





ABSTRACT
The anaerobic digestion of wastes is globally important in the production of methane (CH4) as a biofuel. When sulfate is present, sulfate-reducing bacteria (SRB) are stimulated, competing with methanogens for common substrates, which decreases CH4 production and results in the formation of corrosive, odorous hydrogen sulfide gas (H2S). Here, we show that a population of SRB within a methanogenic bioreactor fed only butyrate for years immediately (within hours) responded to sulfate availability and shifted the microbial community dynamics within the bioreactor. By mapping shotgun metatranscriptomes to metagenome-assembled genomes, we shed light on the transcriptomic responses of key community members in response to increased sulfate provision. We link these short-term transcriptional responses to long-term niche partitioning using comparative metagenomic analyses. Our results suggest that sulfate provision supports a syntrophic butyrate oxidation community that disfavors poly-β-hydroxyalkanoate storage and that hydrogenotrophic SRB populations effectively exclude obligately hydrogenotrophic, but not aceticlastic, methanogens when sulfate is readily available. These findings elucidate key ecological dynamics between SRB, methanogens and syntrophic butyrate-oxidizing bacteria, which can be applied to a variety of engineered and natural systems.
INTRODUCTION
Methane (CH4) biogas produced from anaerobic treatment of organic waste streams has great economic and environmental value (Stillwell, Hoppock and Webber 2010; Lin et al. 2018). Within the anaerobic treatment processes, diverse consortia of Bacteria and Archaea performing hydrolytic, acidogenic, acetogenic and methanogenic processes catalyze the breakdown of complex organic substrates to carbon dioxide (CO2) and CH4 only when no terminal electron acceptors (TEAs) besides CO2 are available in the system (Zinder 1993). When sulfate enters the system as an alternative TEA, competitive interactions between dissimilatory sulfate-reducing bacteria (SRB) and syntrophic methanogenic communities shift carbon flow to favor dissimilatory sulfate reduction (DSR) over methanogenesis as the dominant terminal carbon mineralization process (Oude Elferink et al. 1994; Stams et al. 2005). In addition to decreasing the amount of CH4 biogas formed that could otherwise be used for generating heat and electrical energy (Shen et al. 2015), these competitive interactions also lead to the production of high levels of odorous and corrosive hydrogen sulfide (H2S) gas that can negatively impact reactor function and public acceptance of anaerobic digesters (Muyzer and Stams 2008).
SRB represent a diverse group of metabolically versatile anaerobic microorganisms that are widespread in natural and engineered environments. SRB can couple DSR to the oxidation of a wide variety of electron donors. However, in the absence of sulfate, they are capable of rapidly switching between DSR and syntrophic and fermentative lifestyles (Neretin et al. 2003; Muyzer and Stams 2008; Plugge et al. 2010, 2011; Meyer et al. 2013; Worm et al. 2014; Sedano-Núñez et al. 2018). This metabolic flexibility allows SRB to occupy ecologically broad niches that facilitate the persistence of physiologically distinct populations within anaerobic environments where sulfate levels are at sub-micromolar or non-detectable levels.
Increasing evidence suggests this rapid metabolic switch is due to constitutive expression of large portions of the core DSR machinery and flexibility of syntrophic enzyme systems (Plugge et al. 2011). For example, when growing axenically in butyrate-fed syntrophic dual culture with hydrogen-scavenging methanogens, two deltaproteobacterial SRB, Desulfovibrio vulgaris and Syntrophobacter fumaroxidans, have been shown to constitutively express adenylyl-sulfate reductase (aprAB) and dissimilatory sulfite reductase (dsrAB), while other genes associated with electron transfer and ATP synthesis in DSR are upregulated and genes associated with syntrophy are downregulated (Plugge et al. 2010; Sedano-Núñez et al. 2018). This trend of constitutive aprAB and dsrAB expression has also been observed in SRB from the Firmicutes, specifically Desulfotomaculum (Otwell et al. 2016).
In this work, we performed metagenomic and metatranscriptomic sequencing of a model butyrate-to-methane bioreactor over the course of 48 h after pulse feeding with butyrate and sulfate. We recovered metagenome-assembled genomes (MAGs) for core community members and aligned MAGs to metatranscriptome reads to reconstruct temporal responses of core syntrophic butyrate-oxidizing bacteria, SRB and methanogens responding to sulfate addition. Analyzing the extent to which the metabolic flexibility of SRB is observed in methanogenic bioreactors and its effect on pathways of carbon flow will reveal further insight into the effects of sulfate on the ecophysiology of terminal carbon mineralization in anaerobic digesters and mechanisms of metabolic flexibility within SRB populations.
METHODS
Setup and monitoring of methanogenic bioreactor and sulfate enrichment culture for metagenomic sequencing and microcosms for metatranscriptomic sequencing
The methanogenic bioreactor (SJ1) used was a stirred batch reactor consisting of a 9.1-L Pyrex® bottle (Corning Inc., Corning, NY, USA) closed by a Teflon ®-lined steel top with a three-way stainless steel valve (DuPont, Wilmington, DE, USA) with 5.7 L of culture maintained in a 70% N 2 (High Purity 4.8 Grade Nitrogen; Airgas, Radnor Township, PA, USA) and 30% CO 2 (Bone Dry 3.0 Grade Carbon Dioxide; Airgas, Radnor Township, PA, USA) headspace at 30°C. SJ1 was started as a 20% inoculum in basal medium (Fennell 1998) from a methanogenic, dechlorinating tetrachloroethene (PCE)/butyric acid enrichment culture, DonnaII, that has been studied at Cornell for over two decades without the provision of sulfate (Fennell, Gossett and Zinder 1997; Rowe et al. 2008; Heavner et al. 2013). The culture was pulse-fed with 880 µM butyrate (99%; Acros Organics, Pittsburgh, PA, USA), 40 mg/L fermented yeast extract (FYE) and 0.5% v/v vitamin solution (Fennell 1998). SJ1 was maintained with a 70-day retention time by a fill and draw exchange of 10% of the liquid every 7th day as previously described (Fennell 1998). PCE was omitted from the feeding regimen to rid the culture of chlorinated compounds. After 14 months of enrichment without PCE (7 residence times), a stable methanogenic, non-dechlorinating culture was observed (Fig. 1). A sulfate enrichment subculture (SJ1S) was obtained by filling a 160-mL serum bottle (Corning Inc., Corning, NY, USA) with 100-mL effluent from SJ1. Serum bottles were plugged with a butyl rubber septum (Neta Scientific Inc., Hainesport, NJ, USA) and maintained with a 70% N 2 and 30% CO2 headspace at 30°C while shaking at 150 rpm. SJ1S was fed in the same manner as SJ1 except that sulfate (Sodium Sulfate Anhydrous, Thermo Fisher Scientific, Waltham, MA, USA) was amended and maintained at 2 mM, a molar excess of sulfate given the 880 µM butyrate was provided. SJ1S was likewise maintained for ∼7 residence times after which a stable methanogenic/sulfidogenic culture was observed (Fig. 1). DNA was extracted from SJ1 and SJ1S for metagenomic sequencing after 7 residence times. Also, at this time, effluent from SJ1 was used to seed microcosms for metatranscriptomic sequencing and analysis to study the first exposure response of SJ1 to sulfate (Fig. 1).
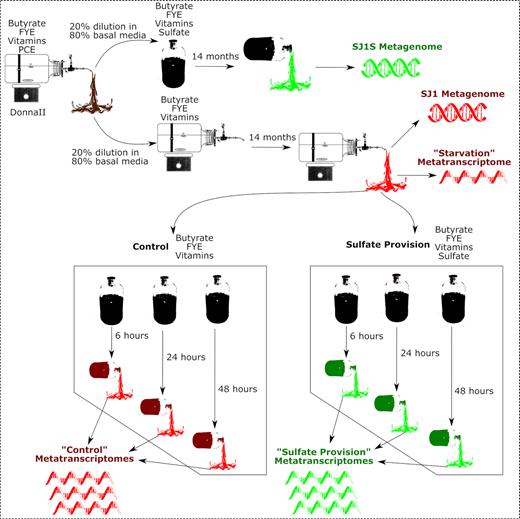
Overview schematic of sampling scheme. Red: control culture and microcosms receiving only butyrate, FYE and vitamin mix. Green: sulfate provision culture and microcosms receiving butyrate, FYE, vitamin mix and sulfate.
Six microcosms containing 100-mL effluent from SJ1 in 160-mL serum bottles with butyl rubber septa were set up to study temporal metatranscriptomic responses of core community members. All microcosms were maintained with the same headspace composition as SJ1 and were fed the same ratio of reagents. Three microcosms also received 2 mM sulfate (‘sulfate provision’), while three did not (‘control’). At 6-, 24- and 48-h post-feeding, microcosms from the control and sulfate provision groups were sacrificed for RNA extraction. RNA extracted from the SJ1 effluent used to seed the microcosms was used as a 0-h ‘starvation’ control. To probe the first exposure response of SJ1 to sulfate, microcosms were sacrificed for RNA extraction after the first feeding (Fig. 1).
Time points were selected based on preliminary studies that demonstrated peak butyrate consumption and methane, acetate and sulfide production during the first 48 h post-feeding (Fig. 2). Methane was measured via headspace injections using a gas chromatograph with a thermal conductivity detector (GC-TCD; Hewlett-Packard 5890 Series II, Palo ALto, CA, USA) with helium (>99.999% purity; Airgas, Radnor Township, PA, USA) as carrier gas. The GC method used was unable to detect hydrogen levels. Acetate and butyrate were measured using anion chromatography (Dionex ICS-2100, Thermo Fisher Scientific, Waltham, MA, USA). Sulfide was measured using the Cline assay (Cline 1969) with a spectrophotometer plate reader (Tecan Infinite 200® Pro, Tecan Group Ltd., Zürich, Switzerland).
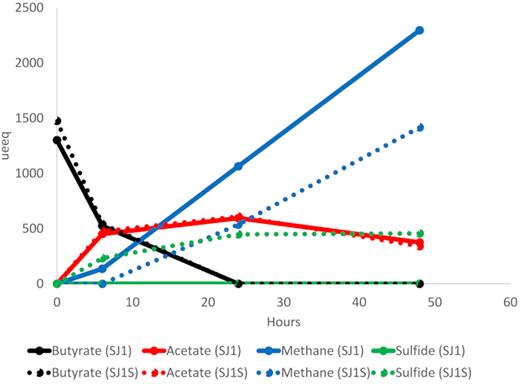
Levels of butyrate, acetate, methane and sulfide in preliminary microcosm incubations used for determining timing of metatranscriptome profiles. Solid lines: control. Dotted lines: sulfate provision. Black: butyrate concentration. Red: acetate concentration. Blue: methane concentration. Green: sulfide concentration. µeeq: microelectron equivalents.
Nucleic acid extraction, sequencing and metagenomic analysis pipeline
Nucleic acids were extracted from cell pellets originating from 50-mL samples that were centrifuged at 4000 rpm for 15 min at 4°C. DNA was extracted from cell pellets of SJ1 and SJ1S and RNA was extracted from the SJ1 microcosm experiments using a Qiagen AllPrep DNA/RNA Mini Kit (Cat. No. 80204; Qiagen, Hilden, Germany). DNA and RNA quality were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
DNA and RNA were sent to the Department of Energy's (DOE) Joint Genome Institute (JGI) for library construction, sequencing and assembly using standard JGI pipelines. Briefly, Illumina libraries were constructed and sequenced using the Illumina HiSeq-2000 1 TB platform (2 × 150 paired-end reads). Reads were subjected to quality control (QC) and filtering using BBTools (sourceforge.net/projects/bbmap/). For metagenomes, QC-filtered raw reads were assembled into contigs using the SPAdes assembler (version 3.11.1) (Nurk et al. 2017) and annotated using standard pipelines in the Integrated Microbial Genomes (IMG) system (Chen et al. 2017). Table 1 contains relevant metadata for all metagenomic and metatranscriptomic sequencing libraries.
GOLD analysis project IDs for metagenomic and metatranscriptomic sequencing libraries. QC: quality control.
| Sequencing Library | GOLD analysis project ID | # Reads (post-QC) | # Contigs | N50 |
|---|---|---|---|---|
| SJ1 Metagenome | Ga0228535 | 222 599 058 | 212 269 | 2212 |
| SJ1S Metagenome | Ga0228536 | 196 244 686 | 171 234 | 2141 |
| ‘Starvation’ Metatranscriptome | Ga0232079 | 30 039 006 | — | — |
| ‘Control’ Metatranscriptome (6 h) | Ga0232080 | 62 907 116 | — | — |
| ‘Control’ Metatranscriptome (24 h) | Ga0247507 | 66 383 206 | — | — |
| ‘Control’ Metatranscriptome (48 h) | Ga0232081 | 49 369 646 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (6 h) | Ga0232082 | 69 051 286 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (24 h) | Ga0247508 | 61 969 624 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (48 h) | Ga0247509 | 50 036 086 | — | — |
| Sequencing Library | GOLD analysis project ID | # Reads (post-QC) | # Contigs | N50 |
|---|---|---|---|---|
| SJ1 Metagenome | Ga0228535 | 222 599 058 | 212 269 | 2212 |
| SJ1S Metagenome | Ga0228536 | 196 244 686 | 171 234 | 2141 |
| ‘Starvation’ Metatranscriptome | Ga0232079 | 30 039 006 | — | — |
| ‘Control’ Metatranscriptome (6 h) | Ga0232080 | 62 907 116 | — | — |
| ‘Control’ Metatranscriptome (24 h) | Ga0247507 | 66 383 206 | — | — |
| ‘Control’ Metatranscriptome (48 h) | Ga0232081 | 49 369 646 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (6 h) | Ga0232082 | 69 051 286 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (24 h) | Ga0247508 | 61 969 624 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (48 h) | Ga0247509 | 50 036 086 | — | — |
GOLD analysis project IDs for metagenomic and metatranscriptomic sequencing libraries. QC: quality control.
| Sequencing Library | GOLD analysis project ID | # Reads (post-QC) | # Contigs | N50 |
|---|---|---|---|---|
| SJ1 Metagenome | Ga0228535 | 222 599 058 | 212 269 | 2212 |
| SJ1S Metagenome | Ga0228536 | 196 244 686 | 171 234 | 2141 |
| ‘Starvation’ Metatranscriptome | Ga0232079 | 30 039 006 | — | — |
| ‘Control’ Metatranscriptome (6 h) | Ga0232080 | 62 907 116 | — | — |
| ‘Control’ Metatranscriptome (24 h) | Ga0247507 | 66 383 206 | — | — |
| ‘Control’ Metatranscriptome (48 h) | Ga0232081 | 49 369 646 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (6 h) | Ga0232082 | 69 051 286 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (24 h) | Ga0247508 | 61 969 624 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (48 h) | Ga0247509 | 50 036 086 | — | — |
| Sequencing Library | GOLD analysis project ID | # Reads (post-QC) | # Contigs | N50 |
|---|---|---|---|---|
| SJ1 Metagenome | Ga0228535 | 222 599 058 | 212 269 | 2212 |
| SJ1S Metagenome | Ga0228536 | 196 244 686 | 171 234 | 2141 |
| ‘Starvation’ Metatranscriptome | Ga0232079 | 30 039 006 | — | — |
| ‘Control’ Metatranscriptome (6 h) | Ga0232080 | 62 907 116 | — | — |
| ‘Control’ Metatranscriptome (24 h) | Ga0247507 | 66 383 206 | — | — |
| ‘Control’ Metatranscriptome (48 h) | Ga0232081 | 49 369 646 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (6 h) | Ga0232082 | 69 051 286 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (24 h) | Ga0247508 | 61 969 624 | — | — |
| ‘Sulfate Provision’ Metatranscriptome (48 h) | Ga0247509 | 50 036 086 | — | — |
Reconstruction, annotation and analysis of MAGs
Reconstruction, annotation and analysis of MAGs were performed using QC-filtered raw reads for each metagenome and metatranscriptome library via the DOE Systems Biology Knowledgebase (KBase) platform (Arkin et al. 2018). JGI-assembled contigs from SJ1 and SJ1S were binned into MAGs using the MaxBin2 (version 2.2.4) app with default parameters and the 40 Bacterial and Archaeal Marker Gene Set (Wu, Simmons and Singer 2016). Genome bins were assessed for completeness and contamination via the CheckM (version 1.0.8) app with default parameters (Parks et al. 2015). For MAGs that were present in both metagenomes, the MAG with the highest completeness was chosen as a consensus MAG. Matching MAGs were identified based on phylogenetic inference, estimated genome size, GC content and average amino acid identity of MAGs.
MAGs were taxonomically classified using the GTDB-Tk classify (version 0.0.2) app, which assigns objective taxonomic classifications to bacterial and archaeal genomes based on a concatenated protein phylogeny against a database of nearly 95 000 genomes (Genome Taxonomy Database, GTDB) comprising all genomes in the RefSeq/GenBank release 80 in addition to nearly 12 000 MAGs recovered from the Sequence Read Archive (Parks et al. 2018).
MAGs were annotated using Rapid Annotation using Subsystems Technology (Aziz et al. 2008) via the Annotate Microbial Assembly (version 0.1.1) app. To estimate relative abundance of MAGs in the metagenomes, QC-filtered raw reads from metagenomic datasets were aligned to individual MAGs via the Bowtie2 (version 2.3.2) app (Langmead and Salzberg 2012). To estimate relative abundance of transcripts within each metatranscriptome, QC-filtered raw reads were aligned to individual MAGs using Bowtie2 and then assembled into transcripts for which relative abundances were calculated via the Cufflinks (version 2.2.1) app (Trapnell et al. 2012). Differences in transcriptional activity of genes were identified by comparing transcript abundances between sampling times and treatments based on fragments per kilobase million reads.
RESULTS AND DISCUSSION
Microbial ecology of the methanogenic bioreactor and sulfate enrichment culture
The microbial community of SJ1 as determined from functional annotation of metagenomic reads using the JGI IMG database is dominated by Proteobacteria, Firmicutes, Bacteroidetes and Euryarchaeota (Fig. 3A), a community composition which is similar to other methanogenic bioreactors where carbon source exerts a strong control on the microbial community (Zheng et al. 2013; Yuan et al. 2016; Xing et al. 2017) and to past studies on the predecessor culture, DonnaII (Rowe et al. 2008; Heavner et al. 2013).
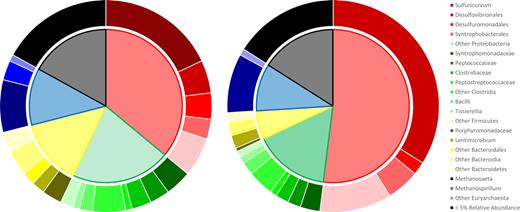
Taxonomic composition of the methanogenic bioreactor (A) and sulfate enrichment (B). Inner chart: phylum abundance; clockwise: Proteobacteria, Firmicutes, Bacteroidetes, Euryarchaeota, phyla with <5% relative abundance. Outer chart: relative abundance of subgroups within phyla.
The Proteobacteria are dominated by populations implicated in internal sulfur cycling including Sulfuricurvum, which are involved in microaerobic or denitrification-driven sulfur or H2 oxidation (Handley et al. 2014; Saia et al. 2016), and Desulfovibrionales and Syntrophobacterales, which contain species of SRB capable of a high degree of metabolic flexibility (Plugge et al. 2011) (Fig. 3A). Together, the presence of both sulfur-oxidizing and sulfate-reducing organisms suggests the possibility of an internal sulfur cycle. This internal sulfur cycle could theoretically be fueled by low levels of sulfide in the media that originate from H2S, which is used as a reductant in the media.
Neither the Firmicutes nor Bacteroidetes are dominated by single genera in the metagenomes (Fig. 3A). These phyla are part of the core microbiota of wastewater treatment facilities (the original seed of the parent culture came from a wastewater treatment facility in Ithaca, NY) and have been shown to have high stability in batch-fed laboratory bioreactors (Mariat et al. 2009; Kampmann et al. 2012). Varied populations are likely maintained by diverse fermentative and respiratory carbon metabolisms driven by the FYE supplement that is supplied to the culture concurrent with butyrate pulse feeding (Fennell, Gossett and Zinder 1997).
The Euryarchaeota are dominated by reads mapping to the aceticlastic genus Methanosaeta and the hydrogenotrophic genus Methanospirillum (Fig. 3A).
In SJ1S, Proteobacteria, particularly Desulfovibrionales, increase substantially in relative abundance while Sulfuricurvum and Methanospirillum effectively reach extinction (Fig. 3B). The disappearance of Methanospirillum under sulfate provision suggests that hydrogenotrophic SRB effectively outcompete Methanospirillum and the persistence of Methanosaeta is consistent with low activity and abundance of acetotrophic SRB.
Description of MAGs
Binning of metagenomic contigs from SJ1 resulted in the assembly of 61 high-quality MAGs (>90% complete, <10% contaminated) (Table S1, Supporting Information). Based on genome content and the alignment of metatranscriptomic reads to MAGs from SJ1 and SJ1S, we identified five highly abundant MAGs involved in DSR (MAG006), syntrophic butyrate oxidation (MAG002 and MAG015) and methanogenesis (MAG004 and MAG010). Genome descriptions of these MAGs and their relative abundances in the metagenomes and metatranscriptomes are provided in Table 2. MAGs of interest were stored in a public KBase narrative (https://narrative.kbase.us/narrative/ws.51077.obj.1).
Genome characteristics of core MAGs. RRA: relative read abundance. MTs: metatranscriptomes.
| MAG | Organism name | Size (Mbp) | GC content | Contigs | Completeness (contamination) | RRA SJ1 | RRA SJ1S | Range of RRA control MTs | Range of RRA sulfate MTs |
|---|---|---|---|---|---|---|---|---|---|
| MAG006 | Desolfovibrionaceae sp. SJ1006 | 3.9 | 65.1% | 32 | 100.00% (0.00%) | 2.14% | 25.99% | 1.52–8.24% | 3.05–10.74% |
| MAG002 | Syntrophomonas sp. UBA5314 | 4.3 | 49.3% | 68 | 97.96% (3.95%) | 7.42% | 2.95% | 33.73–38.90% | 31.62–39.68% |
| MAG015 | Syntrophomonas sp. SJ1015 | 3.2 | 49.3% | 85 | 90.03% (2.93%) | 1.35% | 2.01% | 9.53–12.80% | 11.16–14.03% |
| MAG004 | Methanosaeta concilli | 2.7 | 51.9% | 94 | 100.00% (0.65%) | 3.11% | 3.60% | 7.03–23.91% | 11.82–24.44% |
| MAG010 | Methanospirillum hungatei | 3.4 | 45.4% | 50 | 99.02% (0.98%) | 1.30% | 0.00% | 4.63–12.64% | 4.84–9.91% |
| MAG | Organism name | Size (Mbp) | GC content | Contigs | Completeness (contamination) | RRA SJ1 | RRA SJ1S | Range of RRA control MTs | Range of RRA sulfate MTs |
|---|---|---|---|---|---|---|---|---|---|
| MAG006 | Desolfovibrionaceae sp. SJ1006 | 3.9 | 65.1% | 32 | 100.00% (0.00%) | 2.14% | 25.99% | 1.52–8.24% | 3.05–10.74% |
| MAG002 | Syntrophomonas sp. UBA5314 | 4.3 | 49.3% | 68 | 97.96% (3.95%) | 7.42% | 2.95% | 33.73–38.90% | 31.62–39.68% |
| MAG015 | Syntrophomonas sp. SJ1015 | 3.2 | 49.3% | 85 | 90.03% (2.93%) | 1.35% | 2.01% | 9.53–12.80% | 11.16–14.03% |
| MAG004 | Methanosaeta concilli | 2.7 | 51.9% | 94 | 100.00% (0.65%) | 3.11% | 3.60% | 7.03–23.91% | 11.82–24.44% |
| MAG010 | Methanospirillum hungatei | 3.4 | 45.4% | 50 | 99.02% (0.98%) | 1.30% | 0.00% | 4.63–12.64% | 4.84–9.91% |
Genome characteristics of core MAGs. RRA: relative read abundance. MTs: metatranscriptomes.
| MAG | Organism name | Size (Mbp) | GC content | Contigs | Completeness (contamination) | RRA SJ1 | RRA SJ1S | Range of RRA control MTs | Range of RRA sulfate MTs |
|---|---|---|---|---|---|---|---|---|---|
| MAG006 | Desolfovibrionaceae sp. SJ1006 | 3.9 | 65.1% | 32 | 100.00% (0.00%) | 2.14% | 25.99% | 1.52–8.24% | 3.05–10.74% |
| MAG002 | Syntrophomonas sp. UBA5314 | 4.3 | 49.3% | 68 | 97.96% (3.95%) | 7.42% | 2.95% | 33.73–38.90% | 31.62–39.68% |
| MAG015 | Syntrophomonas sp. SJ1015 | 3.2 | 49.3% | 85 | 90.03% (2.93%) | 1.35% | 2.01% | 9.53–12.80% | 11.16–14.03% |
| MAG004 | Methanosaeta concilli | 2.7 | 51.9% | 94 | 100.00% (0.65%) | 3.11% | 3.60% | 7.03–23.91% | 11.82–24.44% |
| MAG010 | Methanospirillum hungatei | 3.4 | 45.4% | 50 | 99.02% (0.98%) | 1.30% | 0.00% | 4.63–12.64% | 4.84–9.91% |
| MAG | Organism name | Size (Mbp) | GC content | Contigs | Completeness (contamination) | RRA SJ1 | RRA SJ1S | Range of RRA control MTs | Range of RRA sulfate MTs |
|---|---|---|---|---|---|---|---|---|---|
| MAG006 | Desolfovibrionaceae sp. SJ1006 | 3.9 | 65.1% | 32 | 100.00% (0.00%) | 2.14% | 25.99% | 1.52–8.24% | 3.05–10.74% |
| MAG002 | Syntrophomonas sp. UBA5314 | 4.3 | 49.3% | 68 | 97.96% (3.95%) | 7.42% | 2.95% | 33.73–38.90% | 31.62–39.68% |
| MAG015 | Syntrophomonas sp. SJ1015 | 3.2 | 49.3% | 85 | 90.03% (2.93%) | 1.35% | 2.01% | 9.53–12.80% | 11.16–14.03% |
| MAG004 | Methanosaeta concilli | 2.7 | 51.9% | 94 | 100.00% (0.65%) | 3.11% | 3.60% | 7.03–23.91% | 11.82–24.44% |
| MAG010 | Methanospirillum hungatei | 3.4 | 45.4% | 50 | 99.02% (0.98%) | 1.30% | 0.00% | 4.63–12.64% | 4.84–9.91% |
MAG006 is a taxonomically novel member of the family Desulfovibrionaceae with no close relatives at the genus or species level in the GTDB, hereby referred to as Desulfovibrionaceae bacteriumsp. SJ1006. MAG002 has 99.15% average nucleotide identity (ANI) to Syntrophomonas sp. UBA5314 (NCBI: txid1947633), a MAG obtained from the pit mud of a solid-state fermentation reactor used for the production of Chinese liquor (Guo et al. 2014), and is considered to be the same species. MAG015 is a taxonomically novel member of the genus Syntrophomonas with no close relatives at the species level in the GTDB, hereby referred to as Syntrophomonas sp. SJ1015. MAG004 is determined to be the aceticlastic methanogen Methanosaeta concilii based on an ANI of 98.49%. MAG010 is determined to be the hydrogenotrophic methanogen Methanospirillum hungatei based on an ANI of 97.92%.
Temporal transcriptomic response of Desulfovibrionaceae bacterium sp. SJ1006
Principal coordinate analysis (PCoA) and alignment of metatranscriptome reads to Desulfovibrionaceae bacterium sp. SJ1006 reveals a tight cluster of three high-activity metatranscriptome profiles—6 h post-feeding under control conditions and under sulfate provision and 24 h post-feeding under sulfate provision (Fig. 4; Table S2, Supporting Information). These data show that the activity of this MAG is altered by the presence of sulfate to favor prolonged high activity in the presence of sulfate and are consistent with chemical data that show a decrease in methane production in the first 24 h post-feeding with sulfate and a plateau in sulfide levels after 24 h (Fig. 2). When coupled with the increase in relative abundance of reads mapping to the MAG in the long-term sulfate enrichment metagenome (Table 2), these data suggest that the MAG shifts its physiological strategy in response to sulfate. While metatranscriptome profiles at 6- and 24-h post-feeding with sulfate show tight clustering, in-depth analysis of differences in transcriptional activity sheds light on the physiological plasticity of the MAG (Fig. 5; Table S3, Supporting Information).
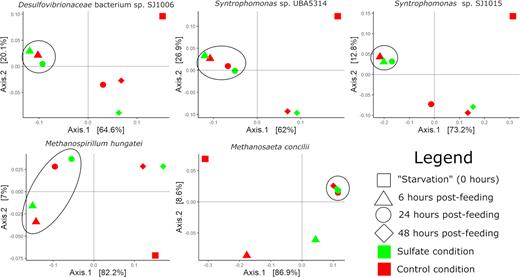
PCoA plots of metatranscriptome profiles for MAGs of interest. High-activity profiles are circled. Shapes and colors represent time points and conditions, respectively, as described in the legend.
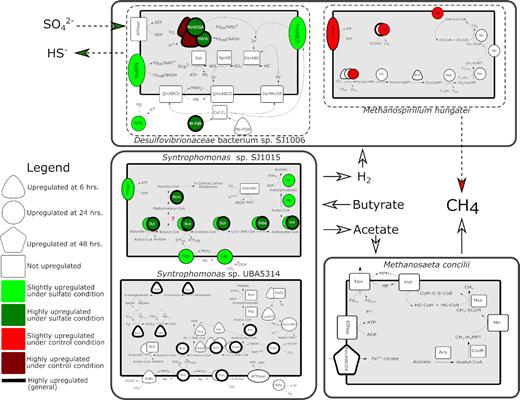
Metabolic reconstruction of MAGs of interest under sulfate provision and control conditions during high activity. Slightly up- or downregulated transcripts correspond to 2- to 10-fold differences in transcript abundance. Highly up- or downregulated transcripts correspond to >10-fold differences in transcript abundance. Coding sequence (CDS) numbers and gene names associated with abbreviations can be found in Table S3 (Supporting Information). Shapes, colors and line width represent time points, conditions and strength of up- or downregulation, respectively, as described in the legend.
The core DSR machinery of Desulfovibrionaceae bacterium sp. SJ1006 is highly transcriptionally active in all high-activity metatranscriptomes, with cytoplasmic components showing higher overall expression than membrane-bound components. This suggests that the MAG maintains high expression of the core DSR machinery in the absence of high sulfate concentrations, which is largely consistent with previous studies that have demonstrated constitutive expression of DSR during both respiratory and fermentative metabolisms of diverse SRB (Neretin et al. 2003; Muyzer and Stams 2008; Plugge et al. 2010, 2011; Meyer et al. 2013; Otwell et al. 2016; Sedano-Núñez et al. 2018). Under both conditions, concomitant high expression of ATP synthase, hydrogenases and various electron transporting complexes (ferredoxin, rubredoxin) further supports that the MAG is generating energy from high levels of electron transport during high-activity conditions irrespective of sulfate provision.
An operon encoding for the one-electron transmembrane transfer complex TmcABCD, which facilitates transmembrane transport of electrons from periplasmic H2 oxidation via TpIc3-type cytochromes (Grein et al. 2013), is highly expressed and upregulated at 24 h under sulfate provision. This complex exhibits high functional redundancy with DsrMKJOP and its upregulation under sulfate provision suggests that it likely serves as an extra pool of electron transport power during instances of high sulfate reduction activity.
Two operons with homology to cytoplasmic selenocysteine-containing electron-bifurcating hydrogenase complexes from Desulfobacterium autotrophicum (Strittmatter et al. 2009) are present, one of which is highly upregulated at 6 h under control conditions, while the other is highly upregulated at 24 h under sulfate provision. The complex contains an Mvh-like hydrogenase that has been suggested to use H2 as an electron donor for reduction of a HdrA-like protein with an NADH binding site (Strittmatter et al. 2009). Bifurcated electrons could reduce both NAD+ and ferredoxin, or the dithiol on DsrC (Grein et al. 2013). The complex, which is upregulated at 24 h under sulfate provision, is located near a proton-translocating NADH:ubiquinone oxidoreductase on the same, which likely contributes to the ATP-generating proton motive force during periods of high activity.
The genome also encodes two highly active periplasmic selenocysteine-containing formate dehydrogenases (FDHs) with homology to molybdenum- and tungsten-containing FDHs from SRB, which are believed to transfer electrons to the cytochrome c3 pool (Pereira, Haveman and Voordouw 2007). One is highly upregulated at 6 h irrespective of sulfate provision, while the other is highly upregulated only at 24 h under sulfate provision. The concomitant high upregulation of an ABC-type tungstate transport system indicates that the operon likely encodes a FDH1-type tungsten containing FDH (W-FDH). Increased expression of a W-FDH under sulfate provision is consistent with previous studies that have found preferential use of W-FDH during formate- and hydrogen-fueled sulfate reduction in Desulfovibrio (da Silva et al. 2011; Mota et al. 2011). This preference has been linked to the increased solubility of tungsten sulfides compared to molybdenum sulfides and is consistent with high rates of DSR under sulfate provision. Differential transcriptional activity of these two homologous electron-bifurcating hydrogenases suggests flexibility to changing redox conditions induced by sulfate provision.
Taken together, these data suggest that Desulfovibrionaceae bacterium sp. SJ1006 responds to sulfate provision by increasing the number of electrons being shuttled to the core DSR machinery (Fig. 5). Additional sources of electrons are provided by a NiFe hydrogenase and a putative W-containing formate dehydrogenase. Aiding in increased transfer of electrons to the core DSR machinery are auxiliary oxidoreductases that are upregulated under sulfate provision. These processes supply the increased demand for electrons by the MAG under DSR conditions.
Absence of sulfur oxidation
Low expression of sulfur oxidation (Sox) genes in a Sulfuricurvum-like MAG (MAG001) recovered from the SJ1 metagenome suggests that this organism is not catalyzing high levels of sulfur oxidation in the culture (Table S4, Supporting Information). Instead, the organism is likely responding to oxidative stress while performing oxidative phosphorylation using oxygen or nitrate as TEA. This is supported by high expression of oxidative stress response and central carbon metabolism pathways in addition to cytochrome c oxidases and a nitrate reductase.
Temporal transcriptomic response of syntrophic butyrate oxidizers
Syntrophomonas sp. UBA5314
PCoA of metatranscriptome profiles for Syntrophomonas sp. UBA5314 reveals clustering by time post-feeding irrespective of TEA availability, suggesting that the activity is largely unaltered by the presence of sulfate (Fig. 4). There is a loose clustering of metatranscriptomes at 6- and 24-h post-feeding. Alignment of metatranscriptomic reads to the MAG suggests highest activity in this loose cluster, which is consistent with preliminary data that show consumption of butyrate by 24 h (Figs 1 and 5; Tables S3 and S5, Supporting Information).
Transcripts encoding for the oxidation of butyrate to acetate are highly abundant at both 6 and 24 h, with all genes leading up to the production of acetyl-CoA being upregulated at 24 h. These data are consistent with the role of Syntrophomonas species in syntrophic butyrate oxidation (Schmidt et al. 2013) and suggest that energy generation from beta-oxidation is most active in the MAG at 24 h post-feeding.
At 6 h, the MAG slightly upregulates an operon encoding for poly-β-hydroxyalkanoate (PHA) storage with homology to PHA storage genes used by Syntrophomonas wolfei. In S. wolfei, PHA synthesis is upregulated during exponential growth for use as an endogenous carbon source when hydrogen and acetate are too high for the fermentation of butyrate to be thermodynamically favorable (Beaty and McInerney 1989; McInerney et al. 1992). At the same time, stress response genes are highly upregulated, including thioredoxin, rubrerythin, superoxide reductase and catalase, which is consistent with previous studies that have linked stress response to PHA storage (Castro-Sowinski et al. 2010). Also highly expressed are enzymes likely involved with cycling reducing equivalents during oxidative stress, including glutamine synthetase and an NADP-specific glutamate dehydrogenase (Castro-Sowinski et al. 2010; Mozejko-Ciesielska, Pokoj and Cieselski 2018). During high PHA synthesis activity, stress response pathways likely function to detoxify peroxide intermediates formed during PHA synthesis (Lee, Li and Yu 2009). These data suggest that oxidative stress response genes play a role during the PHA synthesis stage.
Electrons liberated during beta-oxidation of butyrate flow through a series of oxidoreductases and dehydrogenases to reduce protons to produce H2 and reduce CO2 to formate. At 6 h, there is a slightly upregulated putative dimeric FeFe hydrogenase, while at 24 h, there is a slightly upregulated operon homologous to the multimeric Hyd1ABC hydrogenase. This latter hydrogenase is homologous to a FeFe NADH-dependent, ferredoxin-independent hydrogenase complex (Losey et al. 2017). Its maturation protein complex, HydEFG, is slightly upregulated at 6 h. Another FeFe hydrogenase, which is homologous to a soluble Hyd3 monomeric FeFe hydrogenase and is not predicted to interact with NADH (Poudel et al. 2016), is also highly active within the first 24 h post-feeding. The presence of multiple FeFe hydrogenases is consistent with a role in hydrogen production (Vignais, Billoud and Meyer 2001) and high expression of these hydrogenases at both 6 and 24 h suggests an important role for proton reduction during high transcriptional activity.
There is one slightly upregulated formate dehydrogenase homologous to the periplasmic-oriented FDH-2 at 6 h, while at 24 h, there are four upregulated formate dehydrogenase operons. Two are homologous to the periplasm-oriented FDH-4 and one of the operons also contains a highly upregulated formate efflux transporter, formate dehydrogenase formation protein, FdhE, and NAD-reducing hydrogenase, HoxF. Electrons flowing to these formate dehydrogenases are predicted to originate from beta-oxidation of butyryl-CoA and proceed through a dimeric electron transfer flavoprotein, EtfAB, and its associated Fe-S oxidoreductase, which donate the electrons to the menaquinone pool where they are subsequently transferred to FDH-4 (Sieber et al. 2010; Schmidt et al. 2013). The other two highly upregulated formate dehydrogenases are homologous to the cytoplasmic FDH-5, which is involved with NADH cycling via concomitantly high-expressed NADH oxidoreductases within the same operons (Sieber et al. 2010). Switching of key hydrogenases and formate dehydrogenases from 6 to 24 h suggests differential use of these complexes during PHA storage phase and late-stage beta-oxidation.
Finally, a NiFe hydrogenase homolog typically used for hydrogen oxidation is highly upregulated at 24 h. NiFe hydrogenases have not been identified in the related type species, S. wolfei (Sieber et al. 2010). The presence of a NiFe hydrogenase in the MAG suggests the potential for separation of proton-consuming and proton-generating reactions, which could help fuel a proton motive force.
Taken together, these data suggest that Syntrophomonas sp. UBA3514 does not shift its transcriptomic response under sulfate provision conditions but does exhibit a shift in response over the course of the microcosm incubations (Fig. 5). At 6 h post-feeding, the MAG appears to focus its energy utilization on PHA storage using electrons at 24 h post-feeding, and the MAG upregulates its syntrophic butyrate oxidation machinery, fueling increased production of formate and molecular hydrogen via multiple concomitantly upregulated formate and FeFe hydrogenases.
Syntrophomonas sp. SJ1015
PCoA and alignment of metatranscriptome reads for Syntrophomonas sp. SJ1015 reveal a tight cluster of three high-activity metatranscriptome profiles—6 h post-feeding with and without sulfate and 24 h post-feeding with sulfate (Fig. 4). These data show that the activity of the MAG is indirectly altered by the presence of sulfate to favor prolonged high activity under sulfate provision. When coupled with the increase in relative abundance of reads mapping to the MAG in SJ1S (Table 2), these data suggest that physiological shifts in response to sulfate may provide a long-term competitive advantage to Syntrophomonas sp. SJ1015 under sulfate provision. Within the tight cluster of high-activity metatranscriptomes, in-depth differential expression analysis reveals differences in their expression profiles that shed light on the physiological plasticity of the MAG (Fig. 5; Tables S3 and S6, Supporting Information).
ATP synthase and genes involved in the oxidation of butyrate to acetate are upregulated at 24 h under sulfate provision, suggesting that the MAG increases its ability to catalyze butyrate oxidation under sulfate provision in the reactor. Like Syntrophomonas sp. UBA5314, genes for PHA synthesis are present, however, they are not highly expressed. However, two stress response genes remain highly expressed at 6 h—a putative rubrerythrin-associated Fe-S oxidoreductase and thioredoxin. The decreased reliance of Syntrophomonas sp. SJ1015 on PHA storage could conceivably provide a competitive advantage when sulfate is available if the hydrogenotrophic SRB were able to maintain a low enough H2 partial pressure to drive the thermodynamics of butyrate oxidation forward, thereby favoring a syntrophic butyrate oxidizer that bypasses PHA synthesis and provides a steady source of beta-oxidation end-products to hydrogen scavengers.
There are fewer highly expressed electron transfer complexes and hydrogenases in Syntrophomonas sp. SJ1015 compared to Syntrophomonas sp. UBA5314. There is only one highly expressed multimeric FeFe hydrogenase homologous to Hyd1ABC, which is not differentially expressed in high-activity metatranscriptomes. At 24 h, there is one highly expressed and slightly upregulated periplasmic, FDH-4-like formate dehydrogenase in addition to an associated electron transfer flavoprotein and Fe-S oxidoreductase.
At 6 h, two operons containing genes for arginine biosynthesis are upregulated, as are genes encoding for 2-oxoglutarate oxidoreductase, phosphoenolpyruvate synthase and pyruvate:ferredoxin oxidoreductase. These data suggest high activity of central carbon metabolism during this period. At 24 h post-feeding with sulfate, an operon containing genes for methylmalonyl-CoA mutase metabolism is highly abundant and very highly upregulated. Evidence suggests that butyryl-CoA can be directly converted to methylmalonyl-CoA, though the enzymatic catalysis remains to be elucidated (Vrijbloed et al. 1999; Li et al. 2004). Thus, methylmalonyl-CoA could potentially be important as an intermediate for conversion to butyryl-CoA, either providing an additional source of butyryl-CoA or feeding methylmalonyl-CoA into central carbon metabolism.
Taken together, these data suggest that Syntrophomonas sp. SJ1015 increases its activity under sulfate provision as evidenced by its sustained high expression of its transcriptome profile and the upregulation of core genes for butyrate oxidation, ATP synthesis and central carbon metabolism (Fig. 5).
Temporal transcriptomic response of methanogens
Methanospirillum hungatei
PCoA of metatranscriptome profiles for M. hungatei reveals clustering by time post-feeding, with a loose cluster at 6- and 24-h post-feeding, while TEA availability appears to play a minor role in expression profile clustering (Fig. 4). This suggests that the activity of M. hungatei is minimally altered by the presence of sulfate. The disappearance of M. hungatei in the sulfate enrichment metagenome (Table 2) suggests that M. hungatei is ultimately outcompeted to extinction by hydrogenotrophic SRB. Alignment of metatranscriptomic reads to M. hungatei suggests high activity during the first 24 h post-feeding, consistent with quick release of hydrogen via syntrophic butyrate oxidation by Syntrophomonas sp. UBA5314 and Syntrophomonas sp. SJ1015 (Fig. 5; Tables S3 and S7, Supporting Information).
Transcripts for core enzymes of hydrogenotrophic methanogenesis have different activities throughout the first 48 h post-feeding, which broadly represent sequential stages of hydrogenotrophic methanogenesis (earlier stages have higher transcriptional activity earlier in the incubation). The genes for methyl-coenzyme M reductase, which catalyze the final stage of hydrogenotrophic methanogenesis, are highly slightly upregulated at 24 h under both conditions, while the associated CoB–CoM heterodisulfide reductase genes are upregulated at 24 h under control conditions only. The downregulation of ATP synthase and genes for the CO2 reduction stage of methanogenesis and CoB–CoM cycling under sulfate provision suggest these genes play essential roles in the ability of M. hungatei to compete for electron donors with Desulfovibrionaceae bacterium sp. SJ1006.
There are two highly expressed formate dehydrogenases in M. hungatei. One of these is slightly upregulated at 24 h under control conditions. The other is highly upregulated at 24 h under both conditions. Additionally, there is a highly expressed F420-reducing hydrogenase that is upregulated at 24 h under control conditions but not under sulfate provision conditions. Finally, there is one highly expressed ATP synthase operon that is slightly upregulated at 24 h under control conditions.
Taken together, these data suggest that M. hungatei is highly active during the first 24 h post-feeding under control conditions, with lowered activity of core hydrogenotrophic methanogenesis pathway enzymes, electron transfer complexes and ATP synthase under sulfate provision conditions. This expression difference is ultimately believed to lead to the extinction of M. hungatei in SJ1S (Fig. 5).
Methanosaeta concilii
PCoA of M. concilii reveals tight clustering of four metatranscriptomes—24 and 48 h post-feeding under both sulfate provision and control conditions (Fig. 4), which is consistent with preliminary data that show methane production and acetate consumption continuing through 48 h (Fig. 1). Alignment of metatranscriptomic reads to M. concilii suggests this cluster represents high-activity expression profiles, which is consistent with a slower conversion of the acetate pool to methane than the hydrogen pool. In-depth expression analysis was used to reveal core metabolic activity of M. concilii when activity is high (Fig. 5; Tables S3 and S8, Supporting Information).
During high activity, the core aceticlastic methanogenesis pathway and CoB–CoM heterodisulfide reductase are highly transcriptionally active, as are genes for ATP synthase and an F420H2 dehydrogenase complex (Fpo). This Fpo complex contains methanopherazine subunits but lacks the subunit necessary to interact with F420H2 and is proposed to interact with ferredoxin, which is also highly expressed, and translocate protons to be used by ATP synthase (Welte and Deppenmeier 2011). Additionally, a partial NADH–ubiquinone oxidoreductase complex with homology to NuoEF is highly upregulated at 24 h under both conditions. These data support high activity of aceticlastic methanogenesis beginning by 24 h, which remains unaffected by sulfate provision and continues through 48 h post-feeding.
At 48 h, genes for Fe(III) transport are highly upregulated, including two Fe(III)-ABC transporters, an Fe(III)-dicitrate transport system permease, and an operon with an Fe(III)-dicitrate-binding protein, a methyltransferase, a putative stomatin/prohibitin-family membrane protease, an Fe(III)-dicitrate transport system permease, and a vitamin B12 ABC transporter. The upregulation of Fe(III)-citrate transporters and binding proteins at 48 h suggests an important role for uptaking metal-citrate complexes during the later stages of aceticlastic methanogenesis, likely due to low ferric iron availability. This is consistent with findings from another mineral-poor methanogenic habitat, peatlands, where the addition of chelator citrate was shown to enhance CH4 production in the peat with the lowest metal concentrations (Basiliko and Yavitt 2001).
Limitations
A major methodological limitation of this study is the lack of biological replicates of metatranscriptomes. Without replicates, statistical analyses for differential expression could not be performed. Nevertheless, PCoA clustering of metatranscriptome profiles between treatments and time points for MAGs of interest suggest reproducibility of transcriptomic data. Therefore, we believe the data are reliable and differences in transcript abundances represent real changes in transcriptional activity.
CONCLUSION
We conclude that stimulation of Desulfovibrionaceae bacterium sp. SJ1006 by sulfate provision in our methanogenic bioreactor does not affect the ability of the aceticlastic methanogen, M. concilii, to acquire substrates but reduces the activity of the hydrogenotrophic methanogen, M. hungatei, ultimately leading to its extinction. Furthermore, the competitive interactions shift the ecophysiology of syntrophic butyrate oxidizers to favor Syntrophomonas sp. SJ1015, a species that not appear to rely heavily on the production of PHA.
FUNDING
This work was supported by a microbial/metagenome project (CSP 503205) through the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231. Additional funding was provided by the Cornell University Program in Cross-Scale Biogeochemistry and Climate, which is supported by the National Science Foundation Integrative Graduate Education and Research Traineeship Program (NSF-IGERT) and the Atkinson Center for a Sustainable Future.
Conflicts of interest None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}