





ABSTRACT
Fe-oxidizing bacteria of the family Gallionellaceae are major players in the Fe biogeochemical cycle in freshwater. These bacteria thrive in redox transition zones where they benefit from both high Fe concentrations and microaerobic conditions. We analysed the Gallionellaceae genomic diversity in an artesian hard-rock aquifer where redox transition zones develop (i) in the subsurface, where ancient, reduced groundwater mixes with recent oxygenated groundwater, and (ii) at the surface, where groundwater reaches the open air. A total of 15 new draft genomes of Gallionellaceae representing to 11 candidate genera were recovered from the two redox transition zones. Sulfur oxidation genes were encoded in most genomes while denitrification genes were much less represented. One genus dominated microbial communities belowground and we propose to name it ‘Candidatus Houarnoksidobacter’. The two transition zones were populated by completely different assemblages of Gallionellaceae despite the almost constant upward circulation of groundwater between the two zones. The processes leading to redox transition zones, oxygen diffusion at the surface or groundwater mixing in subsurface, appear to be a major driver of the Gallionellaceae diversity.
INTRODUCTION
Lithoautotrophic microorganisms occupy an essential position in the biogeochemical functioning of aquifers. They mediate a large array of redox element transformations, partly controlling mineral weathering and the chemical composition of water. They also fix inorganic carbon and therefore act as the primary producers for subsurface microbial communities. Microbial activity in aquifers is often limited by the imbalance between concentrations of electron donors and acceptors. Transport of missing reactants along groundwater flowpaths allows creating hot-spots of biogeochemical reactivity where rates of microbially-mediated processes are strongly enhanced.
Iron-rich microbial mats produced by microaerobic iron-oxidizing bacteria (FeOB) represent a clearly visible manifestation of such biogeochemical hot-spots. FeOB are able to conserve energy from the oxidation of dissolved reduced iron (Fe) to sustain their needs for growth and metabolic activity. The mats form at transition zones between a reduced and an oxic compartment. They are typically found in discharge zones of aquifers where iron-rich anoxic fluid comes into contact with atmosphere (e.g. in groundwater seepages; Emerson and Weiss 2004; Krepski, Hanson and Chan 2012; Kato et al. 2014) or mixes with oxygenated water in freshwater streams (Fleming et al. 2014; Quaiser et al. 2014) or hydrothermal vents (Emerson and Moyer 2002). The presence of active FeOB were also detected in subglacial sediments (Hamilton et al.2013). In these environments, FeOB benefit from both an elevated concentration of reduced iron and a low level of molecular oxygen. Microoxic conditions is thought to be especially important for FeOB to thrive because of the FeOB's inability to effectively outpace abiotic oxidation processes when oxygen concentration is high (Druschel et al. 2008; Melton et al. 2014). Wherever these interfacial environments are present, characteristic rust-colored microbial mats resulting from the production by FeOB of extracellular structures encrusted with Fe(III) can be observed (Chan et al. 2016).
In terrestrial environments, microaerobic FeOB are mostly represented by Leptothrix ochracea (Fleming et al. 2011) and the family Gallionellaceae. Leptothrix ochracea appears to be restricted to aboveground environments (Chan et al. 2016). Conversely, members of the family Gallionellaceae have been detected in groundwater of suboxic to anoxic aquifers where they can make up a substantial proportion of microbial communities. Diversity analyses in fractured aquifers of a crystalline basement (Armorican massif, Western France) revealed that Gallionellaceae FeOB predominated in groundwater with long residence time in the bedrock fractures. They were rare or absent in groundwater with short residence time in the alterite layer that covers the bedrock (Ben Maamar et al. 2015). Because of constraints linked to their ecophysiology, the presence of Gallionellaceae in fractured aquifers likely implies the existence of mixing processes between oxygenated recent groundwater from the alterite layer and the anoxic, iron-rich groundwater that fill in fractures in the underneath layer. The ability of the Gallionellaceae to rapidly respond to inputs of electron donors or acceptors was further demonstrated with the injection of CO2-enriched fluids in a basaltic deep aquifer, which led to bedrock dissolution and the release of Fe(II) (Trias et al. 2017) and of nitrate in a shallow alluvial aquifer (Jewell et al. 2016). In both studies, shifts in community composition with an increased abundance of Gallionellaceae were observed following the injections. Higher expression of genes involved in nitrate reduction and sulfur oxidation by Gallionellaceae were also observed in response to the nitrate input (Jewell et al. 2016). These observations add to the body of evidences suggesting that dissimilatory sulfur and nitrogen pathways are part of the Gallionellaceae’s energetic metabolism alongside iron oxidation. Genes involved in S cycling were identified in genomes of representative isolates (Emerson et al. 2013; Kato et al. 2015) while N and S cycling genes were found in draft genomes from assembled metagenomes (Jewell et al. 2016; Kadnikov et al. 2016; Mühling et al. 2016). However, the relative importance of the different dissimilatory pathways in the energetic metabolism of natural communities of Gallionellaceae remained elusive to date.
In this study, we reconstructed genomes of the dominant populations of FeOB present in groundwater of a fractured aquifer in Brittany (Western France). An abundant Fe-rich mat made by FeOB was visible in an observation borehole used to characterize the hydrological functioning of the aquifer. The mat also covered the external wall of the borehole tubing that emerged from the ground. Upward circulations of deep, anoxic, Fe-rich groundwater was measured in the aquifer. Through the fracture network, the reduced fluid mixes with oxygenated groundwater originating from the surficial and weathered part of the aquifer. The mixing process which occurs at fracture intersections, provides favorable conditions for the development and the activity of FeOB in the aquifer. We analyzed the genetic makeup encoded in the reconstructed genomes to assess the diversity and predict the energetic metabolism of FeOB dwelling in the fracture network and in the groundwater-dependent microbial mat that developed outside the borehole.
MATERIAL AND METHODS
Site description
The site study is located near the city of Guidel (Brittany, Western France) and is part of the French network of hydrogeological observatories (H+ hplus.ore.fr/en) and the OZCAR Critical Zone observatories. Previous studies led in Guidel have shown that the aquifer system is separated in two entities: a superficial weathered zone, and a deeper fractured formation (Ayraud et al. 2008). The weathered zone, which is actively recharged, presentsrecent (apparent age ranging from 10 to 20 years), oxygenated groundwater, whereas the deeper fractured area is characterized by ancient (apparent age of about 100 years), anoxic, iron-rich groundwater. The presence of highly-dipping fractures enables mixing of oxygenated and reduced fluids, at fracture intersections and the connections between the two zones (Touchard 1998; Bochet et al. 2020). The productivity of the fractures triggering oxygenated groundwater, and therefore the mixing conditions, vary over time. The main driver of the variations is the alternation of recharge and discharge periods which creates a dynamic environment both spatially and temporally.
Samples used in this study were all collected from a 130 m deep artesian borehole named PZ26. The borehole is located in low topographic area corresponding to the discharge area of the Guidel aquifer. It intercepts fractures at different depth which provide pathways for both a large upward flux of deep anoxic groundwater, which made up the majority of groundwater flowing out the borehole, and for the smaller downward fluxes of oxygenated groundwater from the weathered zone (Fig. 1A).
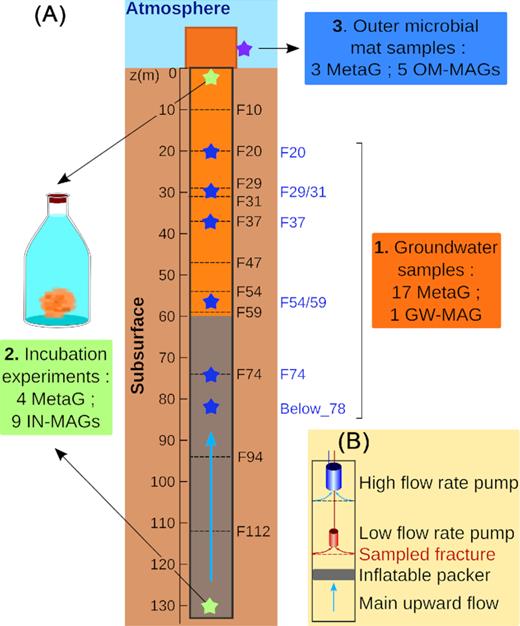
Groundwater and microbial mat samples collected in the PZ26 borehole. (A) Fe-rich anoxic groundwater mostly coming from the fractures deeper than 90 m continuously outflows from the borehole. Rust-colored microbial mat was present on the inner wall of the borehole between 0 m and 60 m depth and on the outer wall of the tubing (see Fig. S1, Supporting Information). Samples for metagenomic analyses were as follows: (1) Groundwater samples collected with the packer system a vertically discrete approach in 5 fractures. A sixth sample representing the main deep groundwater flow was collected below 80 m depth. (2) Groundwater samples for incubation experiments. A Fe-rich microbial mat developed in the bottles. (3) Outer microbial mat samples. The number of sequenced metagenomes (MetaG) and recovered MAGs of Gallionellaceae are indicated for each type of samples. (B) Scheme of the discrete sampling system used to collect groundwater in fractures. Symbol significations: the main upward flow of groundwater is figured with light blue arrows in A and B. Fractures are depicted as dashed lines. Depth is indicated on the left of the borehole. Fracture names are written on the right. Blue stars indicated depth at which groundwater samples were collected with the packer system. Sample names appear in blue. Green stars correspond to sampling depth for the incubation experiments and the purple star represents the outer microbial mat samples.
At the time of this study, a rust-colored microbial mat profusely covered the internal wall of the borehole from the surface down to 60 m depth (Bochet et al. 2020). Below this depth, the internal wall was bare with only traces of the mat in front of the fractures. The massive development of the mat led to the partial obstruction of the borehole in front of fractures located between 20 m and 60 m depth. Flaky, orangish aggregates continuously detached from the mat and were driven to the top of the borehole by the groundwater upward flux. The mat appeared similar to iron-rich microbial mat resulting from the activity of microaerobic iron-oxidizing bacteria (FeOB). The development of FeOB at depth implied the mixing of downward fluxes of oxygenated groundwater with upward circulations of the iron-rich, anoxic deep fluid.
A thin layer (∼ 1 cm thick) of a denser ochreous mat also covered the outer wall of the tubing that protrudes from the well casing (Fig. 1A and Fig. S1, Supporting Information) above the soil. This mat was fed by groundwater directly flowing down along the outer face of the tube and was in direct contact with the atmosphere.
Sampling methods
Metagenomic analyses were carried out on three series of samples (Fig. 1A):
Groundwater samples from 6 fractures or group of fractures intersecting the borehole were collected in June 2015 (Fig. 1A). A depth-discrete sampling using a tailored system was carried out to isolate groundwater fluxes from fractures F20 to F74 (Fig. 1A). The sampling system was made of three different devices: an inflatable hydraulic obturator (packer), a high flow rate pump (35 L/min) and a low flow rate pump (4 L/min) (Fig. 1B). The inflatable packer was placed below the sampled fracture to interrupt the upward flux of deep groundwater. The high flow rate pump was installed close to the opening of the borehole, allowing to divert fluxes from the fractures above the targeted fracture. The low flow rate pump was positioned in front of the targeted fracture to collect groundwater samples. No packer was used to get samples of the deep reduced groundwater (samples below_78, Fig. 1A). Each time, three samples (5 L each) were independently collected and filtered to 0.22 μm with durapor membrane filters.
Four samples of groundwater (1 L each) were also collected at the top (∼ 1 m depth) and at the bottom (∼ 130 m depth) of the borehole (Fig. 1A) for incubation experiments in June 2015. No packer was used for this series of samples. Sampled groundwater was incubated in glass bottles in the dark at 15°C without addition of nutrient and gas. Bottles were sealed with a rubber septum to prevent exchange with the atmosphere. No Fe-rich mat was visible at the beginning of the experiment. Ferrous iron concentration (measured by inductively coupled plasma mass spectrometry) and oxygen concentration (measured by gas chromatography) were followed during the experiment. The incubation lasted until oxygen and Fe(II) were entirely consumed and orangish flocculent deposits, were visible (respectively one week and two weeks for the groundwater of the top and of the bottom of the borehole). Light microscopy observations reveal the presence of numerous twisted stalks and tubular sheaths in the deposits. In freshwater, stalks are often associated with the Gallionellaceae and sheaths are indicative of the presence of Leptothrix ochracea (Chan et al. 2016).
The content of the bottles was filtered to 0.22 μm with durapor membrane filters. All filters were immediately flash-frozen in liquid nitrogen and then stored at −20°C.
Finally, three samples of about 1 g were retrieved from the dense microbial mat growing on the exterior face of the tubing in October 2014 (Fig. 1A and Fig. S1, Supporting Information). They were mixed with 2 ml of RNAlater then flash-frozen in liquid nitrogen and stored at −20°C.
DNA extractions and Illumina sequencing
Filters were placed in sterile tubes with 7 ml of lysis buffer with cetyltrimethylammonium Bromide and polyvinylpyrrolidone. Filters were incubated at 65°C for 30 min (vortexing every 5 min during 20 s) and one volume of chloroform–isoamylalcohol (24:1) was added. Tubes were then vortexed and incubated for 10 minutes at room temperature. After incubations, tubes were centrifuged at 4000 rpm for 30 minutes at 15°C and the aqueous phase was removed. DNA was further purified and concentrated using the NucleoSpin gDNA Clean-up kit (Macherey-Nagel), and the Illustra MicroSpin S-400 HR columns (GE Heathcare). DNA yields were quantified either by Nanodrop or Qbits. DNA concentrations are provided in Table S1 (Supporting Information).
DNA was fragmented with a Covaris M220 Focused-ultrasonicator (ThermoFisher Scientific). Libraries of DNA fragments were then prepared with NEBNext® Ultra DNA Library Prep Kit for Illumina and NEBNext® Multiplex Oligos for Illumina (Index Primers Set 1 and 2) (New England Biolabs, Inc) and pooled after DNA quantification with Quantifluor (Promega). Metagenome libraries for the outer microbial mat and groundwater samples were sequenced with one 300 bp and one 150 bp paired-end MiSeq run (Illumina INC), and the incubation experiment metagenome libraries were sequenced with one 250 bp paired-end MiSeq run. Sequencing runs were carried out by the Human and Environmental Genomics facility of the University of Rennes and the total sequence generated was 24.41 Gbp.
Quality of metagenomic reads was evaluated with FastQC-v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Quality filtration and trimming of the reads were realized with FastQ QC-Trimmer (Galaxy ToolShed; https://toolshed.g2.bx.psu.edu/view/geert-vandeweyer/fastq_qc_trimmer/cba6282b5dc8) for the outer microbial mat and groundwater metagenomes and with CutAdapt-v1.12 (Martin 2011) for the incubation experiment metagenomes. Metagenomes are available from the European Nucleotide Archive (https://www.ebi.ac.uk/ena) with accession number ERP024054.
Taxonomic classification of the metagenomic reads
Metagenomic read sequences were compared to protein sequences of the NCBI-nr database with Diamond-blastx (Buchfink et al. 2014). Results were used to assign reads to taxons with MEGAN 6 (default parameters) (Huson et al. 2016).
Assembly of reads and contig binning
Paired-end reads of all metagenomes were co-assembled in contigs with SPAdes 3.7.0 (Bankevich et al. 2012) and the quality of the assembly was estimated with Quast 4.3 (Gurevich et al. 2013). Protein coding genes were predicted with Prodigal (Hyatt et al. 2010). Contigs were subsequently grouped in metagenome-assembled genomes (MAG) using the CONCOCT binning tool (Alneberg et al. 2014). This was followed by a manual curation of the bins with Anvi'o version 2.1.0 (Eren et al. 2015) in order to increase the percentage of completion and decrease the redundancy of phylogenetic marker genes. Only MAGs displaying at least 60% of completion and no more than 8% of redundancy were retained for analyses described hereafter.
MAGs affiliated to the order Gallionellales were annotated automatically with Prokka v1.12 (Seemann 2014). Protein-coding genes from the Gallionellales MAGs, 5 reference genomes of Gallionellaceae (Gallionella capsiferriformans ES-2, ‘Candidatus Gallionella acididurans’, Sideroxydans lithotrophicus ES-1, Ferriphaselus amnicola and Ferriphaselus sp. R-1) as well as genomes of representative strains of Betaproteobacteria (15 genomes) and Gammaproteobacteria (2 genomes) were classified into groups of orthologs with Orthofinder v2.2.7 (Emms and Kelly 2015). Details about the reference genomes of Gallionellaceae, Betaproteobacteria and Gammaproteobacteria are presented in Supporting Information (Table S2, Supporting Information).
Phylogenomic analyses
A phylogenomic analysis was carried on to confirm the taxonomic affiliation of the Gallionellales MAGs and determine the evolutionary relationships between them. We selected 68 single-copy orthologous genes shared by every MAG, all Gallionellaceae reference genomes and 17 genomes of Beta- and Gammaproteobacteria reference strains. Amino-acid sequences for each group of orthologous genes were aligned with Muscle (Edgar 2004) then concatenated to build up a superalignment of 14 618 positions. A Maximum-Likelihood tree based on the superalignment was generated with PhyML (Guindon et al. 2010) and the atgc-montpellier.fr/phyml website.
Average nucleotide identity (ANI) were calculated with the OrthoANIu algorithm (default parameters) (Yoon et al. 2017) to attempt to delineate species and genus demarcations between MAGs and Gallionellaceae reference genomes. ANI threshold values for delineating species and genera were chosen at 96% and 80%, respectively (Rosselló-Móra and Amann 2015).
Metagenomic read mapping and distribution profiles of the MAGs
Percentage of reads recruited by the MAGs and the reference genomes of Gallionellaceae were calculated to infer their distribution in the metagenomes. Reads from each metagenome were aligned against sequences of MAGs and reference genomes with BBMap-v36.59 (https://github.com/BioInfoTools/BBMap/blob/master/README.md) with default parameters. Only high quality alignments with less than 3 substitutions were considered. Likewise, both reads of a pair were discarded if only one aligned correctly. For each MAG, the percentage of recruited reads was divided by to the total size (i.e. the sum of contig length).
Hydrological and chemical measurements
Oxygen concentration, pH, conductivity and Eh were obtained by using in situ probes in the boreholes. The same parameters were measured during the incubation experiments. Analyses of cations, anions and dissolved organic and inorganic carbon were also carried out, as well as dissolved gas concentration measurements. All details concerning these measurements are available in supporting information.
RESULTS
Hydrological and chemical characterization
Concentrations of oxygen, dissolved iron and sulfate as well as redox potential (Eh) and pH were measured in groundwater samples. Overall, groundwater collected in all fractures was characterized by a circumneutral pH and reducing conditions. Eh increased gradually along the borehole, from 175 mV below 78 m depth to 249 mV at 20 m depth (Fig. 2). An intermediate peak was observed for fractures F54/59 (217 mV). Oxygen concentration was below the detection limit in all samples except in front of fractures F29/31 (0.13 mg/L) and F54/59 (4 mg/L) (Fig. 2). Samples collected in F54/59 were also characterized by higher ChloroFluoroCarbone concentrations (Bochet et al. 2020) indicating groundwater with a short residence time (Ayraud et al. 2008; Roques et al. 2014). This trend is in accordance with an upward flux of reduced groundwater that becomes more oxidizing with the contribution of more oxygenated fluids produced by the fractures crossing the borehole. The minimal dissolved Fe concentration was observed in fracture F20 (0.9 g/L). Dissolved Fe concentration increased with depth to reach 4.2 g/L in the deeper sample (Below_78) (Fig. 2). Sulfate (Fig. 2) and chloride (88.1 mg/L to 106.7 mg/L, not shown) concentrations presented little variations, regardless of the sampled fracture.
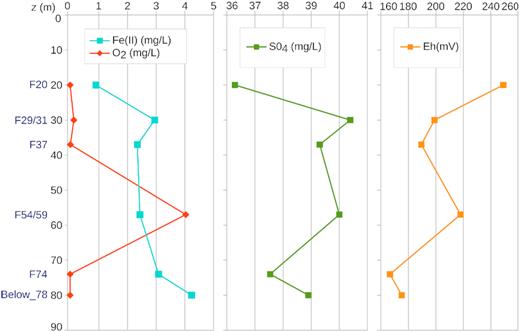
Iron, oxygen and sulfate concentrations and redox potential in groundwater sampled in fractures and below 80 m depth. Depth and name of sampled fractures are indicated on the left.
Assembly of metagenomes and recovery of bacterial genomes
Metagenome sequencing, assembly and binning methods were applied to recover draft genomes of bacteria populations dwelling in the groundwater of the fracture network as well as on the outer surface of the borehole tubing. Taxonomic profiles were obtained by comparing metagenomic reads with protein sequences of the NCBI-nr database. Overall, the percentage of metagenomic reads affiliated to the family Gallionellaceae for the groundwater metagenomes was relatively low with a maximum value of 5.5% for the F37 metagenomes (Table S3, Supporting Information). On the contrary, a much higher percentage of Gallionellaceae-affiliated reads (29.2%) was observed for the outer microbial mat metagenome (Table S3, Supporting Information). Interestingly, virtually no metagenomic read were assigned to the Leptothrix genus although the observation of tubular sheaths at the end of the incubation experiments (data not shown).
Incubation experiments of groundwater samples in closed glass bottles were set up in order to increase the proportion of Gallionellaceae before the preparation of metagenome libraries. At the end of the experiment, fluffy, rust-colored aggregates typical of iron-rich microbial mats, could be seen, suggesting that Gallionellaceae were active and growing in the bottles. Metagenomes were produced from the incubations and taxonomic classification of the reads suggested the enrichment in Gallionellaceae with of 17.05% reads affiliated to this family.
All in all, 30 metagenome-assembled genomes (MAGs) with a minimum of 60% of completion and a maximum redundancy for marker genes of 8% were reconstructed (Table 1 and Table S4, Supporting Information). A total of 19 MAGs came from the incubation experiments (dubbed IN-MAG hereafter), six from the outer microbial mat (OM-MAGs) and five were reconstructed from the groundwater metagenomes (GW-MAGs). It is important to note, however, that all the GW-MAGs came from the F54/59 metagenomes. No satisfactory MAG could be reconstructed from the other groundwater metagenomes. A total of 21 MAGs were classified in the Betaproteobacteria (15 IN-MAGs, 5 OM-MAGs and 1 GW-MAG). Other MAGs were assigned to the Gammaproteobacteria (3 IN-MAGs), Nitrospirae (2 GW-MAGs), Deltaproteobacteria (1 GW-MAG), Acidobacteria (1 IN-MAG), Actinobacteria (1 OM-MAG) and the candidate phylum Desantisbacteria (1 GW-MAG). Among the MAGs belonging to the class of Betaproteobacteria, 15 could be affiliated to the order Gallionellales and are analysed more in details hereafter (Table 1). No MAG related to Leptothrix was recovered from the assembly of metagenomes.
Taxonomic classification and characteristics of the Gallionellaceae MAGs. A total of 15 MAGs with completion greater than or equal to 60% and redundancy less than or equal to 8% were assigned to the order Gallionellales or to a lower taxonomic rank (i.e. the family Gallionellaceae and its associated genera). Abbreviations: % comp, % of completion; % red, % of redundancy.
| MAG | Taxonomic affiliation | Total size (Mb) | No. of contigs | N50 (bp) | GC% | % comp | % red | |
|---|---|---|---|---|---|---|---|---|
| Incubation experiments | IN3 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.1 | 70 | 59 537 | 55.5 | 95.7 | 0.7 |
| IN4 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.3 | 145 | 30 154 | 56.2 | 63.3 | 1.4 | |
| IN5 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.3 | 55 | 107 357 | 52.3 | 71.2 | 3.6 | |
| IN7 | Proteobacteria; Betaproteobacteria; Gallionellales | 4.2 | 417 | 14 115 | 54.7 | 91.4 | 7.9 | |
| IN10 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 369 | 8762 | 55.4 | 60.4 | 2.2 | |
| IN14 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.1 | 212 | 21 888 | 49.8 | 97.8 | 2.2 | |
| IN16 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.4 | 111 | 65 145 | 54.1 | 99.3 | 2.9 | |
| IN18 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.8 | 79 | 80 495 | 55.2 | 95.0 | 1.4 | |
| IN19 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.2 | 71 | 47 124 | 59.6 | 98.6 | 7.9 | |
| Outer microbial mat | OM20 | Proteobacteria; Betaproteobacteria; Gallionellales | 1.9 | 364 | 5888 | 51.4 | 77.7 | 2.2 |
| OM21 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 266 | 14 202 | 52.5 | 85.6 | 1.4 | |
| OM22 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.5 | 185 | 21 221 | 56.2 | 95.7 | 4.3 | |
| OM23 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.0 | 101 | 47 725 | 50.7 | 97.1 | 2.2 | |
| OM25 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.9 | 96 | 44 005 | 48.8 | 81.3 | 2.2 | |
| Groundwater (F54/59) | GW28 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 423 | 6881 | 55.1 | 81.3 | 2.9 |
| MAG | Taxonomic affiliation | Total size (Mb) | No. of contigs | N50 (bp) | GC% | % comp | % red | |
|---|---|---|---|---|---|---|---|---|
| Incubation experiments | IN3 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.1 | 70 | 59 537 | 55.5 | 95.7 | 0.7 |
| IN4 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.3 | 145 | 30 154 | 56.2 | 63.3 | 1.4 | |
| IN5 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.3 | 55 | 107 357 | 52.3 | 71.2 | 3.6 | |
| IN7 | Proteobacteria; Betaproteobacteria; Gallionellales | 4.2 | 417 | 14 115 | 54.7 | 91.4 | 7.9 | |
| IN10 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 369 | 8762 | 55.4 | 60.4 | 2.2 | |
| IN14 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.1 | 212 | 21 888 | 49.8 | 97.8 | 2.2 | |
| IN16 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.4 | 111 | 65 145 | 54.1 | 99.3 | 2.9 | |
| IN18 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.8 | 79 | 80 495 | 55.2 | 95.0 | 1.4 | |
| IN19 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.2 | 71 | 47 124 | 59.6 | 98.6 | 7.9 | |
| Outer microbial mat | OM20 | Proteobacteria; Betaproteobacteria; Gallionellales | 1.9 | 364 | 5888 | 51.4 | 77.7 | 2.2 |
| OM21 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 266 | 14 202 | 52.5 | 85.6 | 1.4 | |
| OM22 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.5 | 185 | 21 221 | 56.2 | 95.7 | 4.3 | |
| OM23 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.0 | 101 | 47 725 | 50.7 | 97.1 | 2.2 | |
| OM25 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.9 | 96 | 44 005 | 48.8 | 81.3 | 2.2 | |
| Groundwater (F54/59) | GW28 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 423 | 6881 | 55.1 | 81.3 | 2.9 |
Taxonomic classification and characteristics of the Gallionellaceae MAGs. A total of 15 MAGs with completion greater than or equal to 60% and redundancy less than or equal to 8% were assigned to the order Gallionellales or to a lower taxonomic rank (i.e. the family Gallionellaceae and its associated genera). Abbreviations: % comp, % of completion; % red, % of redundancy.
| MAG | Taxonomic affiliation | Total size (Mb) | No. of contigs | N50 (bp) | GC% | % comp | % red | |
|---|---|---|---|---|---|---|---|---|
| Incubation experiments | IN3 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.1 | 70 | 59 537 | 55.5 | 95.7 | 0.7 |
| IN4 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.3 | 145 | 30 154 | 56.2 | 63.3 | 1.4 | |
| IN5 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.3 | 55 | 107 357 | 52.3 | 71.2 | 3.6 | |
| IN7 | Proteobacteria; Betaproteobacteria; Gallionellales | 4.2 | 417 | 14 115 | 54.7 | 91.4 | 7.9 | |
| IN10 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 369 | 8762 | 55.4 | 60.4 | 2.2 | |
| IN14 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.1 | 212 | 21 888 | 49.8 | 97.8 | 2.2 | |
| IN16 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.4 | 111 | 65 145 | 54.1 | 99.3 | 2.9 | |
| IN18 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.8 | 79 | 80 495 | 55.2 | 95.0 | 1.4 | |
| IN19 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.2 | 71 | 47 124 | 59.6 | 98.6 | 7.9 | |
| Outer microbial mat | OM20 | Proteobacteria; Betaproteobacteria; Gallionellales | 1.9 | 364 | 5888 | 51.4 | 77.7 | 2.2 |
| OM21 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 266 | 14 202 | 52.5 | 85.6 | 1.4 | |
| OM22 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.5 | 185 | 21 221 | 56.2 | 95.7 | 4.3 | |
| OM23 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.0 | 101 | 47 725 | 50.7 | 97.1 | 2.2 | |
| OM25 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.9 | 96 | 44 005 | 48.8 | 81.3 | 2.2 | |
| Groundwater (F54/59) | GW28 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 423 | 6881 | 55.1 | 81.3 | 2.9 |
| MAG | Taxonomic affiliation | Total size (Mb) | No. of contigs | N50 (bp) | GC% | % comp | % red | |
|---|---|---|---|---|---|---|---|---|
| Incubation experiments | IN3 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.1 | 70 | 59 537 | 55.5 | 95.7 | 0.7 |
| IN4 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.3 | 145 | 30 154 | 56.2 | 63.3 | 1.4 | |
| IN5 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.3 | 55 | 107 357 | 52.3 | 71.2 | 3.6 | |
| IN7 | Proteobacteria; Betaproteobacteria; Gallionellales | 4.2 | 417 | 14 115 | 54.7 | 91.4 | 7.9 | |
| IN10 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 369 | 8762 | 55.4 | 60.4 | 2.2 | |
| IN14 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.1 | 212 | 21 888 | 49.8 | 97.8 | 2.2 | |
| IN16 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Sideroxydans | 3.4 | 111 | 65 145 | 54.1 | 99.3 | 2.9 | |
| IN18 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.8 | 79 | 80 495 | 55.2 | 95.0 | 1.4 | |
| IN19 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.2 | 71 | 47 124 | 59.6 | 98.6 | 7.9 | |
| Outer microbial mat | OM20 | Proteobacteria; Betaproteobacteria; Gallionellales | 1.9 | 364 | 5888 | 51.4 | 77.7 | 2.2 |
| OM21 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 266 | 14 202 | 52.5 | 85.6 | 1.4 | |
| OM22 | Proteobacteria; Betaproteobacteria; Gallionellales; Gallionellaceae; Ferriphaselus | 2.5 | 185 | 21 221 | 56.2 | 95.7 | 4.3 | |
| OM23 | Proteobacteria; Betaproteobacteria; Gallionellales | 3.0 | 101 | 47 725 | 50.7 | 97.1 | 2.2 | |
| OM25 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.9 | 96 | 44 005 | 48.8 | 81.3 | 2.2 | |
| Groundwater (F54/59) | GW28 | Proteobacteria; Betaproteobacteria; Gallionellales | 2.6 | 423 | 6881 | 55.1 | 81.3 | 2.9 |
Evolutionary relationships between the Gallionellales-affiliated MAGs were inferred by the phylogenomic analysis of 68 groups of orthologous protein-coding genes (Fig. 3). In the phylogenomic tree, the 15 Gallionellales MAGs and the Gallionellaceae reference genomes were grouped together in a monophyletic clade therefore confirming the affiliation of the MAGs to the family Gallionellaceae.
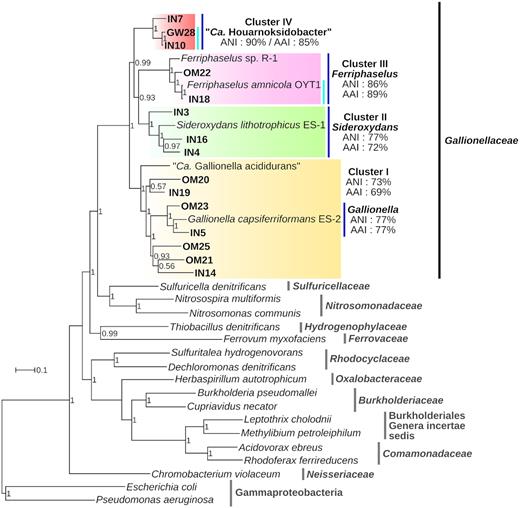
Phylogenomic analysis of the IN-, GW- and OM-MAGs inferred by maximum likelihood. The tree is based on 68 protein families (14 618 positions). Support values on the nodes were calculated with approximate Likelihood-Ratio tests (SH-like). Clusters described in the main text are highlighted with colored boxes. The mean ANI and AAI values for each clusters are indicated. Putative genera and species are shown with dark blue and light blue bars respectively. The name Candidatus Houarnoksidobacter is proposed for the cluster IV (see discussion). ANI and AAI values are provided in Tables S5 and S6 (Supporting Information).
Within the Gallionellaceae, 4 different clusters were delineated, three of them containing both MAGs and reference strains of Gallionellaceae and the fourth consisting only of MAGs from this study (Fig. 3). Average nucleotide identity (ANI) and average amino acid identity (AAI) were measured to attempt specifying the taxonomic rank of these clusters (Tables S5 and S6, Supporting Information).
The cluster IV contained 3 MAGs and no reference strain (Fig. 3). The high ANI and AAI values suggested that members of this cluster formed a distinct genus with the family Gallionellaceae (Tables S5 and S6, Supporting Information). Within the cluster IV, IN10 and GW28 had 98.8% ANI and 95.7% AAI indicating that they belonged to the same species. The cluster III contained two MAGs associated to the two Ferriphaselus reference strains (Fig. 3). The ANI and AAI measurements demonstrated that the cluster III corresponded to the Ferriphaselus genus and that Ferriphaselus amnicola and IN18 were members of the same species (Tables S5 and S6, Supporting Information). The cluster II included the Sideroxydans lithotrophicus ES-1 genome in addition to three IN-MAGs (Fig. 3). The ANI values were all below 80% denoting a lower level of genomic relatedness between members of the cluster II than in clusters III and IV (Table S5, Supporting Information). However, members of the cluster II had higher than 70% AAI and therefore may all belong to the Sideroxydans genus (Table S6, Supporting Information). The cluster I, the largest cluster within the Gallionellaceae clade in the tree, gathered the seven remaining MAGs as well as the two reference strains of Gallionella used in this study: Gallionella capsiferriformans ES-2 and ‘Candidatus Gallionella acididurans’ (Fig. 3). As for the cluster II, the ANI values were less than 80%. Only two MAGs (IN5 and OM23) grouped with Gallionella capsiferriformans ES-2 in the tree. The AAI values showed that these two MAGs were sufficiently similar to G. capsiferriformans ES-2 to be members of the Gallionella genus (Table S6, Supporting Information). All other members of the cluster I (OM20, OM21, OM25, IN14, IN15 and ‘Ca. G. acididurans’) appeared more distantly related to each other as most AAI values ranged from 65 to 70% and were only slightly higher than the AAI estimated between members of different clusters (Table S6, Supporting Information), and we hypothesized that the cluster I encompassed several new genera in addition to the Gallionella genus.
Relative abundances of the Gallionellaceae MAGs
To assess the distribution of the Gallionellaceae along the borehole PZ26, we estimated the percentage of metagenomic reads aligning with a high identity level with sequences of MAGs as well as reference genomes. The percentage was extremely low when considering only the five reference Gallionellaceae since the maximal value did not exceed 0.01% of the reads in the F37 metagenome (Table S7, Supporting Information). In comparison, the Gallionellaceae MAGs were much more abundant in groundwater metagenomes, with percentages ranging from 0.8 to 6.2% of the total reads (Fig. 4B), and even more in the outer microbial mat and in the incubations metagenomes since the percentages of reads summed to 42.8% and 42.2%, respectively (Fig. 4A and Table S8, Supporting Information).
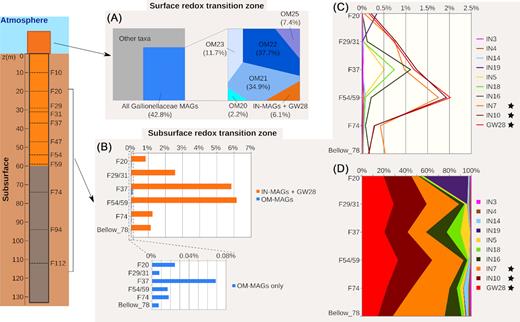
Distribution profiles of the MAGs in the metagenomes. (A) Percentage of reads from the outer microbial mat metagenomes affiliated to the Gallionellaceae MAGs. Left plot: percentages relative to the total read number in the OM metagenomes; right plot: percentages relative to the number of Gallionellaceae-affiliated reads. (B) Cumulative percentages of total reads recruited by IN-MAGs and GW28 versus OM-MAGs in groundwater metagenomes. The zoomed-up inset plot shows cumulative percentages for OM-MAGs only. (C) Detailed profiles of distribution for IN- and GW28 in groundwater metagenomes. (D) Same as B but percentages are expressed relative to the number of Gallionellaceae-affiliated reads. Black stars indicate MAG belonging to ‘Ca. Houarnoksidobacter’. Abbreviation: GW, groundwater
For groundwater metagenomes, reads of Gallionellaceae mostly aligned with the IN- and GW-MAGs. The percentage of total reads recruited by the MAGs (i.e. relative to the total number of reads in each metagenome) showed strong variations depending on the sampled fracture in the borehole (Fig. 4B). It was about five to six times higher in the F54/59 and F37 metagenomes (5.8% and 6.2% of total metagenomic reads respectively) than in the F74 (1.3%) and Below_78 (1.1%) metagenomes. The lowest percentage was observed in F20 (∼ 0.8%) while an intermediate value was found in the F29/31 metagenome (2.5%) (Fig. 4B and Table S8, Supporting Information).
Comparison of the percentages of recruited reads in the groundwater metagenomes showed that the GW-MAG and the IN-MAGs could be grouped into four distribution patterns (Fig. 4C). The first group gathered the three MAGs composing the cluster IV in the phylogenomic tree (IN7, IN10 and GW28). Collectively, they accounted for more than 50% of Gallionellaceae-affiliated reads (i.e. relative to the number of reads recruited by the 15 MAGs) in all groundwater metagenomes (Fig. 4D). The maximum percentage of reads recruited by these 3 MAGs was observed for F54/59 metagenome where it reached up to ∼ 6% of the total reads and 93% of Gallionellaceae-affiliated reads (Fig. 4D). A second group was composed of IN5, IN16 and IN18. These IN-MAGs mostly recruited reads in the F37 metagenome and their relative abundance was negligible in the other groundwater metagenomes (Fig. 4C and D). The third group only contained IN19. The percentage of reads recruited by this IN-MAG was extremely low in groundwater metagenomes except the F20 metagenome (Fig. 4C). In this metagenome, IN19 represented 0.3% of the total reads but 40% of the Gallionellaceae-affiliated reads (Fig. 4D). Finally, the fourth group gathered IN3, IN4 and IN14. These MAGs were very poorly represented in all groundwater metagenomes (Fig. 4C); on the other hand, these IN-MAGs were characterized by a high percentage of recruited reads in the incubation metagenomes (Table S8, Supporting Information).
OM-MAGs were practically absent from groundwater metagenomes (Fig. 4B). Indeed, the percentage of reads recruited with OM-MAGs in groundwater metagenomes were tenth to hundred times lower than with IN- and GW-MAGs. Conversely, Gallionellaceae in the outer microbial mat metagenomes were mostly represented by OM-MAGs (Fig. 4A). Altogether, the OM-MAGs accounted for about 40% of the total reads and about 94% of the Gallionellaceae-affiliated reads (Fig. 4A). None of the IN-MAGs but IN18 recruited more than 1% of the reads in the outer microbial mat metagenomes (Table S8, Supporting Information). Thus, the major difference in the MAG distribution can be seen between groundwater metagenomes from the fractures and metagenomes from the outer microbial mat at the surface.
Energy metabolism and autotrophy
Iron oxidation
Gallionellaceae were first described as chemolithotrophs capable of gaining energy from the oxidation of iron coupled to the reduction of oxygen. Four different systems of proteins involved in extracellular electron transfers (EET), a key step of biological iron oxidation, were encoded in the Gallionellaceae MAGs of PZ26 (Table S9, Supporting Information): (1) the outer membrane cytochrome c Cyc2, two porine-cytochrome c complex encoded by (2) the mtoAB operon and (3) a cluster of genes homologous to the ‘PCC3’ cluster found in Sideroxydans lithotrophicus ES-1 and Leptothrix ochracea and (4) the multicopper oxidase MofA (He et al. 2017). Every MAG had at least one homolog to cyc2 (Barco et al. 2015) (Fig. S2 and Table S9, Supporting Information). All other genes encoding EET systems were variably represented among the MAGs (Table S9, Supporting Information). IN16 possessed all four systems while IN14 and IN18 only encoded the cyc2 gene and lacked all other EET genetic systems. An operon, identical to the mtoCDAB operon of Gallionella capsiferriformans ES-2, was found in IN5 and OM23. Two copies of mtoA and mtoB were found in IN16 and the mto genes were spread in three clusters: two clusters of mtoAB and one cluster of mtoDC. One mtoAB cluster was located on a contig edge hinting that the mtoC and mtoD copies of the operon were missing. The two other mto clusters were located within two different contigs. To our knowledge such genomic organisation for the mto genes has not yet been observed in any Gallionellaceae genomes.
Recently, it has been proposed that the utilization of the alternative complex III (ACIII) to transfer electrons from periplasmic cytochromes to the quinone pool in the inner membrane could be a distinctive attribute of the family Gallionellaceae (Kato et al. 2015; He et al. 2017). All reference strains of Gallionellaceae possessed a seven gene operon (actAB1B2CDEF) to synthesize protein subunits of the ACIII system (Emerson et al. 2013; Kato et al. 2015; Kadnikov et al. 2016). This operon could not be detected in IN4, IN7, IN10 and GW28, casting doubt that ACIII is indispensable for iron oxidation in the family Gallionellaceae (Table S9, Supporting Information). Instead of ACIII, these Gallionellaceae could employ the cytochrome bc1 complex (petABC) to reduce quinones in the inner membrane (Table S9, Supporting Information).
Oxygen reduction
The microaerobic lifestyle of the Gallionellaceae is enabled by the presence of high-affinity terminal oxidases which allow them to respire O2 in microoxic conditions. Genes coding for the cbb3-type cytochrome c oxidase were identified in all MAGs with the exception of OM20 (Table S9, Supporting Information). The lower percentage of completion measured for this MAG (77%) was likely responsible for the absence of these genes. All reference genomes of Gallionellaceae, with the exception of Ferriphaselus sp. R-1, also contained genes cydABX coding for another terminal oxidase: the cytochrome bd oxidase. Despite the prevalence of these genes in the Gallionellaceae reference genomes, only IN4 had the genetic potential to produce the bd-type complex (Table S9, Supporting Information).
Sulfur oxidation
Gallionellaceae have recently been shown to possess genes allowing dissimilatory oxidation of sulfur compounds. All MAGs encoded one to three sulfide: quinone oxidoreductase (sqr) for the oxidation of sulfide in zero-valent sulfur (Table S9, Supporting Information). Phylogenetic classifications showed that all the sqr genes detected in the MAGs belonged to three distinct protein families named SqrA, SqrD and SqrF (Gregersen, Bryant and Frigaard 2011) (Fig. S3, Supporting Information). All MAGs had one gene copy of the SqrA family except OM23 which possessed one gene of the SqrD family. Besides SqrA, six MAGs further encoded either SqrD or SqrF while IN16 possessed a gene copy of each type. Oxidation of sulfide to zero-valent sulfur could also be performed by the flavocytochrome c / sulfide dehydrogenase which was encoded by fccAB genes in MAGs IN7 and IN14 (Table S9, Supporting Information).
All MAGs but four (OM21, OM22, OM23 and IN19) contained the dsrABEFCMKLJOP gene cluster (Table S9, Supporting Information). This cluster also exists in Sideroxydans lithotrophicus ES-1, Ferriphaselus amnicola, Ferriphaselus sp. R-1 (Kato et al. 2015) and ‘Candidatus Gallionella acididurans’ (kadnikov et al. 2016) and is thought to allow the oxidation of elemental sulfur into sulfite based on sequence conservation and shared synteny with sulfur oxidizing proteobacteria (Kato et al. 2015). In addition, ten MAGs contained the soeABC operon which encodes proteins catalysing direct sulfite to sulfate oxidation (Table S9, Supporting Information). Indirect sulfite oxidation into sulfate could also be achieved by the adenylyl-sulfate reductase (aprBA), which converts sulfite into adenosine 5'-phosphosulfate (APS) and sulfate adenylyltransferase (sat) which catalyses the release of sulfate from APS. These genes were found in seven MAGs (Table S9, Supporting Information), most of which possessing both pathways for sulfite oxidation (i.e. direct and indirect oxidation).
Genes needed for thiosulfate to sulfate oxidation (sox genes) were present in S. lithotrophicus ES-1 but not in G. capsiferriformans ES-2 and F. amnicola. These genes were also encoded in OM20 and in three MAGs which were closest to S. lithotrophicus ES-1 in the phylogenomic tree (IN3, IN14, IN16) (Table S9, Supporting Information). However, gene order for the sox operon in these MAGs (soxYZAXB) was not conserved with S. lithotrophicus ES-1 (soxXYZAB; Emerson et al. 2013). A fifth MAG, EN12, also possessed the soxYZ genes but lacked the soxAXB genes which were replaced by two genes coding for the cytochrome subunit of a sulfide dehydrogenase.
Finally, it is also worth noting that homologs of the asrAB genes which code for the dissimilatory sulfite reductase were found in IN4, IN7, IN10 and GW28 (Table S9, Supporting Information). These genes could provide these 4 MAGS the ability to reduce sulfite into sulfide. Interestingly, MAGs lacking the dsr gene cluster (i.e. OM21, OM22, OM23 and IN19) also lacked other S-compound oxidation and reduction genes. The only exception seemed to be the unique copy of the sqr gene in these MAGs (Fig. 5A).
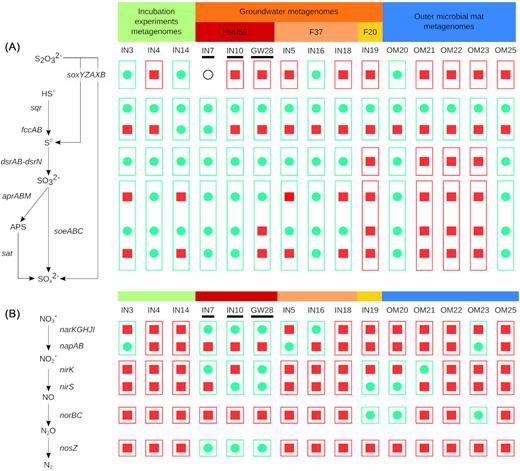
Overview of the genes involved in the S and N dissimilatory pathways. Sulfur oxidation (A) and denitrification pathways (B) are displayed on the left side of the figure. Different enzymes encoded by specific genes can carry on the same reaction (e.g. the periplasmic nitrate reductase NapA and the membrane-bound nitrate reductase NarG). The distribution of the genes in the MAGs is shown on the right side of the figure: a green circle means the presence of the genes and a red square means a lack of the genes. The capacity of the MAGs to carry on the different redox reactions are indicated as follows: a green frame means a feasible reaction. A red frame means that the reaction is not possible. MAGs are grouped according to their maximum abundance in metagenomes. The black circle use for IN7 indicates that only soxY and soxZ were found in this MAG. Black bars indicate MAG belonging to ‘Ca. Houarnoksidobacter’. APS: Adenosine 5'-phosphosulfate
Denitrification
Genes involved in denitrification were recently found in several MAGs of Gallionellaceae that developed in a riverine aquifer following nitrate injection indicating that Gallionellaceae can also play a role in the N cycling in groundwater (Jewell et al. 2016). Similarly, the presence of genes involved in denitrification were observed in the PZ26 MAGs (Table S9, Supporting Information). The narKGHJI gene cluster needed to produce the membrane-bound nitrate reductase was present in IN7, IN10, IN16 and GW28 and the napAB genes coding for the periplasmic nitrate reductase were detected in OM23, IN3 and IN5. Genes coding for nitrite reductase (nirK and nirS) and nitric oxide reductase (norBC) were distributed in 8 and 3 MAGs respectively. The nitrous oxide reductase gene (nosZ), which is necessary for the reduction of N2O into N2, the final step of denitrification, was only found in IN7, IN10 and GW28. Compared to the S-compound oxidation genes, the distribution of denitrification genes in the MAGs appeared more restrained and patchy (Fig. 5B). Hence, no MAG possessed all the genes to realize the entire denitrification pathway from NO3− to N2O or N2.
Inorganic carbon and nitrogen fixation
All Gallionellaceae described previously possess the ribulose-1,5-bisphosphate carboxylase / oxygenase (Rubisco), the key enzyme of the Calvin-Benson-Bassham cycle, to reduce carbon dioxide in organic carbon molecules. The cbbM gene coding for the Rubisco form II was found in all MAGs but GW28 (Table S9 and Fig. S4, Supporting Information). In addition, genes coding for the small (rbcS) and large (rbcL) subunits of the Rubisco form I were identified in OM21, OM22 and OM25 (Table S9 and Fig. S4, Supporting Information). These two forms of Rubisco found in Proteobacteria are distinguished by their specificity for O2 and CO2 with form I allowing to fix CO2 in presence of higher O2 level than form II (Badger and Bek 2008). In addition to being able to grow without organic carbon sources, Sideroxydans lithotrophicus ES-1 and Ferriphaselus amnicola are also able to fix nitrogen into ammonia as they bear the large nif operon which confer the capacity to produce nitrogenase. This operon is missing in both Gallionella capsiferriformans ES-2 and ‘Candidatus Gallionella acididurans’. MAGs related to Sideroxydans (cluster II in Fig. 3) and Ferriphaselus (cluster III Fig. 3) also possessed this operon while it was missing in MAGs member of the cluster IV (IN7, IN10 and GW28) and most MAGs related to Gallionella except OM20 and IN14 (Table S9, Supporting Information).
DISCUSSION
Reconstruction of prokaryotic genomes from metagenomes has considerably enriched our knowledge of microbial diversity by unravelling an amazing number of new prokaryotic candidate divisions. This approach is also extremely powerful to describe the metabolic capacities of natural populations of micro-organisms. Here, we used this approach to recover metagenome-assembled genomes which represented distinct Gallionellaceae populations in a single aquifer. These MAGs correspond to draft assemblies of size comprised between 1.9 and 2.4 Mb with a degree of completion ranging from 60.4 to 99.3%. However, we observed no correlation between the size and the percentage of completion of the MAGs and therefore we assumed that differences of gene composition reflected genuine differences of functional capacities between populations.
Diversity of the Gallionellaceae in the Guidel aquifer
Phylogenomic tree reconstruction confirmed that the 15 MAGs could be unambiguously classified within the family Gallionellaceae. The phylogenomic analysis combined with the pairwise calculations of ANI and AAI between MAGs and reference genomes allowed delineating genus and species boundaries. the representative strains of Gallionella and Sideroxydans to be classified in either genus with certainty. the MAGs clustering in the tree with reference genomes of Ferriphaselus and Sideroxydans could be clearly assigned to this genus. On the other hand, the genus boudaries for the Gallionella genus was less clear. Hence, Gallionellaceae-affiliated MAGs found in this study might represent up to seven new genera that remain to be formally described. The number of recovered MAGs likely corresponds to a lower estimate of the diversity of Gallionellaceae as we only selected the most complete MAGs. This level of diversity for the Gallionellaceae in groundwater from a single borehole is striking and underscores that the diversity within the family Gallionellaceae remains largely unexplored. The use of incubation experiments which allowed to increase the proportion of Gallionellaceae in groundwater microbial communities was decisive in obtaining MAGs since the majority of them were assembled from the incubation metagenomes. The simplicity of this approach makes it particularly interesting to recover new genomes of Gallionellaceae and more generally of microaerobic FeOB in groundwater.
Amongst the MAGs identified in this study, IN7, IN10 and GW28 (cluster IV in Fig. 3) are of particular interest as they are the main representatives of the family Gallionellaceae in all groundwater metagenomes produced in this study. The fact that GW28 is the sole Gallionellaceae MAG recovered directly from groundwater metagenomes (i.e. without incubation) underscores the importance of this group of MAGs in groundwater. The high nucleotide identity measured between these three MAGs indicates that they belong to a single new genus which might have a pivotal role in the biological oxidation of iron and the production of the rust-colored flocky aggregates that are transported with the groundwater up-flow. We propose to name this genus ‘Candidatus Houarnoksidobacter’ (houarn: iron and oksid: oxide in Breton) to acknowledge its significance in the Guidel aquifer and potentially in other aquifers of the Armorican Massif.
Alongside the three MAGs of ‘Candidatus Houarnoksidobacter’ which predominate in all sampled fractures, four others IN-MAGs (IN5, IN16, IN18 & IN19) account for a significant percentage of Gallionellaceae-affiliated reads but only in groundwater metagenomes from a single fracture, suggesting that they have a much more restrained distribution in the fracture network. Furthermore, a series of IN-MAGs assembled from incubation metagenomes were missing in the groundwater metagenomes indicating that they were very rare at the time of sampling. However, the fact that these MAGs were highly abundant in the incubation metagenomes indicates that they were able to respond efficiently to changing environmental conditions. Altogether, these results point to the existence of populations of Gallionellaceae which could be adapted to specific environmental conditions predominating only locally, in some part of the fracture network, or temporally, at some period of the year.
Dissimilatory metabolism of nitrogen oxides and inorganic sulfurs
Annotation of protein-coding genes involved in the energy metabolism revealed that the three ‘Candidatus Houarnoksidobacter’ MAGs correspond to populations of cells which could catalyse the dissimilatory reduction of nitrate, nitrite and nitrous oxide (Fig. 5). These MAGs lack the norBC genes for nitric oxide reduction. However, NO can react abiotically with Fe2+ to produce N2O and Fe3+, thus allowing the full reduction of NO3− to N2. These MAGs could represent real autotrophic nitrate-reducing iron oxidizing bacteria as defined by Bryce et al. (2018). They also possess a full repertoire of genes to oxidize sulfide into sulfate. Thus, the metabolism of ‘Candidatus Houarnoksidobacter’ provides links to connect the N, S, C and Fe cycles in the Guidel aquifer. However, the diversity of possible electron donors and acceptors that can be harnessed for the production of energy raises the question of the main pathway used by these MAGs. Genes involved in nitrate reduction, and iron and sulfur oxidation were co-expressed in growing populations of Gallionellaceae after the injection of nitrate-rich water in an alluvial aquifer (Jewell et al. 2016), suggesting that these Bacteria were able to adapt their metabolism and exploit various electron donors and acceptors at the same time. Such capacity is certainly advantageous in dynamic underground environments where intermittent fluxes of recent groundwater carrying oxygen and or nitrate from surface mix with deep anoxic groundwater.
Genes necessary for denitrification and for the oxidation of sulfur compounds are also encoded in the other MAGs of Gallionellaceae found in the Guidel aquifer (Fig. 5). However, the distribution of these genes in these MAGs displays significant differences depending on the type of redox processes in which they are involved. Sulfur oxidation appear to be a general feature of the Gallionellaceae metabolism. Genes implicated in the oxidation of sulfur are widespread and the corresponding pathways display a low modularity since the ability to produce sulfate from reduced sulfur was either entirely encoded in the MAGs or missing. Conversely, denitrification genes are poorly represented in the MAGs and none of the MAGs possessed the entire denitrification pathway. The sparsity of these genes in the MAGs suggests that denitrification is not a metabolic process shared by all the Gallionellaceae.
Surface and belowground redox transition zones correspond to distinct ecological niches
The Guidel aquifer is characterized by a large upwelling of deep, reduced groundwater with high concentrations of dissolved Fe(II) (see Fig. 2). This upward circulation allows the development of groundwater-dependent redox transition zones which constitute favorable habitats for the Gallionellaceae in subsurface and aboveground.
Belowground, in the deep fractured layer, the oxic-anoxic interface is induced by the mixing of oxygenated and reduced fluids. As O2 inputs in ancient groundwater are likely heterogeneous in time, the interface is probably intermittent. However, the detection of significant proportions of Gallionellaceae at various depth, in all sampled fractures, strongly suggests that this mechanism allows the formation of redox transition zones and the development of Gallionellaceae assemblages dominated by ‘Candidatus Houarnoksidobacter’ over a large width of the fractured bedrock. Hydrological modelling of the Guidel aquifer suggests that the Gallionellaceae habitable zone expands up to several hundred meters below surface (Bochet et al.). This vertical distribution does not extend up to the upper weathered part of the aquifer, where recent groundwater, characterized by high O2 concentration and very little Fe(II), circulates. The absence of Gallionellaceae in the weathered zone of similar aquifer systems of the Armorican Massif (Ben Maamar et al. 2015) strengthens our observations that more frequent oxygen inputs lead to a lack of dissolved Fe(II) and narrow the distribution of the Gallionellaceae in groundwater.
Aboveground, another redox transition zone develops when groundwater comes into direct contact with the atmosphere. The groundwater outflow leads to the growth of a thin iron microbial mat on the external wall of the borehole head. Gallionellaceae in surface environments face specific abiotic stresses such as strong seasonal and diurnal temperature variations, dessication when the groundwater up-flow is strongly reduced at the end of the discharge period and more importantly high oxygen concentrations due to the constant diffusion of O2 from the atmosphere. Compared to the iron mat observed within the borehole, the outer microbial mat looks denser and it is reasonable to assume steep O2 gradients as observed in similar iron-rich mat forming in ferruginous seeps (Emerson and Revsbech 1994). Hence, this mat may serve as a barrier to limit O2 diffusion and help Gallionellaceae maintaining the suitable conditions that are necessary for biological iron oxidation at the groundwater-air interface.
Although, the artesian nature of the Guidel aquifer implies a unidirectional transfer of Gallionellaceae cells and microbial mat fragments from the fractured layer to the surface, we observed a complete change of the Gallionellaceae community structure between the two redox transition zones which suggests that Gallionellaceae thriving in fractures are not adapted to surface conditions and vice versa. Our results highlight that subsurface and aboveground redox transition zones are colonized by Gallionellaceae requiring similar albeit distinct ecological niches. Beyond the well-known requirement for microaerobic conditions, other processes related to the formation of the interface, either groundwater mixing or oxygen diffusion, are likely major drivers of the Gallionellaceae diversity in circumneutral freshwater ecosystems.
ACKNOWLEDGEMENTS
The authors would like to thank the Molecular Ecology facility (UMR 6553 ECOBIO) for the help in extraction and purification of DNA, Olivier Collin and the staff of the Genouest bioinformatic facility (INRIA and university of Rennes 1) for the quality of the computing resources and collaborative services made available to the scientific community in genomics, and the H+ network of hydrogeological observatories (hplus.ore.fr) and the french Critical Zone Initiative OZCAR. The authors are also grateful to Dr. Anniet Laverman for many suggestions that improved the clarity of the manuscript.
FUNDING
This work was supported by the EC2CO initiative of the French National Center for Scientific Research (CNRS). Financial support for Lorine Bethencourt was provided by The French National Research Agency (ANR) projects: Stock-en-Socle (ANR-13-SEED-0009), CRITEX (ANR-11-EQPX-0011) and Subsurface mixing and reactions (ANR-14-CE04-0003), the Region Bretagne and the European Research Council (ERC) project ReactiveFronts (648377).
Conflicts of interest None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}