





Abstract
Mountain glaciers are frequently assessed for their hydrological connectivity from glaciers to proglacial lakes. Ecological process on glacier surfaces and downstream ecosystems have often been investigated separately, but few studies have focused on the connectivity between the different glacial habitats. Therefore, it remains a limited understanding of bacterial community assembly across different habitats along the glacier hydrological continuum. In this study, we sampled along a glacial catchment from supraglacial snow, cryoconite holes, supraglacial runoff, ice-marginal moraine and proglacial lake on the Tibetan Plateau. The bacterial communities in these habitats were analyzed using high-throughput DNA sequencing of the 16S rRNA gene to determine the bacterial composition and assembly. Our results showed that each habitat hosted unique bacterial communities, with higher bacterial α-diversity in transitional habitats (e.g. runoff and ice-marginal moraine). Null model analysis indicated that deterministic processes predominantly shaped bacterial assembly in snow, cryoconite holes and lake, while stochastic process dominantly governed bacterial community in transitional habitats. Collectively, our findings suggest that local environment play a critical role in filtering bacterial community composition within glacier habitats. This study enhances our understanding of microbial assembly process in glacier environments and provides valuable insights into the factors governing bacterial community compositions across different habitats along the glacial hydrological continuum.
Introduction
Glacial and proglacial ecosystems within mountain glaciers are interconnected through hydrological process (Hotaling et al. 2017). Glacier meltwater traverses through englacial conduits, moving from glacier surface into the adjacent moraines (Moorman 2005), and then enters the glacier forefield, eventually streaming into lakes. The supraglacial ecosystem, (e.g. snow, cryoconite holes and supraglacial runoff), experiences substantial influence from solar radiation and inoculated with microbes and essential nutrients via atmospheric deposition (Anesio and Johanna 2012, Boetius et al. 2015). Supraglacial streams drain from glaciers and ice sheets, exporting considerable amounts of nutrients (Hood et al. 2009, Bhatia et al. 2013, Vick‐Majors et al. 2020), and microbes to downstream ecosystems (Boetius et al. 2015, Cameron et al. 2020). For instance, significant dissolved solute and macronutrient (nitrogen, phosphorus and silica) (Hodson et al. 2008, Hawkings et al. 2015, Hauptmann et al. 2016, Wadham et al. 2019) and diverse prokaryotes (Cameron et al. 2017) in Greenland ice sheet were transported to the downstream habitats with increasing meltwater. Mountain glaciers respond strongly to the anticipated global warming, and the amplified melting of glaciers have potential to enhance connectivity between habitats (Hotaling et al. 2017).
Glacial habitats are hydrologically connected through meltwater flows, which transports microbial assemblages to downstream (Cemeron et al. 2020). Except glacier-fed aquatica environments (e.g. stream and lake), meltwater also permeates cryoconite holes through the porous cryoconite material, forming channels within the holes (Edwards et al. 2011). Cryoconite holes, vertical cylindrical melt holes formed by preferential melting of dark dust on the glacier surface, contain a layer of sediment at the bottom and filled with meltwater (Fountain et al. 2008). Besides, ice-marginal moraines are formed along edges of glaciers and created by transportation and deposition of debris by glaciers (Winkler and Atle 1999). The stream connects glacier and downstream lakes tightly, transporting microbiomes from glacier terminus to lake ecosystem (Kohler et al. 2017, Liu et al. 2021).
The level of hydrological and biological connectivity between glacial habitats can vary widely, depending on the physical conditions of the catchment, which in turn can give rise to site-specific microbial assemblages (Dubnick et al. 2017). Despite its hydrological connectivity, glacier ecosystems and forefield habitats host a wide range of microbes, with each habitat hosting distinct microbial assemblages (Simon et al. 2009, Anesio and Johanna 2012, Franzetti et al. 2017, Lutz et al. 2017, Comte et al. 2018, Stibal et al. 2020). For instance, in Ward Hunt Lake in Arctic, higher bacterial species richness was observed in subsurface water tracks comparing with snow (Comte et al. 2018). Similar, the river along the glacial hydrological transect in Lyngmarksbræ Glacier in Greenland demonstrated higher species richness than snow (Cameron et al. 2020). Cyanobacteria dominated snow, while Bacteroidetes and Betaproteobacteria were dominant in moraine lake and stream in a Himalaya glacier (Liu et al. 2011). Cryoconite holes in Forni Glacier had a significantly lower OTUs richness than that in moraine (Franzetti et al. 2017). Additionally, high-Arctic glaciers’ cryoconite holes exhibited higher relative abundance of Cyanobacteria than in tundra and moraine (Edwards et al. 2013). Cyanobacteria is one of the dominant phototrophic bacterial communities, which could fix inorganic carbon in glacier surface environment (Stibal and Tranter 2007, Anesio et al. 2009). Furthermore, the anaerobic Clostridiales (affiliated to Firmicutes) was more abundant in cryoconite holes than those in the moraine and forefield zone (Franzetti et al. 2017).
Within glacial hydrological continuum, snow-originating bacteria contributes to bacterial diversity in downstream habitats (Comte et al. 2018, Cameron et al. 2020, Liu et al. 2021), and proglacial lake is influenced by microbes from upstream environment (Crump et al. 2012). Microbial community compositions in disparate glacier habitats were filtered by the local environment, such as diverse pH and electrical conductivity (Wilhelm et al. 2013, Comte et al. 2018). pH and electrical conductivity in glacial environment were associated with the relative abundance of bacterial taxa (Fierer and Jackson 2006, Wilhelm et al. 2013, Webster-Brown et al. 2015, Kleinteich et al. 2022), which are primary physiological parameters to structure microbial communities (Fierer and Jackson 2006, Brown et al. 2007). With pH being associated with microbial cell structural integrity and cell metabolisms (Jin and Kirk 2018), while electrical conductivity being associated with microbial adaptation to extracellular osmotic pressure (Oren 2008, Zhang et al. 2021).
Deterministic and stochastic processes simultaneously influence microbial community assembly (Stegen et al. 2012). The deterministic process is divided in to homogeneous selection and heterogeneous selection, while stochastic process is divided into homogenizing dispersal, dispersal limitation and undominated (Stegen et al. 2015). Additionally, previous studies have categorized homogeneous selection and homogenizing dispersal as homogenizing, and identify heterogeneous selection and dispersal limitation as differentiating (Yan et al. 2022, West et al. 2024). It has been reported that bacterial assembly in glacial sub-surface snow (Chen et al. 2022a, Liu et al. 2021), and streams (Liu et al. 2021) were primarily governed by stochastic process on the Tibetan Plateau. The analysis was used null-modeling method based on Bray-Curtis matrix, and the average stochastic ratios were more than 50%. In comparison, the deterministic process predominately influenced bacterial communities in cryoconite holes, analyzed with the Zipf and Zipf-Mandelbrot distribution (Edwards et al. 2013). Besides, the proglacial lakes on the Tibetan Plateau and Alps were dominantly shaped by deterministic process (homogeneous selection), based on the analysis of between-community nearest taxon index (βNTI) in bacterial community (Aguilar and Sommaruga 2020, Gu et al. 2021). However, investigation into bacterial ecology in glacier hydrological continuum, including both aquatic and terrestrial habitats, remain limited, with most studies focusing on microbial diversity and community structure in a specific glacial aquatic habitats or distinct habitat.
In this study, we explored the bacterial communities and their assembly in the supraglacial system (snow, cryoconite holes and glacier runoff) and proglacial habitats (ice-marginal moraines and proglacial lake) during the ablation season in Longxiazailongba Glacier (LXZ), using 16S rRNA gene high-throughput sequencing. The objective was to gain insights into the bacterial communities and their assembly dynamics within this complex glacial system. We anticipated that dominant bacterial community in the glacier would be filtered by the local environment to adapt to the unique conditions of the glacier environment.
Materials and methods
Study site and sampling
The LXZ Glacier (33°06′N, 92°04′E) is a valley glacier located in the south-central Tanggula Mountain with a climate influenced by the Indian monsoon and westerlies in summer (June to September) (Zhao et al. 2014). The glacier is 7 km long, with an area of 19.3 km2, and the thickness is 118±10 m. The maximum altitude of the glacier is 6000 m above sea level (a.s.l). The glacier equilibrium line altitude was 5463 m in 1989. With glacial retreat, the equilibrium line altitude has been up to 5600 m a.s.l (Huang et al. 2013, Yao 2014). The ablation season begins in early June and lasts until October. Due to the lack of meteorological data in this study area, we use the climate data from adjacent glaciers Da Dongkeamadi (DD) (33°04′N, 92°04′E) and Xiao Dongkemadi Glacier (XD) (33°04′N, 92°05′E). The annual precipitations were 658 mm and 302 mm in DD and XD glacier, respectively (Shi et al. 2016, He et al. 2022). Negative annual mass balance of DD (−125 mm w.e. [millimeter water equivalent]) (Liang et al. 2019) and XD glacier (−233 mm w.e.) (Pu et al. 2008) has been observed.
Sampling was carried out in July 2017. A total of 35 samples were collected from five different habitats (Fig. 1), including snow (n = 9), cryoconite holes sediment (n = 13), supraglacial runoff (n = 5), ice-marginal moraine (n = 3), and proglacial lake water (n = 5) (Table S1). We collected fresh snow after removing surface snow (∼2 cm) in a 1 m × 1 m area, using a sterile Whirk-Pak bag with a sterilized shovel with 75% alcohol to prevent cross-contamination. The cryoconite samples (mixed water and sediment) were collected aseptically from the holes using sterile syringes and stored in 50 ml sterile bottles, with about 30 ml water and about 20 g cryoconite sediment slurry. Glacier runoff samples were supraglacial meltwater, not impacted by the subglacial environment, which is located about 100 m far away from the terminus. Then glacier runoff was collected with a sterile Nalgene bottle. The ice-marginal moraine was connected with the glacier, and the sampling sites were located about 3 m from the glacier terminus. The ice-marginal moraine was collected into a sterile Whirk-Pak bag with a sterilized shovel. Glacial melting water routes through terminal moraines, and moraines are wet in the melting season (Fig. 1E). Following the flow path of the meltwater, the runoff is mixed into a proglacial lake (Fig. 1A and C). The proglacial lake sampled in this study receives water from our studied area and other adjacent glaciers. The lake water (surface) was collected into 1 L sterile Nalgene bottles. As mentioned above, this glacial continuum is hydrologically connected in the melting season. All samples were kept at 4 °C and transported to the laboratory within 24 h. Once in the laboratory, the cryoconite samples were centrifuged at 11 000 rpm to collect the sediment for DNA extraction. About 500 mL of snowmelt water, 1 L of glacier runoff water and 1 L of lake water were filtered through a 47 mm polycarbonate membrane (0.22 µm, Millipore, USA) to collect the microbial material using a vacuum pump (Model GM-1.0A, Tianjin Jinteng, China, 60 L/min, 0.08Mpa). The filters, cryoconite holes sediment, and ice-marginal moraine were frozen at −80°C until further processing.
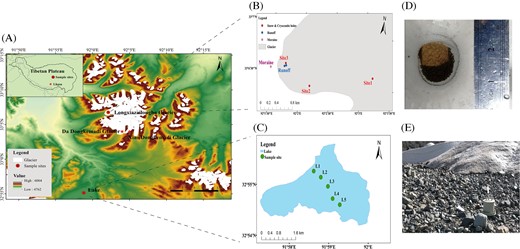
(A) Location and distribution of sample sites on glacier ecosystem. The insert map indicated the sample site (red point) on the Tibetan Plateau. The red asterisks represent the locations of Da Dongkemadi Glacier (DD) and Xiao Dongkemadi Glaciers (XD). (B) Sample sites on the glacier surface ecosystem, including site 1, site 2, and site 3 that collecting snow and cryoconite holes samples, glacier runoff samples and ice-marginal moraine samples. (C) Sample sites in the glacier fed lake. (D) Cryoconite holes in LXZ glacier surface, consisting of sediment and meltwater. (E) Ice-marginal moraines in LXZ glacier. Sampling sites were located about 3 m from glacier terminus. The moraines are wet during melting season.
DNA extraction, amplification, and MiSeq sequences
Approximately 0.5 g of cryoconite sediment, ice-marginal moraines, and one piece of filters were placed into a Fastprep bead-beating tube to extract DNA. The solid samples, including cryoconite sediments and moraines, were placed into the tube with a sterile spoon. Meanwhile, we got two filters at each liquid sample (snow, runoff and lake samples). One piece was cut into pieces with sterile scissors for DNA extraction, such that the filter pieces diameter was about 5 mm, and the other spare piece of filter was stored at −80°C. Genomic DNA was extracted by Fast DNA®SPIN Kit for Soil (MP Biomedicals, Santa, CA) according to manufacturer's instructions. Concentrations and purity of extracted DNA were measured with a NanoDrop 1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
About 50 ng DNA of each sample was used for the Illumina sequence. The V4 region of 16S rRNA genes was amplified with the primer pair 515F (5′-GTGCCAGCMG CCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAA T-3′) combined with Illumina adapter sequences, a pad and a linker of two bases, as well as barcodes on the reverse primers (Caporaso et al. 2012). Each PCR reaction contained 25 µL 2 × Premix Taq DNA polymerase (Takara Biotechnology, Dalian Co.,Ltd., China), 3 µL DNA template (20 ng µL −1), 1 µL each primer (10 µM), and 20 µL nuclease-free water. An aliquot of 10 ng purified DNA template from each sample was amplified in triplicate in a 50 µL reaction system with the following conditions: 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 30 s, with a final extension at 72°C for 10 min. Positive PCR products were confirmed by agarose gel electrophoresis.
PCR products from triplicate reactions were combined and quantified with PicoGreen staining (ThermoFisher Scientific Inc., Waltham, MA, United States). PCR products from the samples sequenced in the same MiSeq run were pooled at equal molality. The pooled mixture was purified with a QIAquick Gel Extraction Kit (QIAGEN Sciences, Germantown, MD, USA) and re-quantified with PicoGreen. Sample libraries were generated from purified PCR products. The MiSeq 500 cycles kit was used for 2 × 250 bp paired-end sequencing on the MiSeq machine (Illumina, San Diego, CA).
Sequence processing
Pair-end reads were first assembled with FLASH (Tanja and Salzberg 2011). Overlapped and poor-quality sequences were filtered out before demultiplexing based on barcodes. We processed the sequences using QIIME (Quantitative Insights Into Microbial Ecology) pipeline (v1.8.0) (Caporaso et al. 2010). The sequences were clustered into Operational Taxonomic Units (OTUs) with a criterion of 99% identity of 16S rRNA gene. OTUs that affiliated with archaeal, chloroplast, and unclassified sequences and singleton sequences (sequences appearing only once in the whole data) were removed. The taxonomic identity of each representative sequence was determined using the reference database SILVA (version 132 NR) (Pruesse et al. 2007).
Computational analyses
The sequences were sub-sampled to the smallest sequencing depth (24 800) for each sample before further bacterial α- and β-diversity analysis. The α-diversity estimates, including Shannon index and Chao1 richness, were calculated using the R package (“picante”). Bray-Curtis distance was used to estimate taxonomic dissimilarity between samples and was visualized by non-metric multidimensional scaling (NMDS) analysis, which calculated by “vegan” package. Bacterial taxonomic composition among habitats based on Bray-Curtis dissimilarity metrics was tested with ANOSIM (Analysis of Similarities). We carried out UpsetR (https://github.com/hms-dbmi/UpSetR) for data exploration and generation of set visualizations (Conway et al. 2017), showing the unique and shared OTUs among the habitats.
To assess the contributions of various ecological processes in structuring bacterial metacommunity, we employed the null model method described by Stegen et al. (2012). Within this model, the ecological process is divided into heterogeneous selection (HeS), homogeneous selection (HoS), dispersal limitation (DL), homogenizing dispersal (HD) and undominated. The mean nearest taxon distance (βMNTD) represents the pairwise phylogenetic turnover between communities, and β-nearest taxon index (βNTI) represents the difference between observed βMNTD and the mean of the null distribution. βNTI based on the phylogenetic information was used to quantify the community assembly process in each habitat (Stegen et al. 2013, Stegen et al. 2015). Absolute βNTI values greater than 2 (|βNTI| > 2) indicate selection strongly influences community composition, or βNTI > 2 and βNTI < -2 were identified as homogeneous selection and heterogenous selection, respectively. Moreover, the |βNTI| < 2 were then analyzed, and the action dispersal and drift was calculated based on the taxonomic turnover with the Raup-Crick metric using Bray-Curtis dissimilarities (RCbray). The |βNTI|<2 and RCbray<−0.95 or the |βNTI|<2 and RCbray ≥ 0.95 RCbray were identified as homogenizing dispersal and dispersal limitation, respectively. Besides, When the |βNTI|<2 and |RCbray|<0.95 were identified as “Undominated”.
Results
Dominant bacterial assemblages among habitats
After cleaning and quality processing, a total of 1251321 high-quality sequences were obtained, with 24866 to 43014 sequences (34759 ± 4029, mean ± s.e., n = 35) per sample (Table S1). To avoid sample size-based artefacts, the dataset was subsampled to 24800 sequences per sample (corresponding to the smallest sequencing effort across all samples). The phylum-level taxonomic classification illustrated that most OTUs belonged to Proteobacteria, Bacteroidetes, Actinobacteria, and Cyanobacteria (Fig. 2). Generally, Proteobacteria gradually increased in relative abundance from snow to downstream habitats (26% to 59% of each habitat). Beta- and Alphaproteobacteria were the most dominant classes within Proteobacteria. Bacteroidetes showed a contrasting pattern and decreased from 31% in the snow to 5.6% in the lake samples. Cyanobacteria were significantly abundant in snow and cryoconite holes, with an average relative abundance 21% and 25%, respectively. On the other hand, the relative abundance of Alphaproteobacteria was significantly higher in runoff (13%), moraine (13%) and lake samples (16%) than that in snow (6.7%) and cryoconite holes (3.3%). Actinobacteria were less abundant in cryoconite holes (8%, versus > 17.6% in the other sample types, P < 0.05).
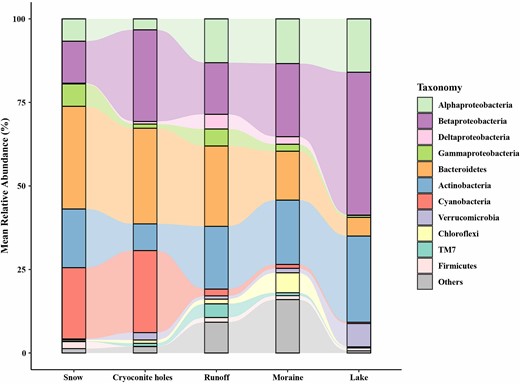
Taxonomic compositions in snow, cryoconite holes, runoff, moraine, and lake samples based on the mean relative abundance of bacterial 16S rRNA gene sequences. The relative abundance < 1% in all samples were grouped together in “Others”.
At the genus level, Hymenobacter (psychrophilic bacteria) in Bacteroidetes and Pseudanabaena in Cyanobacteria dominated the bacteria community in snow, which accounted for 26% and 10%, respectively. The genus Flavobacterium in Bacteroidetes and the genus Phormidium in Cyanobacteria dominated cryoconite holes, accounting for 10% and 9.6% of all sequences in that habitat, respectively. Samples of glacier runoff were dominated by the genus Flavobacterium of the phylum Bacteroidetes, accounting for 12%. Hymenobacter in Bacteroidetes also dominated the bacterial community in the moraine, accounting for 5.2%. Bacteria in lake samples were dominated by the genus Polynucleobacter, accounting for 17%.
Bacterial biodiversity among habitats
OTUs richness (number of OTUs) varied along the glacial continuum, with the highest values in moraines and the lowest in cryoconite holes (Table 1). Furthermore, bacterial α-diversity (Shannon index and Chao1 richness) varied greatly along the glacial continuum, with significantly higher values in runoff and moraine, and significantly lower in snow, cryoconite holes and the proglacial lake (Fig. 3). The NMDS ordination analysis, based on Bray-Curtis dissimilarity, showed that bacterial community structures were clustered by habitats (Fig. 4). Besides, bacterial community compositions significantly varied among all habitats based on ANOSIM (global r = 0.712, P = 0.001, Permutation = 999) (Table S2). This indicates higher bacterial connectivity in transitional habitats.
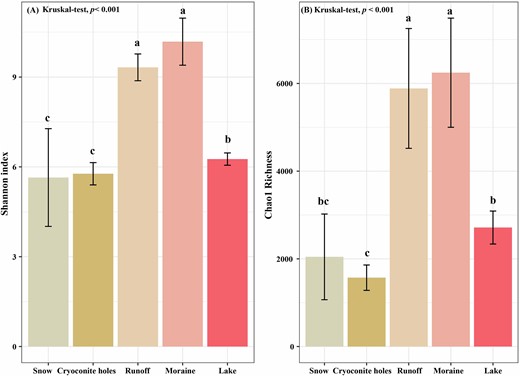
The mean Shannon index (A) and Chao1-richness (B) in snow, cryoconite holes, runoff, moraine, and lake samples. Significant difference (P < 0.05) among habitats is indicated by different letters. The significant analysis is conducted by Kruskal-Wallis test. Error bars indicate Standard Error.
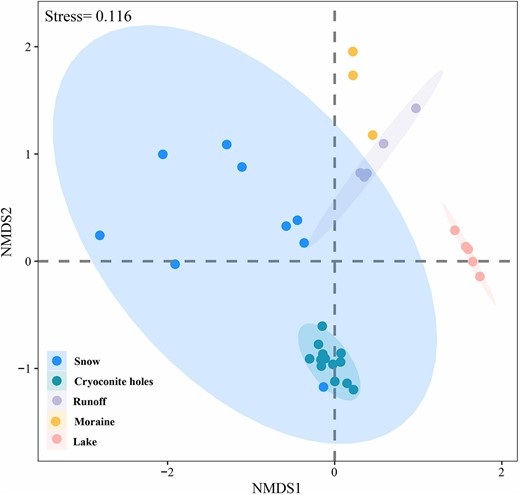
The NMDS ordination analysis based on Bray-Curtis distance of the bacterial community composition in snow, cryoconite holes, runoff, moraine, and lake.
Species richness of the communities retrieved from snow, cryoconite holes, runoff, moraine, and lake. The data represent the mean ± standard deviation. Different letters indicate significant difference among habitats (Wilcox-test, P < 0.05).
| Habitats | OTUs number | Sequences number | Unique OTUs (%) | Unique sequences (%) |
|---|---|---|---|---|
| Snow | 1411 ± 541b | 35622 ± 4,996a | 414 ± 197 (31.8 ± 18.9)b | 1156 ± 861(4.7 ± 3.5)b |
| Cryoconite holes | 858 ± 142c | 36154 ± 3,568a | 127 ± 73 (14.1 ± 5.7)c | 207 ± 174 (0.8 ± 0.7)c |
| Runoff | 3703 ± 641a | 33305 ± 3,647a | 1144 ± 352 (30.5 ± 5.0)a | 2970 ± 1352 (12.0 ± 5.5) a |
| Moraine | 4292 ± 934a | 30225 ± 3,910a | 936 ± 105 (22.4 ± 4.7)a | 3359 ± 929 (13.6 ± 3.8)a |
| Lake | 1344 ± 146b | 33174 ± 2,393a | 428 ± 27 (32.1 ± 3.2)b | 1307 ± 215 (5.3 ± 3.5)b |
| Habitats | OTUs number | Sequences number | Unique OTUs (%) | Unique sequences (%) |
|---|---|---|---|---|
| Snow | 1411 ± 541b | 35622 ± 4,996a | 414 ± 197 (31.8 ± 18.9)b | 1156 ± 861(4.7 ± 3.5)b |
| Cryoconite holes | 858 ± 142c | 36154 ± 3,568a | 127 ± 73 (14.1 ± 5.7)c | 207 ± 174 (0.8 ± 0.7)c |
| Runoff | 3703 ± 641a | 33305 ± 3,647a | 1144 ± 352 (30.5 ± 5.0)a | 2970 ± 1352 (12.0 ± 5.5) a |
| Moraine | 4292 ± 934a | 30225 ± 3,910a | 936 ± 105 (22.4 ± 4.7)a | 3359 ± 929 (13.6 ± 3.8)a |
| Lake | 1344 ± 146b | 33174 ± 2,393a | 428 ± 27 (32.1 ± 3.2)b | 1307 ± 215 (5.3 ± 3.5)b |
Species richness of the communities retrieved from snow, cryoconite holes, runoff, moraine, and lake. The data represent the mean ± standard deviation. Different letters indicate significant difference among habitats (Wilcox-test, P < 0.05).
| Habitats | OTUs number | Sequences number | Unique OTUs (%) | Unique sequences (%) |
|---|---|---|---|---|
| Snow | 1411 ± 541b | 35622 ± 4,996a | 414 ± 197 (31.8 ± 18.9)b | 1156 ± 861(4.7 ± 3.5)b |
| Cryoconite holes | 858 ± 142c | 36154 ± 3,568a | 127 ± 73 (14.1 ± 5.7)c | 207 ± 174 (0.8 ± 0.7)c |
| Runoff | 3703 ± 641a | 33305 ± 3,647a | 1144 ± 352 (30.5 ± 5.0)a | 2970 ± 1352 (12.0 ± 5.5) a |
| Moraine | 4292 ± 934a | 30225 ± 3,910a | 936 ± 105 (22.4 ± 4.7)a | 3359 ± 929 (13.6 ± 3.8)a |
| Lake | 1344 ± 146b | 33174 ± 2,393a | 428 ± 27 (32.1 ± 3.2)b | 1307 ± 215 (5.3 ± 3.5)b |
| Habitats | OTUs number | Sequences number | Unique OTUs (%) | Unique sequences (%) |
|---|---|---|---|---|
| Snow | 1411 ± 541b | 35622 ± 4,996a | 414 ± 197 (31.8 ± 18.9)b | 1156 ± 861(4.7 ± 3.5)b |
| Cryoconite holes | 858 ± 142c | 36154 ± 3,568a | 127 ± 73 (14.1 ± 5.7)c | 207 ± 174 (0.8 ± 0.7)c |
| Runoff | 3703 ± 641a | 33305 ± 3,647a | 1144 ± 352 (30.5 ± 5.0)a | 2970 ± 1352 (12.0 ± 5.5) a |
| Moraine | 4292 ± 934a | 30225 ± 3,910a | 936 ± 105 (22.4 ± 4.7)a | 3359 ± 929 (13.6 ± 3.8)a |
| Lake | 1344 ± 146b | 33174 ± 2,393a | 428 ± 27 (32.1 ± 3.2)b | 1307 ± 215 (5.3 ± 3.5)b |
Shared and Unique OTUs within habitats
Among all the habitats, glacier runoff harboured the highest level of unique OTUs (3275 OTUs), followed by moraine (2711 OTUs) and snow (22687 OTUs) (Fig. S1). At the same time, proglacial lake and cryoconite holes hold relative lower unique OTUs (949 and 909, respectively). Besides, glacial runoff and moraine shared more OTUs (1994 OTUs). Although the bacterial community compositions differed among habitats, they shared many OTUs (427 OTUs). Meanwhile, a small proportion of the unique OTUs was observed in cryoconite holes (14%) (Table 1). This suggests that the bacterial taxa were shared among cryoconite holes and adjacent habitats. In contrast, glacier runoff and moraine had higher unique OTUs to other habitats.
Ecological processes
The relative contributions of community assembly processes differed in different glacier habitats (Fig. 5). A deterministic process, especially the homogenizing selection (59.3%), was the most important assembly process in glacier snow. Similar with snow, deterministic process primarily shaped bacterial community in cryoconite holes, where the relative contribution of homogenizing selection was 60.5%. Moreover, deterministic process dominantly governs proglacial lake, with homogenizing selection contributing to 70.1% to bacterial assembly. On the contrary, the transitional habitats were primarily shaped by stochastic process, contributing 59.2% and 65.3% to bacterial community assembly in runoff and moraine, respectively (Fig. 5). Homogenizing process, including homogeneous selection and homogenizing dispersal, dominate bacterial community in snow (59.5%), cryoconite holes (60.5%) and proglacial lake (70.3%), exhibiting less effect on bacterial assembly in runoff (40.9%) and moraine (34.0%). Differentiating, including heterogeneous selection and dispersal limitation, contributed largely to bacterial assembly in moraine (57.3%) (Fig. 5B).
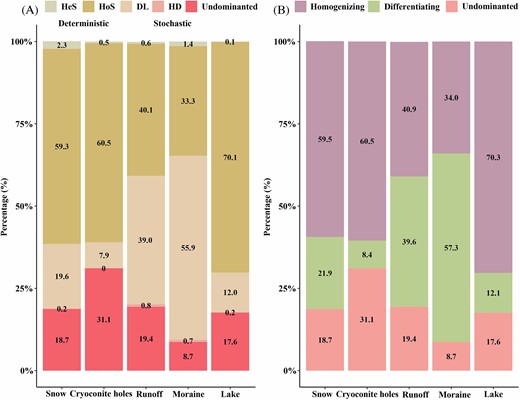
The ecological processes in bacterial community assembly estimated by null model analysis. (A) The relative contribution of Homogenizing dispersal (HD), Dispersal limitation (DL), Undominated, Heterogeneous selection (HeS) and Homogenizing selection (HoS) in glacier habitats. (B) The relative contribution of homogenizing, differentiating, and undominated in glacier hydrological continuum.
Discussion
Different ecological process primarily shaped bacterial community in glacial hydrological continuum
Bacterial community composition in glacier surface snow can be seed by aeolian deposition, where microbes of aeolian origin faced intense competition once they were deposited on the glacier surface (Musilova et al. 2015). Consequently, bacterial community in snow were more homogenous compare to transitional habitats (Peter and Sommaruga 2016, Cameron et al. 2020, Dindhoria et al. 2021, Liu et al. 2021). Meanwhile, our calculations further support the conclusion that the deterministic process, particularly homogeneous selection, predominantly drives bacterial structure in snow (Fig. 5). Microorganisms in snow may be filtered by low temperature, excessive UV radiation and nutrients limitation in the local glacier environment (Siddiqui et al. 2013, De Maayer et al. 2014).
Within cryoconite holes, the bacterial community hold lower α-diversity (Fig. 3). This finding aligns with the previous studies that have also observed reduced biodiversity in cryoconite holes than those in ice-marginal habitats (Edwards et al. 2013, Segawa et al. 2014, Franzetti et al. 2017), indicating that cryoconite holes is a selective environment. Consistent with these findings, the pattern in bacterial assembly in cryoconite holes on the Tibetan Plateau (Chen et al. 2022b) and Svalbard (Edwards et al. 2013) suggests that deterministic processes play a significant role in shaping the bacterial community structure (Fig. 5). Bacteria wrapped in the particles in cryoconites, with longer residence time (Langford et al. 2010, Stabil et al. 2012), and filtered by environmental selection (Franzetti et al. 2017).
Higher bacterial α-diversity occurred in glacial runoff and ice-marginal moraine (Fig. 3). Our result is in agreement with previous studies in the Arctic (Comte et al. 2018, Cameron et al. 2020) and Alpine regions (Franzetti et al. 2017), where transitional habitats (e.g, water tracks, mixing zone or moraines) in glacier ecosystem had greater biodiversity. Glacier runoff and moraine contained diverse bacterial taxa, and shared a higher proportion of OTUs (Fig. S1). Frequent dispersal occurring in transitional habitats contributes to the exchange of bacterial taxa, and alters bacterial diversity and compositions (Evans et al. 2017, Albright and Martiny 2018, Shen et al. 2018). Furthermore, our results also indicated stochastic process also dominantly shaped bacterial community in glacier runoff and moraine (Fig. 5B). Especially dispersal limitation had an important effect on glacier runoff (39.0%) and moraine (55.9%) (Fig. 5A). Random dispersal and immigration between supraglacial habitats and forefield with increasing glacier meltwater (Wojcik et al. 2021), resulting in more dissimilar structures among communities. Higher turnover of bacterial community in transition habitats (Wang et al. 2022) also increase stochasticity (Wang et al. 2013).
Previous studies suggested that bacterial community assembly in proglacial lakes is predominantly governed by environmental filtering (Wilhelm et al. 2013, Milner et al. 2017) and homogeneous selection (Aguilar and Sommaruga 2020, Gu et al. 2021). This deterministic assembly pattern in proglacial lakes can be attributed to the longer residence time of water in these habitats. With lower interference frequency, microbial communities would be more stable over time (Stegen et al. 2016, Ji et al. 2022), and local environmental filtering deterministically governs microbial community compositions (Stegen et al. 2012).
Dominant bacterial communities in glacial hydrological continuum
Our findings demonstrated a clear directional structuring of the bacterial communities in this glacier hydrological continuum, revealing a sequential of bacterial taxa from glacier surface snow and cryoconite holes to the proglacial lake, through glacial runoff and ice-marginal moraine. That the dominant bacterial group shifts among different habitats (Fig. 2). Glacier meltwater moves directionally from glacier surface to downstream habitats, transporting microbial assemblages (Cameron et al. 2020). Compared to cryoconite holes, snow shared more OTUs with downstream habitats (Fig. S1). This probably due to snow could be the microbial source for downstream habitats through meltwater (Comte et al. 2018, Cameron et al. 2020, Liu et al. 2021). Besides, the result demonstrated that few taxa were unique in local species pool (Table 1), indicating that large proportion of taxa were shared among different habitats in this glacier continuum. This result corroborates the findings of previous study that few taxa were unique to a given type of habitat (Ruiz-Gonzalez et al. 2015, Comte et al. 2018, Liu et al. 2021).
Despite the glacial hydrological connectivity in this glacial continuum, the dominant bacterial assemblage differs across different habitats (Fig. 2). Local environment plays an important role in filtering the dominant bacterial group within these habitats. For instance, the strong solar radiation on glacier surface stimulates a higher proportion of Cyanobacteria in glacier surface snow and cryoconite holes (Fig. 2). Additionally, a higher relative abundance of Flavobacterium in glacier runoff (12%) aligns with previous study indicating the presence of psychrotolerant Flavobacterium in cold lakes and glacial meltwater (Liu et al. 2006). Furthermore, polynucleobacter, which has adapted to various freshwater ecosystems, is abundant in various freshwater ecosystems (Nuy et al. 2020), exhibited relative dominance in proglacial lake in present study (∼ 17%).
Bacterial community compositions in the glacier hydrological continuum on the Tibetan Plateau is not significantly different from the composition observed in other geographical regions (Comte et al. 2018, Cameron et al. 2020). Betaproteobacteria are distributed globally in freshwater lakes (Buck et al. 2009, Newton et al. 2011). The Betaproteobacteria in present study was assigned to Burkholderiales, that are similar to previously found in cryoconite holes (Cameron et al. 2012, Webster-Brown et al. 2015, Liu et al. 2017) and moraines (Mapelli et al. 2011, Franzetti et al. 2017). Besides, Betaproteobacteria is also known for their ability to rapidly use anaerobic decomposition products of organic matter in cryoconite holes in Antarctic glaciers (Lutz et al. 2017). In contrast, Bacteroidetes exhibited an opposite distribution pattern, with a higher relative abundance in snow and cryoconite holes (Fig. 2). This lineage was identified to be more abundant in snow. Bacteroidetes have also been more abundant following cyanobacterial blooms, as they are favored during periods of high heterotrophic activity and growth enhancement (Newton et al. 2011).
Conclusion
Alpine glacier ecosystems on the Tibetan Plateau received relatively less attention than their polar counterparts. Here, we explore the bacterial community and its assembly across a glacial catchment, aiming to compare the similarities and differences between a range of glacier-related habitats (snow, cryoconite holes, runoff, moraine and lake). Our results illustrated the relative abundance of dominant bacteria across these habitats was different. Besides, the harsh glacier environment (e.g. high UV and cold environment) filters dominant bacteria in glacier habitats, thus deterministic process shaped bacterial community in snow, cryoconite holes and proglacial lake. The transition habitats in this hydrological continuum hold higher bacterial α-diversity, and stochastic process primarily shaped bacterial community.
Acknowledgements
We appreciate Prof. Alexandre M. Anesio from Aarhus University for his comments and suggestions about our paper. The 16S rRNA sequences were submitted to the NCBI SRA database (BioProject accession number: PRJNA699337).
Conflict of interest
The authors have no conflict of interest to declare.
Funding
This work was supported by the State Key Program of National Natural Science Foundation of China (42330410) and Joint Funds of the National Natural Science Foundation of China (U21A20176).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}