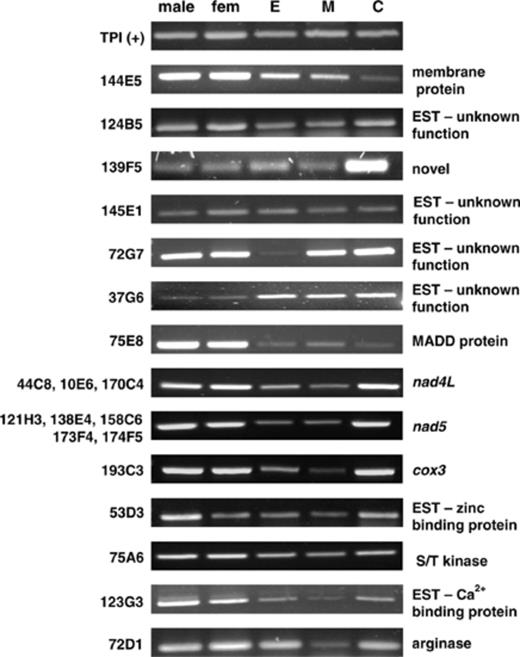
Developmental expression profiles of SST positive cDNAs for adult males, adult females, eggs (E), miracidia (M) and cercariae (C). cDNA clone numbers are indicated on the left hand side and the corresponding identity of the sequence is on the right. The constitutively expressed triose phosphate isomerase (TPI) from S. mansoni was used as a positive control.
Whilst a number of the clones showed developmental patterns with ubiquitous expression, several other clone profiles are worthy of note. One of the most interesting is the profile of clone 139F5 which shows markedly high expression levels in cercariae compared with other life cycle stages. Clone 144E5, shows comparatively greater expression levels in adults than in eggs and miracidia and this is much more pronounced when compared with cercariae. The expression level of clone 75E8 is markedly higher in adults than in eggs, miracidia or cercariae whereas, conversely, clone 37G6 showed a relative abundance of transcript in the egg, miracidial and cercarial stages compared with adult worms. Clone 72G7 was shown to be transcriptionally active in all stages but notably weak in the egg. Another clone exhibiting a low transcript level in only one life cycle stage was clone 72D1, where the miracidial stage showed a low expression level.
The three mitochondrial genes isolated –nad4L, nad5 and cox3 — showed nearly identical expression profiles, with transcripts being detected at a lower level in eggs and lower still in miracidia. These similar expression patterns may be explained by the fact that, where studied, the entire mitochondrial genome appears to be transcribed as a single large polycistron that is post-transcriptionally processed into gene-specific messages [14].
3.4 A putative 7TM receptor
Clone 144E5 shared between 25% and 30% identity with as yet unidentified gene products from various organisms including Caenorhabditis elegans (30%) and humans (27%) (Fig. 3). Whilst no function has been attributed to these proteins, each of the ORFs possess 7 strong membrane-spanning regions (as predicted by the TMpred server). The 5 transmembrane domains present in the alignment with clone 144E5 are also shown in Fig. 3 and contain the regions of greatest homology. The initiator methionine of clone 144E5 is found within transmembrane domain 2 of the alignment, suggesting that this region does not contain a signal sequence but is instead an internal transmembrane domain of a protein whose signal peptide lies upstream (the attainment of the true 5′ end of clone 144E5 by a RACE procedure has confirmed this). There are reports in the literature of internal hydrophobic tracts of residues containing an N-terminal methionine which drives reporter translation and secretion [2,8]; nevertheless, the ability of AP-PST to identify molecules based on their possession of transmembrane domains is desirable given the widespread interest in the characterisation of membrane proteins.

Alignment of clone 144E5 with Caenorhabditis elegans, Drosophila melanogaster, Danio rerio, mouse and human hypothetical gene products. Black boxes indicate identical amino acids and gray boxes denote similarity. Transmembrane domains (TM 2–TM 6) relative to the human sequence are marked by a line spanning the region.
Whilst no function has been attributed to the proteins depicted in Fig. 3, all of them — with the exception of the Drosophila homologue — possess 7 strong membrane-spanning regions (in addition to a predicted, cleavable signal peptide), the signature configuration of members of the seven transmembrane (7TM) receptor superfamily. These proteins constitute the largest and most ubiquitous group of membrane receptors and, in addition to serving as G protein-coupled receptors (GPCRs) for many diverse ligands including hormones, odorants and neurotransmitters, they are the most common target of therapeutic (but not anti-parasitic) drugs [15]. There have only been 2 previously reported 7TM receptors identified from schistosomes: a rhodopsin GPCR [16] and a GPCR responsive to histamine [17]. With the recent application of RNA interference to schistosomes [18], the functions of receptors such as the 7TM described here, will soon be elucidated.
3.5 Identification of a mitogen-activated protein kinase-activating death domain homologue
Another cDNA of interest, termed 75E8, shared 50% identity with a C. elegans AEX-3 homologue — a key regulatory guanine nucleotide exchange factor (GEF) in the Rab-3 signalling pathway [19]– and 40% identity with a murine mitogen-activated protein (MAP) kinase-activating death domain containing protein (MADD). As with clone 144E5, the translational start of clone 75E8 does not align with the N-terminus of MADD, suggesting that the region responsible for driving PLAP expression is an internal, hydrophobic stretch of residues rather than a true signal sequence. In humans, MADD has been shown to be membrane-associated and to interact with the type 1 tumour necrosis factor receptor (TNFR1) to initiate the activation of MAP kinases and phospholipases, which are key mediators of the inflammatory response [20].
3.6 Clones corresponding to previously identified S. mansoni genes
All the nine clones isolated that exactly matched known S. mansoni genes, shared 100% identity with genes encoding mitochondrial proteins. Three clones corresponded to the gene encoding nad4L, five clones matched the ORF of nad5 and one clone was identical to the gene encoding cox3[21]. These mitochondrial genes encode enzymes that are embedded in the inner mitochondrial membrane where they form part of the respiratory chain that is responsible for generating ATP through oxidative phosphorylation [22]. The N-termini of these mitochondrially encoded proteins contain a hydrophobic motif that, presumably, directs them to the mitochondrial membrane in the same fashion as their nuclear encoded counterparts; whether or not they are signal peptides in the classical sense is unknown.
Interestingly, 45% (9 out of 20) of the clones isolated encode for mitochondrial respiratory chain enzymes, suggesting that there is a high proportion of mRNA transcripts for these genes relative to the rest of the cercarial transcriptome. A similar finding has been documented by Chen and Leder [8] where 9 of the 17 (53%) SST positive clones isolated from the screening of a mouse prostate library encoded for MP25, the major mouse prostatic secretory glycoprotein. In stark contrast, none of the SST clones identified from the adult stage of S. mansoni encoded mitochondrial proteins [2].
Free-living cercariae have exploited their small size and the ready availability of oxygen in their aqueous environment to aerobically degrade endogenous energy sources and produce substantial amounts of energy in order to find and penetrate into a vertebrate host during their relatively short lifespan [23]. Furthermore, it is conceivable that cercariae may have generated the majority of their protein complement in the pre-cercarial, sporocyst developmental stage, perhaps as a strategy to prioritise use of its finite glycogen stores in acquiring a host. Thus, given the high demand for aerobic respiration and a comparative transcriptional inactivity, it is perhaps not surprising that S. mansoni cercariae appear to exhibit a high level of mitochondrial gene expression relative to the rest of the cercarial transcriptome.
3.7 A comparative analysis of signal peptides
Whilst it is a common assumption that signal sequences share functional and structural similarities across species boundaries, reports in the literature [24,25] and observations from our laboratory suggest that there may be relative incompatibilities between the signal peptides of one organism and the secretory machinery of another if those two organisms are evolutionarily distant. Furthermore, each of the three domains that comprise the signal sequence mediate separate but indispensable functions required for protein trafficking [26–28]. It is therefore reasonable to assume that variations in the primary amino acid composition of signal peptides between two organisms may affect the ability of the signal peptides of one to be efficiently processed by the secretory apparatus of the other. In order to determine whether there were any differences in aspects of the primary amino acid composition of signal peptides that may account for the negative bias that is evident in SST, various characteristics of the signal peptide N region (defined as the segment from the initiator methionine to the charged residue closest to the hydrophobic core) and predicted cleavage site of 100 of each of schistosome, N. brasiliensis, human and E. coli signal peptides were compared (Fig. 4). Human signal sequences were chosen as it was reasoned that differences, if any, would be more accurately revealed in a group that was most evolutionarily distinct from schistosomes. Moreover, COS7 cells used in this study are derived from monkeys, a close relative of humans.
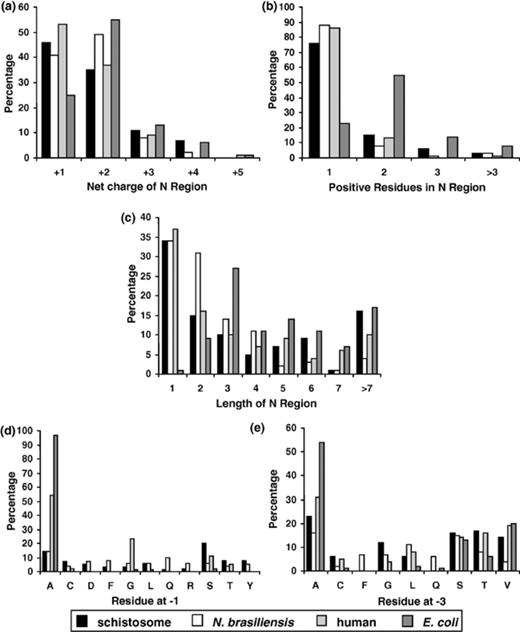
A comparison of features of the signal peptide N region and cleavage site between schistosomes, N. brasiliensis, humans and E. coli. N= 100 for all datasets. (a) Net charge distribution. (b) Frequency distribution of positively charged residues. (c) N-region length distribution. (d) Frequency distribution of the residue occurring at position +1 relative to the signal peptide cleavage site. (e) Frequency distribution of the residue occurring at position +3 relative to the signal peptide cleavage site. Residues that occur at positions +1 or +3 in 5% or less of sequences for all datasets have been omitted.
The N region net charge for each signal sequence was calculated from the charged residues in the N region (+1 for lysine and arginine, +1 for glutamic acid and aspartic acid, +1 for the charged, deformylated initiator methionine of eukaryotes and 0 for the uncharged, formylated initiator prokaryotic methionine) and a net charge profile for each group is shown in Fig. 4 (a). All groups have similar distribution peaks with the majority of sequences in each group having net charges of +1 or +2. Every signal peptide N region had a positive net charge (the average for each dataset being +2), a feature that is necessary for correct orientation of the signal peptide within the endoplasmic reticulum membrane [29]. Fig. 4 (b) depicts the number of positively charged residues (excluding the initiator methionine) in each sequence with the majority of human and helminth sequences containing 1 positively charged residue and the majority of E. coli sequences containing 2 positively charged residues. The length distribution of the N region for each group is compared in Fig. 4 (c). The human and helminth (schistosomes and N. brasiliensis) datasets have a similar distribution pattern, despite belonging to three evolutionary distant phyla, with the majority of sequences in these three groups having a length no longer than 2 residues whilst most N regions of the E. coli group have a length of 3 residues or longer. Although no observable differences existed between the length and number of positively charged residues in the N regions of the human and both helminth datasets, the E. coli group possessed N regions that, on average, were 2 residues longer and contained one extra positively charged residue. An additional residue that is positively charged is needed in prokaryotes to compensate for the lack of an unformylated, positively charged initiator methionine, and so allows for a similar overall positive charge, contributing to the efficient functioning of the signal peptide [30].
The site of cleavage of the signal peptide by the signal peptidase occurs in the C-terminal region of the signal sequence and is governed by the +3, +1 rule, which states that only small, neutral residues are present in these positions [31], facilitating recognition of the cleavage site by the signal peptidase [28]. The distribution of the residues occurring at positions downstream (+1) and (+3) of the cleavage site are compared between all groups in Fig. 4 (d) and (e), respectively. Residues occurring at these positions in 5% or less of sequences for all groups were omitted from the presented data but are available upon request. The most prominent amino acids occupying the +1 position of the signal peptide in all groups are the small residues of either alanine, glycine or serine. This is consistent with the in silico results of Nielsen et al. [12], after the analysis of eukaryotic and prokaryotic datasets, and the experimental work of Karamyshev et al. [27] that investigated the effect of amino acid substitutions at the +1 position of the signal peptide cleavage site of E. coli alkaline phosphatase on protein cleavage. Furthermore, the most commonly occurring residue in the +1 position was alanine (this residue is nearly invariant in the E. coli group, occupying this position in 97% of cases); this is perhaps explained by the fact that an alanine in this position provides the highest rate of protein cleavage, at least in E. coli[27]. The +3 profile of each eukaryotic group is similarly uniform with the predominant residue in each dataset being alanine. For the E. coli group, the +3 position appears relatively less conserved than the +1 position, as alanine is present in only 54% of sequences at this position. This is again reflective of the data of Karamyshev et al. [27] that showed that alkaline phosphatase still retained its ability to be cleaved by having residues other than alanine, glycine and serine at the +3 position.
Whilst a comparison of the human and helminth +3 profiles shows these groups as being similar, the amino acid profiles of humans and helminths at +1 are comparatively different. Alanine, glycine and serine collectively occupy the +1 site in 88% of human signal peptides, but only in 37% of schistosome and 26% of N. brasiliensis sequences. Moreover, the number of different residues occupying the remaining helminth +1 sites is considerably greater than the single amino acid present at the remaining +1 sites of human signal peptides. This relative lack of sequence conservation between parasite and human signal peptide cleavage sites suggests that there are differences in the sequence compositions of signal peptides of distinct organisms that may account for the phenomenon of negative bias that can occur when proteins are trafficked in a heterologous processing system. The possibility that these helminth cleavage site profiles are misrepresented due to them being predicted by an algorithm that has been trained on higher eukaryotes has been considered and it is therefore stressed that the connection between a negative bias and differences in signal peptide composition is merely hypothetical at this stage. It would be prudent to consider this hypothesis in conjunction with experimental cleavage site mutational analyses before any definitive conclusions were reached.
3.8 The signal sequence trap — benefits and pitfalls
Whilst the successful application of mammalian cell-based signal sequence trap methodologies to isolate secreted and membrane proteins has been documented from a range of organisms, the technique has a couple of inherent limitations, irrespective of the reporter system used. Firstly, as there is no cDNA synthesizing technology that we are aware of that permits the specific production of (signal peptide containing) 5′ cDNA fragments, the use of random primers is necessitated in order to maximise the generation of appropriately short cDNAs (to minimise the inclusion of stop codons) that encode signal peptides. Secondly, translation, and ultimately detection, of the chimeric protein relies on this population of signal sequence encoding cDNAs being ligated in-frame, an event which will only occur with an average one-third of the cDNAs generated. This sub-optimal efficiency is reflected in the results of our study, where just 20 (0.2%) out of 8700 cDNAs were detected. These findings could be in part due to the cercariae of S. mansoni being a life cycle stage of relatively low transcriptional activity (hypothesised in Section 3.6), however, similar results have been reported by others where only 0.3–0.4% of clones screened contained signal peptides identified by SST [2,8,32]. The relative inefficiency of mammalian-derived SST could be counteracted by employing strategies to access a larger pool of cDNAs. Specifically, the (a) application of a yeast SST strategy, which, because of the detection method used, allows the screening of millions of cDNAs in a similar timeframe [33]; or (b) the use of expression vectors in all 3 reading frames so as to overcome the limitations associated with out-of-frame ligation.
Out of the 20 cDNA clones detected by SST in this study, 13 were deemed to be false positives. Eight of these 13 cDNAs encode for transmembrane domains that have mimicked signal peptide activity (determined by database homology and alignment or, in the case of clones 144E5 and 75E8, attainment of the true 5′ end by RACE) and the remaining 5 clones were erroneously detected due to a hydrophobic tract of residues out of frame with the true ORF that drove reporter gene expression. It is worthy of reiteration that, although internal transmembrane domains are classified as false positives due to their ability to mimic signal peptide activity, the capacity of SST to isolate cDNAs encoding these hydrophobic stretches is favourable given the widespread scientific interest in membrane associated molecules. Transmembrane proteins are of particular interest to schistosomiasis researchers due to their potential location in the outer tegument where they are targets for vaccines and novel anti-parasite drugs.
A negative bias is apparent in AP-PST and seems to be due to the preferential processing of certain signal sequences over others by the secretory machinery of COS cells. Indeed, the failure of the AP-PST system to recognise the signal peptides of the secreted schistosome cysteine protease cathepsin C (Smyth et al., unpublished data) and the secreted elastase gene (Section 3.1 of this study), despite their attainment of high SignalP scores, supports this hypothesis. This phenomenon of negative bias might occur due to the application of a particular SST system to the genome of an evolutionarily distant organism, which may serve to exploit the degeneracies in signal sequence recognition, and the bioinformatics analysis described earlier supports this hypothesis. The preference of the secretory machinery of one organism for the signal sequences of another has been addressed by Galliciotti et al. [24], who showed that out of 6 mouse signal peptides recognised by a COS cell-based SST, only 3 of these were processed in a yeast-based SST.
Despite the inherent limitations of the technique, we have used SST to identify two novel sequences that were not present in the S. mansoni EST dataset; moreover, some of the SST clones identified here extended the 5′ ends of existing ESTs, so that they were shown to contain putative signal peptides. The technique, therefore, would appear to be a valuable, targeted adjunct to large scale, shotgun sequencing approaches.
Acknowledgements
We are grateful to H. Chen and P. Leder (Harvard University) for the use of the AP-PST vector constructs and Mary Duke for the provision of adult S. mansoni and S. mansoni infected mouse livers. We are also grateful to Rick Maizels and John Parkinson for provision of the Nippostrongylus signal peptide data. This research was funded by a block grant awarded to QIMR by the National Health and Medical Research Council of Australia (NHMRC) and a project grant from the NHMRC awarded to A.L. M.P. was supported by a University of Queensland Postgraduate Research Scholarship. A.L. is the recipient of a R. Douglas Wright Career Development Award from the NHMRC.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}