





ABSTRACT
Wolbachia has deeply shaped the ecology and evolution of many arthropods, and interactions between the two partners are a continuum ranging from parasitism to mutualism. Non-dispersing queens of the termite Cavitermes tuberosus are parthenogenetically produced through gamete duplication, a mode of ploidy restoration generally induced by Wolbachia. These queens display a bacteriome-like structure in the anterior part of the mesenteron. Our study explores the possibility of a nutritional mutualistic, rather than a parasitic, association between Wolbachia and C. tuberosus. We found a unique strain (wCtub), nested in the supergroup F, in 28 nests collected in French Guiana, the island of Trinidad and the state of Paraíba, Brazil (over 3500 km). wCtub infects individuals regardless of caste, sex or reproductive (sexual versus parthenogenetic) origin. qPCR assays reveal that Wolbachia densities are higher in the bacteriome-like structure and in the surrounding gut compared to other somatic tissues. High-throughput 16S rRNA gene amplicon sequencing reveals that Wolbachia represents over 97% of bacterial reads present in the bacteriome structure. BLAST analyses of 16S rRNA, bioA (a gene of the biosynthetic pathway of B vitamins) and five multilocus sequence typing genes indicated that wCtub shares 99% identity with wCle, an obligate nutritional mutualist of the bedbug Cimex lectularius.
INTRODUCTION
Endosymbiosis is a particular case of association between microorganisms living within eukaryotic cells. Endosymbiotic bacteria deeply shaped the ecology and evolution of many arthropods. Some of these associations are occasional and display spatial and temporal variations, others are essential and integrated in the host's core physiology (Weeks et al. 2007; Werren, Baldo and Clark 2008; Zug and Hammerstein 2015; Diouf et al. 2018). The most striking examples are mitochondria and plastids, two essential membrane-bound organelles of eukaryotic cells, that evolved from bacteria by endosymbiosis (Maynard Smith and Szathmáry 1995; Embley and Martin 2006). According to the fitness costs and benefits conferred to the organisms involved in the association, interactions can be perceived as a continuum ranging from parasitism to mutualism (Zug and Hammerstein 2015).
One of the most studied endosymbionts are the reproductive parasites, which have evolved various strategies to manipulate host reproduction in order to enhance their own transmission to the next generation: (i) by generating cytoplasmic incompatibility (CI) between sperm and egg depending on the infection status of both partners, (ii) by killing males, (iii) by feminizing genetic males into functional females and (iv) by inducing thelytokous parthenogenesis leading to the production of females only (Weeks, Reynolds and Hoffman 2002; Werren, Baldo and Clark 2008; Hurst and Frost 2015; Zug and Hammerstein 2015). As they are maternally transmitted, such manipulations increase the proportion of infected females in the population of hosts, and thus of the reproductive parasites in them. Although they are usually vertically transmitted, these reproductive parasites can also move horizontally within a host species or between different species, thereby explaining the success of their pandemics (Zug, Koehncke and Hammerstein 2012). Among them, the bacterium Wolbachia (Rickettsiales: Rickettsiaceae) is the most common and infects a considerable fraction of arthropod species (Zug, Koehncke and Hammerstein 2012; Weinert et al. 2015).
At the extreme end of this continuum of interactions, parasitism may evolve towards less harmful relationships, and even mutualisms (Zug and Hammerstein 2015). For instance, the parasitoid wasp Asobara tabida is infected by three Wolbachia strains: two manipulate host reproduction (CI), while the third is essential for host oogenesis (Dedeine, Boulétreau and Vavre 2005). Beside reproductive phenotypes, other types of obligate mutualism can be selected in the case of long-term stable relationships, such as nutritional symbioses (Hosokawa et al. 2010; Zug and Hammerstein 2015). In the pea aphid Acyrthosiphon pisum, the bacterium Buchnera provides specific amino acids that are lacking in the host's diet (Shigenobu et al. 2000). Additionally, the bacterium only lives in specialized cells, the bacteriocytes, localized in the vicinity of the gut (Simonet et al. 2018). In the most integrated associations, bacteria are concentrated in dedicated permanent organs, the bacteriomes. In the tsetse flies, the bacterium Wigglesworthia lives in a midgut-associated bacteriome. It provides vitamins lacking in the fly's diet and are essential to its survival (Akman et al. 2002; Pais et al. 2008). Bacteriomes may take various shapes and locations, and are sometimes associated with the digestive tract or to the gonads (Heddi et al. 2005; Kuechler et al. 2012; Douglas 2015).
Numerous studies have reported the evolution of complex symbiotic relationships in ants, social bees, wasps or termites (Ohkuma and Brune 2011; Treanor, Pamminger and Hughes 2018). From the perspective of endosymbionts, eusocial insects are primary targets as colonial organization and close interactions between nestmates help to maintain the presence of endosymbionts in the nests and favour their transmission in the hosts. From the perspective of eusocial insects, their ancient origin (ant and termite fossils indicate advanced sociality 100 million years ago; Rust and Wappler 2016), the diversity of their life history strategies (Lach, Parr and Abbott 2009; Jones and Eggleton 2011), and their high speciosity (Bolton et al. 2007; Krishna et al. 2013) confer a long evolutionary time and strong opportunities to develop intimate relationships with endosymbionts.
In the termite Cavitermes tuberosus, parthenogenesis is an integral part of the lifecycle: the queen uses sex to produce the altruistic castes (workers, soldiers) and the dispersers (alates), and thelytokous parthenogenesis to produce female neotenics, i.e. non-dispersing queens (Fournier et al. 2016) (Fig. 1). Two peculiar traits of this species attracted our attention. First, these neotenic queens are parthenogenetically produced through gamete duplication (Fournier et al. 2016), a mode of ploidy restoration rare among animals and generally induced by Wolbachia (Suomalainen, Saura and Lokki 1987; Weeks and Breeuwer 2001; Ma and Schwander 2017). Second, these neotenic queens display an empty digestive tract except from an enlarged pouch in the anterior part of the mesenteron (visible as a black dot; Fig. 2E). If a more or less distinct hump was described in the anterior end of the midgut of workers from higher termites (Noirot 2001), an hypertrophy of this structure (Fig. 2C and D) has never been reported so far, and could be regarded as a bacteriome playing a role in the nutrition by housing mutualistic symbionts.
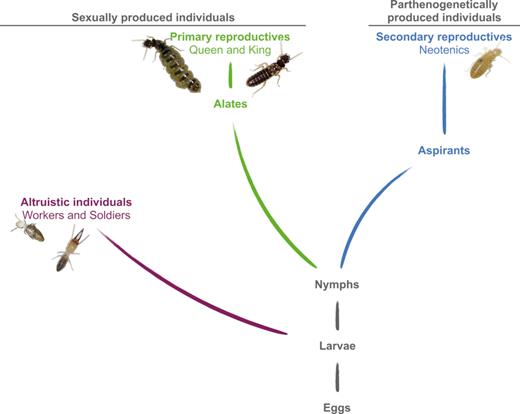
Simplified developmental pathways and reproductive modes in C. tuberosus. After an undifferentiated larval instar, a bifurcation between the altruistic and reproductive castes occurs. Individuals in the altruistic caste (workers and soldiers) are sexually produced (purple). In the reproductive caste, a difference stems from the mode of reproduction: sexually produced individuals develop predominantly through five nymphal instars to the alate stage (84%) while some differentiate into secondary reproductives (neotenics; 16%); parthenogenetically produced females (blue) develop predominantly into neotenic queens (93%) while some reach the alate imago (7%) (Fournier et al. 2016). We call aspirants the instars that differentiate into neotenics. Alates initiate new colonies after a nuptial flight and become primary reproductives while neotenics stay in the nest to replace primaries upon their death. Adapted from Hellemans et al. (2017b). To enhance its own transmission, Wolbachia could induce thelytokous parthenogenesis in C. tuberosus.

Worker digestive tract of C. tuberosus in dorsal (A) and left (B) view; with focus on the foregut-midgut junction in left view of worker (C) and neotenic queen (D), highlighting a pouch-shaped expansion at the antero-dorsal part of the mesenteron. Primary king with numerous neotenic queens (E): the pouch-shaped expansion is filled with particles and visible as a black dot through the cuticle of all neotenic queens. Foregut: Cr, crop; G, gizzard. Midgut: M, mesenteron (stippled). Hindgut: P1, ileum; P2, enteric valve; P3, paunch; P4, colon; P5, rectum. Scale bar for drawings = 0.5 mm. Nomenclature to describe the digestive tract follows Noirot (1995) and Noirot (2001).
We tested in the termite C. tuberosus the hypothesis of an infectious induction of parthenogenesis and/or of a mutualistic relationship with endosymbionts. First, over a range of 3500 km, we screened C. tuberosus colonies for the presence of Wolbachia, as well as Cardinium and Rickettsia, the two other bacteria known to induce parthenogenesis in insects (Zchori-Fein et al. 2004; Perlman, Hunter and Zchori-Fein 2006; Engelstädter and Hurst 2009). Wolbachia was found in all tested nests of C. tuberosus, but Cardinium and Rickettsia were absent. We also estimated the prevalence of infection in the populations, and sequenced and identified the strain(s) infecting C. tuberosus. Secondly, we determined the phenotype(s) of Wolbachia in C. tuberosus. In order to link Wolbachia to parthenogenesis induction, we estimated the prevalence of infection according to the sex and the reproductive origin of the individuals (i.e. sexually or parthenogenetically produced). To determine if the expansion connected to the mesenteron of C. tuberosus could be considered as a bacteriome, we investigated spatial aggregation of Wolbachia in different tissues by performing comparative qPCR to quantify expression levels of a diagnostic fragment of the Wolbachia 16S rRNA gene, and we identified the bacterial communities present in this bacteriome-like expansion and in the gut by performing a targeted Illumina high-throughput sequencing of the 16S rRNA gene. To highlight a potential nutritional symbiosis, and as Wolbachia may fulfil a role of dietary provisioner of biotin (vitamin B7) (Nikoh et al. 2014; Gerth and Bleidorn 2016), we amplified one of the five genes of the biotin operon.
MATERIAL AND METHODS
Samples collection
Whole nests of the Neotropical termite Cavitermes tuberosus (Termitidae: Termitinae) were collected in 2012, 2014, 2015, 2016, 2017 and 2018 in three regions of French Guiana (the Petit Saut dam area: N 05.07°, W 52.87°; Kaw Mountain: N 04.53°, W 52.15°; and Saül: N 03.62°, W 53.20°), the island of Trinidad (Lopinot Road: N 10.73°, W 61.32°) and in Brazil (campus of the Federal University of Paraíba, João Pessoa: S 7.14°, W 34.84°) (see details in Supplementary Table S1). Our sampling covers over 3500 km of the distribution of this species.
Termite species identity was ascertained using the mitochondrial barcode gene cytochrome oxidase II (COII). DNA was extracted using a NucleoSpin Tissue kit (Macherey–Nagel). A fragment of approximately 780 bp of the COII was amplified using the modified forward primer A-tLeu (Miura, Roisin and Matsumoto 2000) and the reverse primer B-tLys (Simon et al. 1994). Amplification and cycling conditions are given in Supplementary materials SM-1. A fraction of the amplification products was screened by electrophoresis on a 1% agarose gel and another fraction was then purified with the Nucleofast PCR purification kit (Macherey–Nagel). Purified amplicons were sequenced with BigDye Terminator Cycle Sequencing kit v3.1 (Applied Biosystems) according to the manufacturer's recommendations (see SM-1). Sequencing products were purified with an ethanol/EDTA/sodium acetate method. Sequence data were obtained with an ABI 3730 Genetic Analyzer (Applied Biosystems), and were visualized and edited using the software CodonCode Aligner (CodonCode Corporation, Dedham, MA.).
Screening of endosymbionts
We selected 20 nests of C. tuberosus representative for the presence of each caste and sex to screen for infection by Cardinium, Rickettsia and Wolbachia. DNA extraction was performed on termite heads of two individuals per nest (Supplementary Table S1) using a NucleoSpin Tissue kit (Macherey–Nagel). To avoid contamination and PCR inhibition from microorganisms present in the gut, and because our preliminary studies on C. tuberosus showed that infection can be detected using heads as well as gonads, we selected heads for the rest of this study.
We used species-specific primers targeting the 16S rRNA gene for each bacterium: the primer pair CLOf/CLOr1 specific to Cardinium (Weeks, Velten and Stouthamer 2003), the pair Rb-F/Rb-R specific to Rickettsia (Gottlieb et al. 2006) and the pair Wspecf/Wspecr (Werren and Windsor 2000), which proved to be the most specific to Wolbachia (16S-2; Simões et al. 2011). Positive controls (Cardinium-infected false spider mite Brevipalpus phoenicis (Acari), Rickettsia-infected whiteflies Bemisia tabaci (Hemiptera) and Wolbachia-infected mosquito Culex pipiens pipiens (Diptera)) and negative controls (PCR-grade water) were used for each amplification. Cycling conditions followed specifications of each primer pair in the original publications. Amplification products were screened by electrophoresis on a 1% agarose gel. In order to discard false negatives, DNA extracts that did not amplify were tested at the mitochondrial gene cytochrome oxidase I (COI) using the universal forward LCO (LCO1490) and reverse HCO (HCO2198) primers (Folmer et al. 1994). Cycling conditions are given in Supplementary materials SM-2. Low-quality samples that did not amplify were discarded from the analyses (8 individuals, 1.8% of the data).
Infection prevalence in the nests and among castes and sexes
Prevalence of Wolbachia was tested for a subset of 10 nests (420 individuals; Supplementary Table S1), all collected in October 2014. DNA was extracted from termite heads by a Chelex-based method (Walsh, Metzger and Higuchi 1991) and was screened with amplification, cycling conditions and gel screening as above.
The developmental scheme of C. tuberosus outlined an altruistic caste composed of workers and soldiers, and a reproductive caste composed of male and female dispersers (alates), primary king and queen, aspirants (i.e. nymph-like pre-neotenics) and female neotenics (Fig. 1). We analysed the infection rate for all nests, and for each caste and sex under a generalized linear model framework with a binomial error structure and a logistic link function to model the dichotomous infection status (0, non-infected; 1, infected). Analyses were performed with the R software v3.0.1 (R Development Core Team 2013).
Strain identification of Wolbachia and supergroup assignment
Wolbachia strain was identified in all 20 nests by sequencing genes of the multilocus sequence typing (MLST), i.e. the conserved bacterial housekeeping genes coxA, ftsZ, gatB, hcpA and fbpA (Baldo et al. 2006), and the 16S rRNA gene. Wolbachia strain determination was carried on DNA from heads of one individual per nest extracted using a NucleoSpin Tissue kit (Macherey–Nagel). Amplifications and cycling conditions of MLST genes are given in Supplementary materials (SM-3). Sequences were obtained with an ABI 3730 Genetic Analyzer and visualized with CodonCode Aligner. All sequences were deposited in GenBank (see Supplementary Table S2).
From the 16 described supergroups of Wolbachia (A–F and H–Q; Lindsey et al. 2016a; Lindsey et al. 2016b), we retrieved sequences of coxA, fbpA, ftsZ, gatB and hcpA from representatives of supergroups A, B, C, D, F, and H from the Wolbachia MLST (https://pubmlst.org/wolbachia/) and GenBank databases (Supplementary Table S3). We used Ehrlichia ruminantium as an outgroup for phylogeny reconstruction (Gerth et al. 2014). Each gene was aligned individually using the MUSCLE algorithm (Edgar 2004) implemented in CodonCode Aligner. The best nucleotide-substitution models were determined using jModelTest 2 v2.1.10 (Darriba et al. 2012) according to the best Bayesian Information Criterion (BIC) for each gene (coxA: HKY+I; fbpA: HKY+I+G; ftsZ: TrN+G; gatB: GTR+I+G; hcpA: TIM+G).
Phylogenetic analyses with Bayesian inference were performed using the software BEAST 2 (Bouckaert et al. 2014) with the method *BEAST (Heled and Drummond 2010) for inference from multilocus data. Because Wolbachia strains may recombine (Jiggins et al. 2001) or there may be horizontal transfers between bacterial species (Duplouy et al. 2013; Duron 2013), molecular clocks and gene trees were maintained unlinked between markers to allow for recombination. The ploidy level was set to ‘Y or mitochondrial’, strict molecular clocks were used and rates were assigned with gamma-distribution priors (shape = 2, scale = 2), the species tree was modelled under a Yule model and birthrate set to 1/X, and the species tree population size was set to ‘linear’ with population mean estimated during the run. All other priors and operators were adjusted after a first run.
We ran five independent runs of 10 million generations and trees were sampled every 10 000 generations. We followed traces with Tracer v1.7 (Rambaut et al. 2018) to ensure that all effective sample sizes were >200. We combined the five different runs and removed 20% of sampled trees for each run as burn-in (leaving a total of 5000 trees) using LogCombiner v1.8.3 (Bouckaert et al. 2014). Bayesian posterior probabilities for the support of nodes were computed as maximum clade credibility retaining median branch lengths using TreeAnnotator v2.4.2 (Bouckaert et al. 2014). The resulting tree was visualized with the package GGTREE v3.5 in R (Yu et al. 2017).
Wolbachia infection and impacts on phenotype
In order to highlight a link between Wolbachia and the induction of parthenogenesis, we analysed the infection status of males (n = 91) and females (n = 186) from the reproductive caste, distinguishing females according to their reproductive origin, sexual (n = 100) or parthenogenetic (n = 86), using Fisher's exact tests. Reproductive origin was determined from genetic data retrieved from previous datasets (Fournier et al. 2016; Hellemans et al. In press), in which individuals were genotyped at 17 microsatellite markers (Fournier, Hanus and Roisin 2015). Additional genotyping was performed in this study using the same molecular procedures. All genotypes used in this study are available in Appendix S1 (see Electronic Supporting Information). We performed a factorial correspondence analysis (FCA) using GENETIX v4.05.2 (Belkhir et al. 1998) in order to visualize the distribution of alleles according to the reproductive origin and the infection status of females.
Because the sex ratio in eggs (primary sex ratio, PSR) is an index of reproductive manipulation by endosymbionts (Werren, Baldo and Clark 2008), we studied PSR in 11 nests of C. tuberosus (Supplementary Table S1). Sex determination was carried out using the diagnostic microsatellite locus Ctub-94, at which females are systematically homozygous for one allele, whereas males are heterozygous (Fournier et al. 2016). The presence of Wolbachia in eggs was confirmed from the two tested colonies (Supplementary Table S1).
Wolbachia is a nutritional mutualist that may supply B vitamins to its host (Nikoh et al. 2014). We investigated whether Wolbachia could be associated in a nutritional mutualism with C. tuberosus by testing the presence of the bioA gene involved in the biotin (vitamin B7) biosynthetic pathway (Nikoh et al. 2014; Gerth and Bleidorn 2016). Amplification and cycling conditions are given in Supplementary materials (SM-4). Sequences were obtained with an ABI 3730 Genetic Analyzer and visualized with CodonCode Aligner.
Quantification of Wolbachia densities across tissues using quantitative PCR
Wolbachia densities were assessed in five tissues—head, hindleg, ovaries, bacteriome-like structure and the rest of the gut (from P3 to P5; see Fig. 2)—of five neotenic queens from nest M_82 (Supplementary Table S1) using quantitative PCRs (qPCRs). qPCRs were performed on a Rotor-Gene Q cycler (Qiagen). Total DNA was extracted using a NucleoSpin Tissue kit (Macherey-Nagel). We used Wolbachia-specific primers (INTF2 and INTR2; (Sakamoto, Feinstein and Rasgon 2006) amplifying a small fragment of the 16S rRNA gene. Amplification and cycling conditions are given in Supplementary materials (SM-5). Each sample was run in triplicate, and we used mean threshold cycle (Ct) values of triplicates for each sample. Melt curves were run after the last cycle for each reaction and always produced single products, ensuring primer specificity. We used controls (elution buffer from the DNA extraction kit) to ensure that amplification did not occur in samples without template DNA. For each tissue, primer efficiency (E) was determined using standard curves with template dilutions of 1X, 2X, 4X and 8X. Optimal reaction efficiency occurred at 0.150 ng/µl of stock template DNA for hindlegs and at 0.400 ng/µl for the other tissues. DNA was quantified in each sample using Qubit dsDNA HS assay kit (Invitrogen). Each sample was diluted at the above stated optimal reaction efficiency for the final qPCR assay. All analyses were performed using the Rotor-Gene Q software v2.3.1.49.
Results are reported as expression ratios slightly modified from Pfaffl (2001) (see SM-5). We used hindlegs as a control sample to which we compared all other tissues. We analysed expression ratios as log10 transformed data in order to ensure normality, sphericity and homoscedasticity. Transformed ratios were compared between tissues (head, ovaries, bacteriome-like structure and the rest of the gut) using one-way within subject (i.e. neotenic identity) ANOVA with tissue as the between-subject factor. Post-hoc comparisons were performed using multiple paired t-tests and p values were corrected for multiple comparisons using the False Discovery Rate (fdr) method (Benjamini and Hochberg 1995). Analyses were performed with the R software v3.0.1.
Illumina 16S rRNA gene sequencing-based microbial community profiling
The microbial community composition of digestive tubes of workers and neotenic queens from nest A_147 (Supplementary Table S1) was investigated by means of Illumina sequencing of 16S rRNA gene. Individuals were cold-immobilized and surface-sterilized using 80% ethanol and 1 X PBS. Using sterile forceps, whole guts were dissected and pooled from 20–30 workers and directly preserved in liquid nitrogen. In the case of the neotenics, after dissection of whole guts from 20 individuals, bacteriome-like structure located at the anterior part of the mesenteron (Fig. 2) were separated, pooled and preserved. All samples were collected in triplicates. PowerViral Environmental RNA/DNA Isolation Kit (MO-BIO) was used for DNA extraction following the manufacturer's protocol with introduction of cell lysis step by bead-beating with 0.1 mm glass beads at 20 Hz for 2 min. The extracted DNA was treated with 1 μl of 10 μg/mL RNase A (Sigma) at room temperature for 30 min and quantified. The microbial 16S rRNA gene libraries were prepared using an Illumina platform-optimized protocol (Marynowska et al. 2017). More specifically, universal modified primers S-D-Bact-0909-a-S-18 and S-*-Univ-*-1392-a-A-15 (Klindworth et al. 2013), targeting a 484 bp fragment of the V6–V8 region of the bacterial 16S rRNA gene, were used in the first round PCR where 1 ng of template DNA was subjected to 22 cycles of amplification. Subsequently, 1 μl of the purified and quantified reaction was used as template in a second step 8 cycle-PCR, which allowed for the incorporation of index barcodes (Nextera XT Index Kit V2, Illumina) and Illumina adaptors. Purified (Agencourt AMPure XP) and quantified (KAPA SYBR FAST Universal qPCR Kit, KapaBiosystems) libraries were pooled in equimolar ratios, spiked with PhiX control (Illumina) and sequenced using MiSeq Reagent Kit V3–600 on the Illumina Platform. After demultiplexing, quality trimming and chimera and singletons removal, resulting sequencing reads were assigned to Operational Taxonomic Units (OTUs) at 97% similarity with Usearch pipeline v7.0.1090_win64 (Edgar 2010) and further taxonomically annotated with SILVA database v.128 (Pruesse et al. 2007) using the naive Bayesian classifier (Wang et al. 2007) as implemented by the mothur software v.1.38.0 (Schloss et al. 2009). The resulting dataset is available within GenBank repository under accession numbers MG958889 - MG962339 (OTU sequences) and in Sequence Read Archive (SRA) under accession number SRP148861 (raw sequencing reads).
Termite genera phylogenetically close to Cavitermes and reported cases of facultative parthenogenesis in higher termites
We tested for the presence of the three endosymbionts in other termite species from the Cavitermes lineage sensu Hellemans et al. (2017a): Palmitermes impostor (Termitidae: Termitinae) that facultatively reproduces asexually through gamete duplication (heads and gonads of 25 neotenic queens distributed in five nests), and Spinitermes trispinosus (Termitidae: Termitinae), a species known for the occurrence of neotenic queens (Carrijo 2009), and whose female aspirants arise from gamete duplication (Hellemans et al., unpublished data) (head and gonads of one primary queen, and heads of two soldiers). Additionally, we tested Inquilinitermes inquilinus of the Termes lineage, the sister lineage to Cavitermes (Hellemans et al. 2017a), in which female aspirants and neotenic queens are also produced through gamete duplication (Hellemans et al., unpublished data) (heads of six neotenic queens).
We also screened for the presence of the three endosymbionts in all other reported cases of facultative parthenogenesis in the Neotropics: Embiratermes neotenicus and Silvestritermes minutus (Termitidae: Syntermitinae), both restoring ploidy through central fusion (Fougeyrollas et al. 2015; Fougeyrollas et al. 2017) (heads and gonads of 25 neotenic queens distributed in five nests).
RESULTS
Species identity
All 20 nests used for the screening of endosymbionts and strain identification belonged to C. tuberosus: sequences obtained from 17 nests collected from the Petit Saut dam area, Kaw and Saül have a 100% identity with the COII from the full mitochondrial genome of C. tuberosus (accession KP026294; E-value = 0); sequences from individuals of the nest K_100 from the Petit Saut dam area differed by two nucleotide substitutions; sequences of the individuals collected in Trinidad (nest TT18–59) and Brazil (nest BR18–06) showed, respectively, 98.8% and 97.4% identity with the COII of the full mitochondrial genome (see Supplementary Table S2 for GenBank accessions).
Infection prevalence in the nests and among castes
Wolbachia was found in all tested nests of C. tuberosus, but Cardinium and Rickettsia were not detected. Infection rates were analysed on 10 nests under a GLM framework in which the best model (according to the minimum theoretical Aikake Information Criterion; AIC; Supplementary Table S4) was the one considering nests and reproductive origin (i.e. sexually or parthenogenetically produced). GLM analysis determined a significant effect of nests on the infection rate (Likelihood-ratio Chi-Squared test, χ2 = 24.29, df = 9, p = 0.004), with nest K_100 strongly differing from all others (post-hoc pairwise comparison using the fdr method, all p < 0.004). In nest K_100, Wolbachia infected 54.76% of individuals (Supplementary Table S1, Fig. 3), whereas for all other nests, virtually all individuals (372 out of 378, 98.41%) were positive for Wolbachia (mean per nest ± SD = 98.20% ± 3.24; Supplementary Table S1, Fig. 3), whether they were from the altruistic or reproductive caste (97.22% ± 4.54 and 98.70% ± 2.86 respectively; Wilcoxon rank sum test, W = 35, p = 0.576).
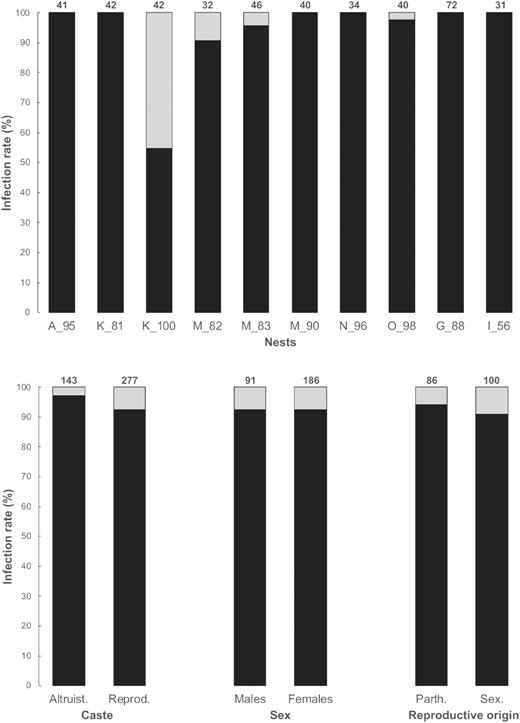
Infection rate of Wolbachia per nest, between the altruistic and the reproductive castes, between males and females from the reproductive caste and between parthenogenetically and sexually produced females from the reproductive caste, in 10 nests of C. tuberosus. The total number of individuals tested is indicated above each bar.
Strain determination and supergroup assignment
All six sequenced genes (16S rRNA, coxA, ftsZ, gatB, hcpA and fbpA) were identical in all 20 tested nests (except for one substitution at base #364 and #352 for coxA and fbpA respectively for the sample TT18–59 collected in Trinidad) showing that one single strain of Wolbachia (hereafter referred to as wCtub) infects C. tuberosus. BLAST and phylogenetic results showed that all sequenced genes were highly similar to those of the Wolbachia endosymbionts of the bedbug Cimex lectularius (99% of identity, E-value = 0 for all genes), and belongs to supergroup F (Fig. 4; Supplementary Tables S2 and S3).
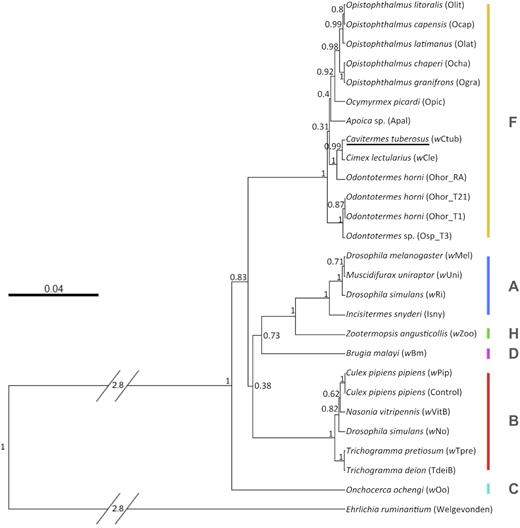
Rooted Wolbachia species tree estimated under Bayesian Inference using MLST genes (coxA, fbpA, ftsZ, gatB and hcpA) and highlighting the position of the new strain described from the termite C. tuberosus (wCtub) in supergroup F. Wolbachia strains are represented by the host species’ name followed by the Wolbachia strain in parentheses. Letters (A–D, F, H) stand for Wolbachia supergroups. Bayesian posterior probability support values are shown for each clade.
Wolbachia infection and impacts on phenotype
In order to detect a possible connection between Wolbachia and the induction of parthenogenesis in C. tuberosus, we analysed the infection status of males (n = 91 individuals) and females (n = 186) from the reproductive caste. We distinguished females according to their reproductive origin, sexual (n = 100) or parthenogenetic (n = 86). There was no association between sex and the infection status (Fisher's exact tests, two-tailed; p = 1), nor between the infection status of females and their reproductive origin (Fisher's exact tests, two-tailed; p = 0.580). Accordingly, infection rates differed neither between males and females from reproductive castes (93.09% ± 14.41 and 93.47% ± 18.57, respectively; Wilcoxon rank sum test, W = 45, p = 0.65; Fig. 3), nor between sexually and parthenogenetically produced females (92.08% ± 21.01 and 95% ± 15.81, respectively; Wilcoxon rank sum test, W = 55, p = 0.58; Fig. 3). FCA revealed segregation of allelic frequencies between (i) all nests and nest K_100 and (ii) in nest K_100, between infected and uninfected individuals (Supplementary Fig. S1). All tested eggs were infected by Wolbachia. The primary sex ratio was balanced in all nests (Supplementary Table S1; mean ± SD = 0.527 ± 0.077, min–max: 0.431–0.700; one sample t-test: t = 1.176, df = 10, p = 0.267).
To determine if Wolbachia could be involved in a nutritional mutualism by supplying B vitamins to its host, we tested for the presence of the bioA gene involved in the synthesis of the biotin. The bioA gene amplified in all 20 nests of C. tuberosus. Its sequence was 99.5% identical to the bioA gene from the endosymbiotic Wolbachia of the bedbug C. lectularius (E-value = 0; accession numbers MK053590 and AP013028, respectively; Supplementary Table S2).
Quantification of Wolbachia densities across tissues using quantitative PCR
The density of infection by Wolbachia significantly differed among tissues (one-way ANOVA, F3,12 = 18.63, p < 0.001). The highest densities of Wolbachia were detected in gonads, the lowest in the head. Pairwise comparisons showed that bacteriome-like structure and digestive tract have similar densities of Wolbachia (post-hoc pairwise t-test with fdr correction, p = 0.180; Fig. 5).
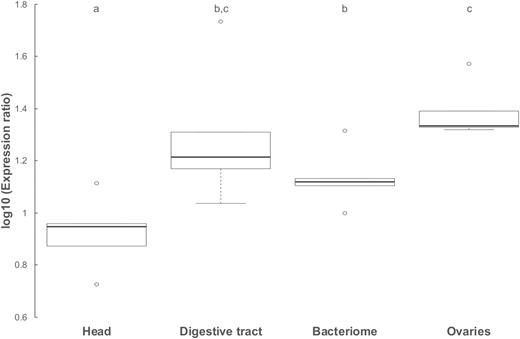
Boxplots showing the density of infection of Wolbachia across four tissues (head; ovaries; bacteriome-like structure: digestive tube from P3 to P5; see Fig. 2) of five neotenic queens from nest M_82 of C. tuberosus using qPCR. Density of infection was estimated as the log10 of the expression ratio using hindlegs as a control sample to which we compared all other tissues (see SM-5). The black line within the box represents the median, the bottom and top of the box are the lower (Q1) and upper (Q3) quartiles respectively (the range gives the interquartile distance, IQR), the lower and upper whiskers of the box represent 1.5*IQR below Q1 and above Q3 respectively, and outliers are found outside the range of whiskers. Letters denote significant differences between tissues (post-hoc multiple paired t-tests with fdr correction of p values).
Microbial community profiling of whole guts and bacteriome of C. tuberosus
Fifty-one OTUs were identified from the bacteriome-like structure of the neotenics. Strikingly, one specific OTU (OTU_29) constituted over 97.3% ± 0.7 reads in the three tested replicates. It was assigned with 100% identity to Wolbachia sp. of Proteobacterium phylum (Fig. 6, Supplementary Table S5). Additionally, OTU_29 was the only one assigned to this species. This points towards the fact that the pouch-shaped expansion at the antero-dorsal part of the mesenteron in neotenics of C. tuberosus is a bacteriome that hosts endosymbiotic Wolbachia. The remaining OTUs identified were characterized by very low abundance (Fig. 6, Supplementary Table S5), and might result from a possible cross-contamination from the surrounding gut tissue.
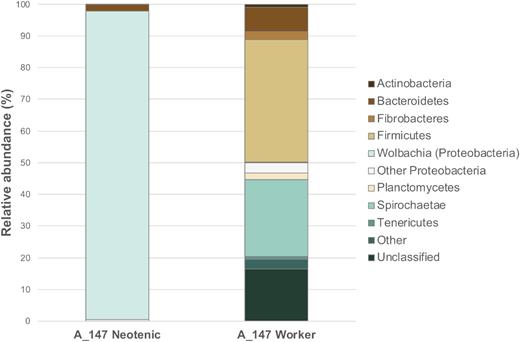
Taxonomic distribution into major phyla of prokaryotic OTUs from the guts of neotenic queens (bacteriome) and workers (whole guts) from nest A_147 of C. tuberosus. Relative abundances are derived from the percentage of reads assigned to specific OTUs (Supplementary Table S4). Microbial community profiles are based on the 16S rRNA gene amplicon high-throughput sequencing and annotation is done according to SILVA database v.128.
Analyses of the whole gut of workers of C. tuberosus indicated a more diverse and evenly distributed microbial community (Fig. 6, Supplementary Table S5). In total, 3457 OTUs were identified and assigned to multiple prokaryotic taxa. The dominant phyla identified were: Firmicutes, Spirochaetae and Bacteroidetes, with 38.8% ± 6.4, 24.2% ± 5.3 and 7.6% ± 0.6 relative abundance, respectively. The Wolbachia-related OTU constituted a minor percentage of the total diversity (0.17% ± 0.04).
Termite genera phylogenetically close to Cavitermes and reported cases of facultative parthenogenesis in higher termites
Endosymbionts (i.e. Wolbachia, Cardinium and Rickettsia) were totally absent in two species from the Cavitermes lineage (i.e. in P. impostor and S. trispinosus), and in the two species where facultative parthenogenesis has been reported, i.e. E. neotenicus and S. minutus (Syntermitinae). The quality of DNA extractions was verified by amplifying COI, which excluded false negatives. On the other hand, Wolbachia was detected in all six neotenic queens of I. inquilinus from the Termes lineage, the sister lineage of Cavitermes. The Wolbachia strain infecting I. inquilinus (wIinq) shows 99.6%, 100% and 99.1% similarity with wCtub at loci coxA, gatB and hcpA, respectively (fbpA and ftsZ could not be amplified) (see Supplementary Table S2 for GenBank accessions).
DISCUSSION
Infection by Wolbachia was found in all nests of C. tuberosus, both altruistic and reproductive castes, males and females, sexually and parthenogenetically produced individuals. One single strain (wCtub), belonging to the supergroup F, infects all nests on a wide geographic range (at least 3500 km). Wolbachia was found at different densities in reproductive and somatic tissues of C. tuberosus and represents over 97% of bacterial reads in the bacteriome associated with the mesenteron of neotenics, a structure here described in termites for the first time. One nest of C. tuberosus (K_100), differs from all others: nearly one out of two individuals is uninfected while the mean infection rate for all other nests reaches 98.20%. While it shares the same strain wCtub with all other nests, our data showed that nest K_100 is in striking contrast to them regarding allele frequencies at 17 nuclear microsatellite and mitochondrial profiles. These differences could be in line with K_100 belonging to a distinct population in the course of losing its symbiosis with Wolbachia (Sicard et al. 2014; Zug and Hammerstein 2015; Correa and Ballard 2016).
Termite-infecting Wolbachia strains usually belong to supergroups H and F, with basal termites infected by supergroup H and higher termites by supergroup F (Lo and Evans 2007). Wolbachia from supergroups A and B sporadically infect Cubitermes spp. (Roy and Harry 2007), supergroup B was reported in Odontotermes horni and Coptotermes heimi (Salunke et al. 2010) and a divergent clade within supergroup A was reported for Neotermes luykxi, N. jouteli and Serritermes serrifer (Lo and Evans 2007). While other termite-infecting Wolbachia are frequently reported from the supergroup F, wCtub is most closely related to the strain infecting the bedbug C. lectularius (wCle) (Fig. 4). The lack of phylogenetic congruence between Wolbachia and its hosts evidences common horizontal transmission of Wolbachia; the mechanisms driving these transmissions have mostly remained unclear (Oliver et al. 2010; Himler et al. 2011; Balvín et al. 2018).
Wolbachia, a mutualistic partner of C. tuberosus
Our data show that wCtub is 99% similar to the Wolbachia strain infecting the bedbug C. lectularius (wCle). In this species, Hosokawa et al. (2010) demonstrated that Wolbachia (i) resides in bacteriomes located adjacent to the gonads and (ii) is an obligate nutritional mutualist: clearing of Wolbachia by using antibiotics led to sterility and reduced growth, which was rescued by B vitamin supplementation. Nikoh et al. (2014) later demonstrated that wCle genome possesses operons involved in the biosynthesis of B vitamins. Unfortunately, such direct proofs using antibiotics are practically impossible to obtain in termites because their digestion depends on a whole and complex community of symbiotic microorganisms (Ohkuma and Brune 2011). Interestingly, our results show that wCtub harbours the bioA gene involved in the biotin (vitamin B7) synthesis pathway. This hints that wCtub and wCle may share a similar genetic background of mutualism with their host (Gerth and Bleidorn 2016).
Dissection of the digestive tract revealed that neotenic queens of C. tuberosus display a bacteriome-like structure in the anterior part of the mesenteron (Fig. 2). Quantitative PCRs showed that Wolbachia densities are higher in this structure and in the surrounding gut compared to other somatic tissues (Fig. 5). Furthermore, high-throughput sequencing of the 16S rRNA gene showed that Wolbachia represents over 97% of bacterial reads in the bacteriome associated with the mesenteron of C. tuberosus, while it represents less than 1% in the gut (Fig. 6). In Cimex lectularius, Wolbachia specifically inhabits the bacteriome and the gonads of its host (Hosokawa et al. 2010), but in the leaf-cutting ant Acromyrmex octospinosus, Wolbachia occurs abundantly in the gut lumen (Andersen et al. 2012). In both cases, Wolbachia plays a mutualistic nutritional role, by providing essential B vitamins for the former, or by interacting in the ant-fungus cultivation for the latter. As interactions between two genomes may lead to an extended phenotype shaping the morphology of the host (Hughes 2014), comparison with these two species may suggest an ongoing evolution of a specialized structure for which the intracellular bacterium Wolbachia has a peculiar affinity. This structure could house and maintain the endosymbiotic bacteria before that it passes on to the host's digestive tract.
Diouf et al. (2018) reported that the relative abundance of Wolbachia in the gut of the wood-feeding termite Nasutitermes arborum is caste- and age-dependent and negatively correlated with the diversity of the microbiota: Wolbachia was less abundant in the gut (fully colonized by true gut-resident bacteria) of (old) castes directly feeding on their substrate, while it was dominant in the gut of young individuals which are fed with salivary regurgitates of workers. In C. tuberosus, neotenic queens display an empty digestive tube and receive nutriments through mouth-to-mouth exchanges (i.e. trophallaxis) with workers (Hellemans et al. 2017b). Composition of the gut community of workers shows a potential capacity allowing to feed on diverse lignocellulose rich biomass (Fig. 6), but the nutritional quality of the fluids transferred to neotenics is unknown and may be deficient in some nutrients. Furthermore, C. tuberosus lives in arboreal nests constructed by other species (Mathews 1977) and is not known to forage outside. Cavitermes tuberosus, as most of inquiline species, feeds on their hosts’ nest material (Martius 1997; Florencio et al. 2013), which may be, once again, deficient in some nutrients that Wolbachia would synthesise. Actually, in every known case, insects carrying bacteriome-associated endosymbionts have peculiar diet depleted in essential nutrients (Moran, McCutcheon and Nakabachi 2008). Buchnera provides essential amino acids to aphids, as Carsonella does to psyllids (Hemiptera) (Shigenobu et al. 2000; Nakabachi et al. 2006); Portiera is a source of carotenoids in whiteflies (Hemiptera) (Sloan and Moran 2012); Wiggleworthia synthesizes essential vitamins for the tsetse flies (Akman et al. 2002); and Blattabacterium has allowed cockroaches, the closest relatives of termites, to subsist successfully on nitrogen-poor diets (Sabree, Kambhampati and Moran 2009).
Wolbachia is not associated with parthenogenesis in C. tuberosus and related species
While we described here for the first time a potential nutritional mutualism between Wolbachia and a termite, it is also the first example of the occurrence of this bacterium in a diplo-diploid organism restoring ploidy through gamete duplication during parthenogenesis. In haplo-diploids, Wolbachia was shown to restore ploidy through gamete duplication in Hymenoptera (Rabeling and Kronauer 2013; Ma and Schwander 2017), and via functional apomixis in several species of phytophagous mites of the genus Bryobia (Weeks and Breeuwer 2001). For diplo-diploids, induction of parthenogenesis by Wolbachia is extremely difficult to formally demonstrate, but has been suggested for the female-only springtail Folsomia candida in which ploidy is restored by terminal fusion (Pike and Kingcombe 2009; Ma and Schwander 2017). However, the induction of parthenogenesis by Wolbachia is not obvious in C. tuberosus as Wolbachia infects all individuals, whether they are male or female, sexually or parthenogenetically produced. At the same time, two other reproductive manipulations possibly induced by Wolbachia—male-killing and feminization—can be definitively discarded as a same proportion of males and females occur in eggs, and infected males remain phenotypic males.
Contrary to C. tuberosus, P. impostor and S. trispinosus, two species also restoring ploidy through gamete duplication and belonging to the Cavitermes lineage, are not infected with Wolbachia. At first glance, these observations could discard Wolbachia as the inducer of parthenogenesis in species of the Cavitermes lineage. However, species with secondary loss of Wolbachia can remain capable of thelytoky. For example, in the asexual lineage of the thrips Aptinothrips rufus, Wolbachia is present in only 69% of females (van der Kooi and Schwander 2014), which can probably be explained by the transfer of genes involved in parthenogenesis from the Wolbachia genome into the host's genome (Dunning Hotopp et al. 2007; Feldhaar and Gross 2009).
Wolbachia and the inquiline lifestyle
Taken together, the high similarity between Wolbachia strains in C. tuberosus and Cimex lectularius, the high prevalence of a single strain of Wolbachia over a wide geographical area, the fact that Wolbachia shows a high affinity for the bacteriome-like structure located at the anterior part of the mesenteron of C. tuberosus, combined to the behaviour and ecology of the species, suggest that the two partners are likely engaged in a nutritional mutualism. Whole genome sequencing of these partners may be useful to elucidate the exact role of Wolbachia in the nutrition processes of C. tuberosus, and to reveal horizontal gene transfers. Further analyses on species with similar inquiline lifestyles, such as the Termitidae I. inquilinus (this study) and the Serritermitidae S. serrifer (Lo and Evans 2007), respectively infected by Wolbachia from supergroups F and A, will be conducted to investigate whether these intimate relationships evolved multiple times independently from different Wolbachia supergroups and in unrelated termite lineages.
ACKNOWLEDGMENTS
We are grateful for logistic support during fieldwork to the late Philippe Cerdan and to Régis Vigouroux and the staff of the Laboratoire Environnement HYDRECO of Petit Saut (EDF-CNEH) in French Guiana; to Christopher K. Starr in the Republic of Trinidad and Tobago (University of the West Indies); and to Alexandre Vasconcellos and Alane Ayana Vieira de Oliveira Couto in Brazil (Federal University of Paraíba). We thank Xavier Goux for help in the field; Johannes A. J. Breeuwer and Martha S. Hunter for the supply of positive controls for endosymbionts screening; Robert Hanus and Virginie Roy for the supply of termite samples from the Syntermitinae subfamily; and Sarah Chérasse for help in quantitative PCR laboratory work. We are grateful to Johannes A. J. Breeuwer and Roman Zug for comments on an early version of this manuscript, and to the two anonymous referees for their constructive comments.
Data archiving
Sequences produced for this study have been deposited in GenBank repository under accessions MF953226 to MF953242, MH522792, MH522793, MK053589 to MK053591 and MK064568 to MK064570 (Sanger sequencing; see Supplementary Table S2 for details), MG958889 to MG962339 (Illumina sequencing; OTU sequences); and in Sequence Read Archive (SRA) under accession number SRP148861 (Illumina sequencing; raw sequencing reads).
Authors’ contribution
SH and DF designed the study. SH, NK, MM, YR and DF collected the material. SH and NK performed the microsatellite genotyping, single gene sequencing and qPCR. MM and MC performed Illumina sequencing and subsequent analyses. SH, NK and DF carried out the statistical analyses. All authors contributed significantly to the manuscript and approved the final version.
FUNDING
This work was supported by a PhD fellowship and a travel grant from the KMDA Royal Zoological Society of Antwerp (S.H.), grants from the Belgian National Fund for Scientific Research FRS-FNRS (D.F. and Y.R.: FRFC grant nos. 2.4594.12; D.F.: J.0110.17), from the Fonds Defay (D.F.) and by an FNR 2014 CORE project OPTILYS (Exploring the higher termite lignocellulolytic system to optimize the conversion of biomass into energy and useful platform molecules/C14/SR/8286517).
Conflicts of interest. None declared.
REFERENCES
Author notes
These authors contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}