





Abstract
Gut microbiota play a key role in host health. Mammals acquire gut microbiota during birth, but timing of gut microbial recruitment in birds is unknown. We evaluated whether precocial chicks from three species of arctic-breeding shorebirds acquire gut microbiota before or after hatching, and then documented the rate and compositional dynamics of accumulation of gut microbiota. Contrary to earlier reports of microbial recruitment before hatching in chickens, quantitative PCR and Illumina sequence data indicated negligible microbiota in the guts of shorebird embryos before hatching. Analyses of chick feces indicated an exponential increase in bacterial abundance of guts 0–2 days post-hatch, followed by stabilization. Gut communities were characterized by stochastic recruitment and convergence towards a community dominated by Clostridia and Gammaproteobacteria. We conclude that guts of shorebird chicks are likely void of microbiota prior to hatch, but that stable gut microbiome establishes as early as 3 days of age, probably from environmental inocula.
INTRODUCTION
Gut microbiota contribute to maintaining organismal health through nutrient uptake (Leser and Mølbak 2009), detoxification of digestive byproducts (Kohl 2012), energy and fat metabolism (Velagapudi et al.2010), and interactions with the host immune system (Rescigno 2014). Environmental exposure early in life can shape the gut microbiota and aid in defending against pathogens while the immune system is immature (Bar-Shira, Sklan and Friedman 2003). In mammals, maternal vaginal and fecal microbe transmission are crucial in the recruitment and establishment of microbiota in the digestive tract of a neonate (Palmer et al.2007). Thus, maternal effects strongly control the initial composition of gut microbiota in mammalian offspring (Stevens and Hume 1998).
It remains unclear whether birds can acquire gut microbiota while inside the egg, and how microbial communities accumulate after initial recruitment. Bacterial colonization can theoretically occur before or after hatching, and we propose two alternative hypotheses to describe the process of microbial colonization: the ‘head start’ and the ‘sterile egg’ hypothesis. The ‘head start’ hypothesis posits that microbes enter the gastrointestinal tract of avian embryos through transovarian transmission during oogenesis as a maternal effect, or possibly from the environment by penetration through eggshell pores and embryonic membranes after egg laying (Gantois et al.2009; Cox et al.2012; Martelli and Davies 2012). The ‘sterile egg hypothesis’ predicts that microbiota recruitment in the avian gut occurs after hatch because the chorion membrane maintains a sterile environment within the egg (van der Wielen et al.2002; Kohl 2012). The development of the avian embryo within the membrane-enclosed yolk supports the latter hypothesis, but enteric bacteria have been cultured from the guts of embryos of domestic chicken (Gallus gallus domesticus; Kizerwetter-Świda and Binek 2008), and vertical transfer of pathogenic Salmonella enterica has been documented between mother and embryo in domestic chickens (Guard-Petter 2001; De Buck et al.2004). The two hypotheses differ in the maternal contribution to gut microbiota of offspring and in the source of inoculum. The ‘head start’ hypothesis predicts that maternal or environmental inoculation occurs before hatching, possibly resulting in metabolic and immunological advantages at hatching. In contrast, if gut microbiota are absent in embryos, as posited by the ‘sterile egg’ hypothesis, chicks must recruit microbiota from the environment after hatch.
Developmental strategies after hatching vary within birds and range from chicks hatching with complete dependence on their parent(s), like most altricial species, to superprecocial species that are independent of parental care after hatching. Altricial and precocial birds likely differ in the degree of maternal influence on microbial inoculation of their chicks. An altricial bird species, the Barn Swallow (Hirundo rustica) showed slight maternal, but not paternal, effects on the fecal microbiota of their offspring (Kreisinger et al.2017). Altricial birds have undeveloped young that require parental feeding and brooding until fledging, whereas precocial chicks hatch fully developed with invaginated yolk sacs, leave the nest immediately, forage independently, but rely on parental brooding and defense until able to thermoregulate (Sibly et al.2012). Thus, altricial birds have higher potential for parental influence on gut microbiota through salivary transfer and a longer period of nest occupation, whereas precocial parents have little direct influence on their offspring's microbiota aside from exposure to bacteria on feathers during brooding, and leading young to brood-rearing areas.
In addition to uncertainty about the timing of initial recruitment of microbiota, the early successional dynamics of the avian gut microbiota are unclear. For example, it remains unknown how rapidly chick guts are colonized, in what order bacterial taxa become established and when convergence towards an adult gut microbiome occurs. Precocial young of most Arctic-breeding shorebirds are nidifugous, self-feeding and dependent on parents for thermoregulation, and could benefit from recruiting gut microbiota before hatching, because symbionts can promote nutritional uptake and growth immediately after hatch (Angelakis and Raoult 2010). Efficient use of nutrients may be particularly important in the Arctic because a short and synchronous breeding season requires rapid chick development (Schekkerman et al.2003), favoring the recruitment of microbiota via the ‘head start’ hypothesis.
We investigated the ontogeny of the gut microbial community in three species of Arctic-breeding shorebirds. Our objectives were to (i) test the ‘head start’ and ‘sterile egg’ hypotheses and determine if precocial chicks of arctic shorebirds recruit gut microbiota before or after hatching, and (ii) assess successional trajectories in the gut microbiota of shorebird chicks after hatching. To address our two objectives, we combined quantitative PCR (qPCR) and Illumina MiSeq techniques to analyze the gut microbiota of embryos prior to hatch and in fecal samples collected from shorebird chicks from hatch to up to 10 days of age.
MATERIALS AND METHODS
Sample collection and preparation
We collected 27 eggs from seven clutches of dunlin (Calidris alpina) and 18 eggs from five clutches of semipalmated sandpipers (Calidris pusilla), at Utqiaġvik (formerly Barrow), Alaska (71°17΄26 ΄N, 156°47΄19 ΄W) in June–July 2013. We estimated incubation stage via changes in egg buoyancy that occur with embryonic development (Liebezeit, Smith and Lanctot 2007) and collected eggs 1–3 days before the predicted hatch date to ensure near complete embryo development. After collection, embryos were removed from eggs, euthanized and kept frozen at –20°C until dissection. Before dissection, embryos were washed in a weak solution of 0.6% sodium hypochlorite (10% bleach) to minimize contamination by external microorganisms. We aseptically removed the lower intestinal tract between the gizzard and cloaca and stored samples at –80°C. We also collected the invaginated yolk sacs from the embryos of five dunlin and four semipalmated sandpipers.
To investigate gut microbiota of mobile broods after hatching, we selected dunlin and red phalaropes (Phalaropus fulicarius) as study species because dunlin use mesic terrestrial habitat, whereas red phalaropes use freshwater habitat (Cunningham, Kesler and Lanctot 2016). Different habitats and associated microbiomes could affect the recruitment of gut microbiota in chicks. We fit the attending adults with 1.55 g very high frequency (VHF) radios, and, for dunlin, one chick per brood with 0.26 g VHF radios (Holohil Systems Ltd., Carp, Ontario, Canada; Fig. S3.1, Supporting Information). Red phalarope chicks were too small at hatch to apply transmitters so we located young by tracking the attending male. We attempted to locate mobile broods every 3 days, using radio telemetry and by searching areas where broods were last seen. Once broods were located, chicks were captured and feces were collected by placing the chicks in individual compartments in an insulated, thermally heated bag, lined with sterile wax paper. Chicks were held for <5 min to minimize stress and then released together. At several capture sites, we also collected environmental samples consisting of a mix of water and soil in a 1.5-ml Eppendorf tube using a flame-sterilized spatula. All samples were stored in 100% ethanol at –20°C until further analyses.
To remove ethanol from fecal samples, we centrifuged samples for 10 min at 10 000 rpm and discarded the supernatant. We repeated this step twice with 1 ml of RNase/DNA-free molecular grade water to minimize ethanol contamination (Grond et al.2014; Ryu et al.2014). We shredded embryonic gastrointestinal tissues, and extracted DNA from embryo and chick fecal samples using the MoBio Power Lyzer/Power Soil kit following the manufacturer's instructions (Mo Bio Laboratory, Carlsbad, CA, USA), except for replacing the bead beating step with 15 min vortexing at high velocity for tissue homogenization. Yield of genomic DNA was determined using a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Waltham, MA, USA).
Conventional PCR
To test for the presence of bacteria in the embryonic gut, we PCR-amplified total bacterial communities using general bacterial primers 515F and 806R to generate 16S rRNA gene amplicons (Caporaso et al.2012). Primer sequences are provided in the Supplemental Materials (Table S3.1, Supporting Information). PCR reactions were conducted in a 25-μl reaction volume, using AmpliTaq Gold DNA polymerase (Applied Biosystems, Waltham, MA, USA) and 5 μl of DNA template (5 ng-DNA/μl). PCR conditions consisted of 30 cycles of 15 s at 95°C, 30 s at 55°C and 30 s at 72°C, preceded by an initial denaturing step of 10 min at 95°C, and followed by a final extension step of 5 min at 72°C.
To ensure that the bacterial DNA within the embryo lower gastro-intestinal (GI) tract was not degraded by our disinfection procedures, we amplified avian DNA from 24 random samples with primers 2550F and 2718R designed for molecular sexing of non-ratite birds, following PCR conditions of Fridolfsson and Ellegren (1999). Had the sodium hypochlorite compromised the DNA, we should not have been able to acquire avian or bacterial amplicons. Validation tests confirmed the integrity of the lab procedure and our ability to amplify the bacterial DNA.
Quantitative PCR
We estimated 16S rRNA gene copy numbers in embryo gut, yolk, fecal and control samples in triplicate qPCR reactions on a BioRad CFX96 Touch Real-Time PCR thermocycler (Bio-Rad Laboratories, Inc., Berkeley, CA, USA). We used a TaqMan® qPCR assay (Applied Biosystems) targeting the bacterial 16S rRNA gene in combination with 2X TaqMan Gene Expression Master Mix with two general bacterial primers at 100 nM final concentrations (F_Bact1369 and R_Prok1492; Furet et al.2009; primer sequences can be found in Table S1). Cocktails included 4 μl of DNA template (5 ng-DNA/μl) and conditions consisted of 2 min at 50°C and an initial denaturing for 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The negative controls were RNase/DNA-free molecular grade water and the positive controls was Semipalmated Sandpiper fecal DNA at 5 ng-DNA/μl. We included our positive and negative controls in triplicate in the two qPCR runs we conducted. Standard curves were generated using 2 to 2 × 106 16S rRNA gene copies of Staphylococcus aureus subsp. aureus (efficiency = 105–116%, R2 = 0.984–0.990). To control for the possibility of PCR inhibition and to establish minimum detection levels, we spiked a subset of samples (n = 15) with a dilution series of the S. aureus rRNA gene ranging from 100 to 106 copies.
Illumina sequencing and sequence analyses
We used general bacterial 515F and uniquely barcoded 806R primers to generate multiplexed 16S rRNA gene amplicons from embryo guts and fecal samples, following protocols of the Earth Microbiome Project (Caporaso et al.2012). PCR reactions were performed in a 25-μl reaction volume, using AmpliTaq Gold DNA polymerase (Applied Biosystems) and 5 μl of DNA template (5 ng-DNA/μl), and were run using 25 cycles, opposed to the 35 cycles described in the Earth Microbiome Project (EMP) protocols. We removed primers from our PCR product using the Agencourt AMPure XP PCR purification system (Beckman Coulter, Brea, CA) following the manufacturer's instructions except AMPure volume ratio was adjusted to 1:1 and the ethanol washes were repeated three times instead of two. We sequenced the V4 region in 2 × 250 bp paired-end runs using the Illumina MiSeq platform. Each Illumina run included a 15% PhiX spike. Embryo samples and fecal samples from chicks of 0–3 days old were sequenced twice because of low-sequence yields.
Bacterial 16S rRNA gene sequences were quality filtered, contiged and demultiplexed using the QIIME program (Caporaso et al.2010), and aligned against the GreenGenes 16S rRNA gene reference database using PyNAST algorithms (v.13_8;DeSantis et al.2006). For full QIIME pipeline, we refer to the QIIME Code supplement. We identified chimeras—artifacts that combine multiple different sequences—using CHIMERASLAYER (Haas et al.2011). Sequences were clustered to operational taxonomic units (OTUs) at 97% sequence similarity, and assigned to taxa using the Naive Bayesian Classifier with an RDP reference (Wang et al.2007). After taxonomy assignment, we identified and removed non-target sequences of chimeras, singletons, non-aligned sequences, archaea, chloroplasts and mitochondria.
Prior to subsequent analyses, we rarefied chick samples to 5000 sequences per sample due to low-sequence yields in samples of chicks from 0–2 days of age. All embryo samples yielded far fewer than 5000 sequences, and were analyzed without rarefication.
STATISTICAL ANALYSIS
Embryo gut and yolk microbiota
Statistical analyses were conducted in R (ver. 3.3.1, R Development Core Team 2016). To test for differences in 16S rRNA gene copy numbers between embryos and chicks, we performed a one-way analysis of variance (ANOVA) and assessed pairwise differences using a post hoc Tukey HSD test with functions of the base Program R. We compared bacterial richness (number of OTUs) among our embryo gut, yolk and negative controls using an ANOVA, followed by a post hoc Tukey HSD test. To compare microbial communities of embryo gut and yolk samples and the negative control, we applied non-metric multidimensional scaling (NMDS) of Bray-Curtis distance matrices using the metaMDS function in the ‘vegan’ package.
Chick gut microbiota
To look for natural breaks in bacterial abundance in our chick qPCR data, we used the segmented function from the ‘Segmented’ package (Muggeo 2008). Large changes in the slope represent changes in the accumulation of 16S rRNA copies in the chick gut and were used to determine whether we could pool our chick microbiota data into naturally occurring broader age classes. To test for significant differences among slopes in bacterial abundance versus chick age, we ran a Davies test using the davies.test function from the ‘Segmented’ package.
We calculated diversity indices (observed number of OTUs, Simpson's (1-D) and Shannon's H) using the QIIME alpha_diversity.py script. In addition, we calculated evenness (Pielou's J) using the diversityresult function in the ‘BiodiversityR’ package (Kindt and Coe 2005). We estimated beta diversity to compare variance between age groups (0–2 days versus 3–10 days) and environment. We tested for pairwise differences among age groups using post hoc Tukey HSD tests.
We determined shared core OTUs among young and old chicks and environmental samples, and constructed a scaled Venn diagram using EulerAPE (Micallef and Rodgers 2014). Core OTUs were defined as OTUs that were present in >50% of our samples within a group, and were determined using the QIIME compute_core_microbiome.py script. We used the core community instead of the entire bacterial community as peripheral low abundance OTUs could skew our results.
To investigate potential differences among microbial communities in chicks and the environment, we applied NMDS. We tested for treatment differences through an analysis of similarity using the anosim function in the ‘vegan’ package. Means are shown ± standard error.
To determine whether variation in gut communities differed among samples from dunlin and red phalarope chicks, or if samples could be pooled for further analyses, we analyzed homogeneity of variance using the betadisper function from the ‘vegan’ package (Oksanen et al.2015). To test for pairwise differences in variance between young and old chicks and environmental samples, we performed a permutation F test using the permutes function in the ‘vegan’ package.
Because we sampled multiple chicks within the same brood, we tested for potential effects of brood identity on OTU richness and bacterial abundance (log10 16S rRNA copy numbers). We compared Akaike Information Criterion (AIC) values generated by linear mixed effect models using the lmer and glm function in the ‘vegan’ and ‘lme4’ packages (Bates et al.2015; Oksanen et al.2015). Our null model included a fixed effect of chick age, and our full model included an additional random effect of Brood ID.
Data accessibility
The nucleotide sequences and metadata have been made available through Figshare at https://doi.org/10.6084/m9.figshare.4587394.v1 and https://doi.org/10.6084/m9.figshare.4587379.v1.
RESULTS
Timing of recruitment
Conventional and qPCR
Conventional PCR assays yielded no visible 16S rRNA gene amplification from gut or yolk samples of shorebird embryos collected 1–3 days before hatching. Consistently, the high fidelity and high sensitivity Taqman qPCR assays suggested low copy numbers of 16S rRNA genes in the intestinal tract and yolk sac of embryos. The qPCR detected bacterial amplification in our positive control after 12.6 ± 1.1SE cycles, compared to 33.8 ± 0.2 and 33.2 ± 0.3 cycles for the guts of dunlin and semipalmated sandpiper embryos, 34.1 ± 0.2 and 33.7 ± 0.2 cycles for yolk samples, and 33.9 ± 0.5 cycles for our negative control. We detected negligible copy numbers of the bacterial 16S rRNA gene in all embryo samples, and we pooled samples from semipalmated sandpipers and dunlin for statistical analyses. Our gut and yolk samples did not differ from the negative controls in either threshold cycle or copy numbers (Tukey HSD; P > 0.22; Fig. 1a), and all samples and negative controls had higher threshold cycle and lower copy numbers than our positive controls (Tukey HSD; P < 0.001).
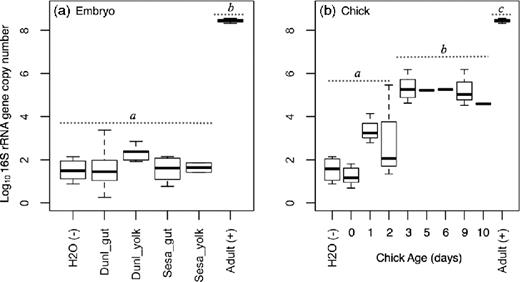
Median (thick, black line), 25% and 75% quartiles (boxes), and 90% confidence intervals (whiskers) for the log10 16S rRNA gene copy numbers. (a) Samples included lower gastrointestinal tracts (gut) and yolk sacs from developed embryos of dunlin (DUNL) and semipalmated sandpipers (SESA) at Utqiaġvik, Alaska, 2013. The negative control was RNase/DNA-free molecular grade water, whereas the positive control was a fecal sample from an adult SESA (adult (+); 5 ng DNA/μl). Dashed lines and letters above boxes represent significant differences in 16S rRNA copy number (Tukey HSD). The microbial community from the positive control was not included in further analyses, but is shown here for reference purposes. (b) 16S rRNA gene copy numbers in fecal samples from REPH and DUNL chicks from 0–10 days after hatch.
Avian DNA from all 24 gut samples successfully amplified in conventional PCR, further confirming that disinfecting the embryo's external surfaces did not compromise DNA within the GI tract of shorebird embryos. We detected 10 copies of the 16S rRNA of S. aureus after 33.7 ± 0.21 cycles, which was a similar detection threshold as found for our embryo and negative control samples. Thus, our conventional and qPCR assays indicate that low copy numbers of bacterial rRNA were present in our embryo samples, which represented the entire gastrointestinal system from the gizzard to cloaca.
Sequencing
Illumina MiSeq libraries acquired from 42 of our 45 embryo samples contained an average of 108.6 ± 15.9 sequences (range 0–427), whereas the positive control contained over 7 million sequences. Three samples did not provide any sequencing output and were not included in further analyses. A second sequence library generated from the gastrointestinal tracts and yolk samples from shorebird embryos resulted in similarly low yields. The number of OTUs in the embryo samples was not significantly different from the negative control (F2,38 = 101.6, P = 0.13), and richness of embryo gut samples did not differ from yolk samples (TukeyHSD, P = 0.71). Our NMDS showed that the minimal number of sequences obtained samples from Semipalmated Sandpiper and Dunlin embryos, and our negative control overlapped in ordination space (Fig. S2, Supporting Information). Overall, our sequence data indicated that minimal microbiota were present in the gastrointestinal systems or yolk sacs of shorebird embryos before hatching of the eggs.
Establishment of the microbiota
We collected 45 fecal samples from 1–10 day old chicks of dunlin and red phalaropes at Utqiaġvik, AK, in 2013. Despite the different habitats used by dunlin and red phalaropes to rear their chicks, fecal samples of chicks did not differ in the abundance and variance of their microbiota (F2,27 = 5.5, P = 0.16). We pooled samples for further analyses to increase sample sizes. We did not detect an effect of brood identity on the bacterial abundance and OTU richness (AICnull = 285.6 and 66.7; AICfull = 283.6 and 68.7), and we therefore regarded samples of chicks from the same brood as independent.
Abundance
We observed an increase in 16S rRNA gene copy numbers from age 0–2 days. After 3 days post hatch, copy numbers stabilized at on average 395 852 ± 98 048 per sample (Fig. 1b). Our broken-stick regression indicated a natural break at 3 days post hatch, and the slopes between 0–2 and 3–10 days post hatch differed significantly (Davies’ test, P < 0.001; Fig. S3, Supporting Information). The slope from day 3–10 did not differ from 0, suggesting stability with no change in microbial abundance over this period (t = 0.095, P = 0.925). As a result, we pooled our samples into a ‘young’ (0–2 day age) and ‘old’ (3–10 day age) group for further analyses.
Richness and composition
After quality filtering, we retained 1689 081 high-quality sequences (90.2%) from our total of 1860 751 raw sequences for further analyses. After rarefaction to 5000 sequences per sample, we included five samples of 0- to 2-day-old chicks, 19 samples of 3- to 10-day-old chicks and six environmental samples.
Overall, environmental samples were highest in bacterial richness on all phylogenetic levels, followed by young chicks and old chicks (Table 1; Fig. 2; Fig. S4, Supporting Information). We identified a total of 36, 723 and 5715 unique bacterial phyla, genera and OTUs in our samples. Observed OTU richness was lower in samples from old chicks than in samples from young chicks (Tukey HSD, P = 0.03), consistent with environmental/habitat filtering occurring as chicks aged. In both young and old chicks, the observed OTU richness was lower than in the environmental samples (Tukey HSD, P < 0.001). Simpson's (1-D) and Shannon's (H) diversity indices indicated that environmental samples had the highest diversity scores (0.98 ± 0.01; 8.10 ± 0.39), and old chicks the lowest (0.56 ± 0.04; 1.98 ± 0.14). In addition, evenness of bacterial communities in the gut was highest in environmental samples (0.81 ± 0.03), followed by young chicks (0.53 ± 0.11) and old chicks (0.30 ± 0.02; Fig. S4).
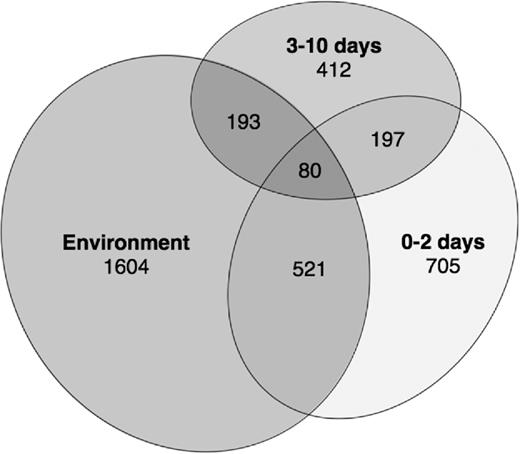
Scaled Venn diagram of the core bacterial OTUs isolated from fecal samples of young (0–2 days) and old (3–10 days) dunlin and red phalarope chicks, as well as environmental samples of water and soil. Numbers represent numbers of unique or shared core OTUs.
Total and per sample bacterial richness at different phylogenetic levels in guts of young chicks (young; 0–2 days old), old chicks (old; 3–10 days), and in environmental samples (environment).
| Total phyla | Phyla/sample | Total genera | Genera/sample | Total OTU | OTU/sample | |
|---|---|---|---|---|---|---|
| Young | 23 | 16.2 ± 1.5 | 432 | 132.2 ± 32.9 | 1279 | 308.4 ± 80.1 |
| Old | 19 | 9.5 ± 0.4 | 379 | 42.5 ± 3.4 | 1005 | 105.7 ± 7.7 |
| Environment | 34 | 25.2 ± 1.2 | 568 | 304.7 ± 23.1 | 4116 | 1060.5 ± 122.7 |
| Total phyla | Phyla/sample | Total genera | Genera/sample | Total OTU | OTU/sample | |
|---|---|---|---|---|---|---|
| Young | 23 | 16.2 ± 1.5 | 432 | 132.2 ± 32.9 | 1279 | 308.4 ± 80.1 |
| Old | 19 | 9.5 ± 0.4 | 379 | 42.5 ± 3.4 | 1005 | 105.7 ± 7.7 |
| Environment | 34 | 25.2 ± 1.2 | 568 | 304.7 ± 23.1 | 4116 | 1060.5 ± 122.7 |
Total and per sample bacterial richness at different phylogenetic levels in guts of young chicks (young; 0–2 days old), old chicks (old; 3–10 days), and in environmental samples (environment).
| Total phyla | Phyla/sample | Total genera | Genera/sample | Total OTU | OTU/sample | |
|---|---|---|---|---|---|---|
| Young | 23 | 16.2 ± 1.5 | 432 | 132.2 ± 32.9 | 1279 | 308.4 ± 80.1 |
| Old | 19 | 9.5 ± 0.4 | 379 | 42.5 ± 3.4 | 1005 | 105.7 ± 7.7 |
| Environment | 34 | 25.2 ± 1.2 | 568 | 304.7 ± 23.1 | 4116 | 1060.5 ± 122.7 |
| Total phyla | Phyla/sample | Total genera | Genera/sample | Total OTU | OTU/sample | |
|---|---|---|---|---|---|---|
| Young | 23 | 16.2 ± 1.5 | 432 | 132.2 ± 32.9 | 1279 | 308.4 ± 80.1 |
| Old | 19 | 9.5 ± 0.4 | 379 | 42.5 ± 3.4 | 1005 | 105.7 ± 7.7 |
| Environment | 34 | 25.2 ± 1.2 | 568 | 304.7 ± 23.1 | 4116 | 1060.5 ± 122.7 |
At a community level, our NMDS (k = 3, stress = 0.083) distinguished environmental from chick samples (ANOSIM, R = 0.853, P = 0.003; Fig. 3). Young chick sample communities differed from old chick communities (ANOSIM, R = 0.487, P = 0.006), but were not significantly different from environmental samples (ANOSIM, R = –0.052, P = 0.599). However, microbial communities of old chick samples were highly differentiated from environmental samples (ANOSIM, R = 0.999, P < 0.001).
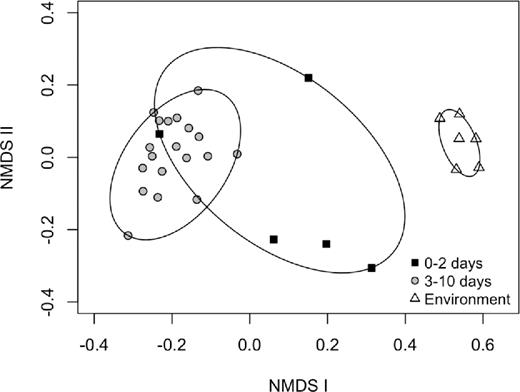
NMDS of the OTUs in the fecal samples of young chicks (0–2 days, n = 5) and old chicks (3–10 days, n = 19) of dunlin and red phalaropes, and environmental samples (n = 6), at Utqiaġvik, Alaska, 2013.
The gut microbiota composition of chicks was dynamic over time. Relative abundance of Firmicutes increased from 37.7 ± 14.2% in young to 73.4 ± 5.9% in old chicks (Fig. 4). The increase was mainly driven by increases in abundance of the classes Clostridia and Bacilli (Fig. 5). Within the class Clostridia, Clostridium colinum (sequence similarity = 98%; Genbank Accession LC011040.1)—an opportunistic avian pathogen—was responsible for 73.6 ± 8.6% of sequences among young chicks and 88.1 ± 4.1% in old chicks. The second most abundant OTU within the Clostridia belonged to the genus Candidatus Arthromitus with a relative abundance of 6.1 ± 2.7% in young and 10.3 ± 4.1% in old chicks. In contrast to Firmicutes, Proteobacteria relative abundance decreased from 45.5 ± 14.4% in young to 22.3 ± 6.0% in old chicks. Within Proteobacteria, 43% of sequences belonged to the genus Rickettsiella within the Gammaproteobacteria. The relative abundance of Actinobacteria and Bacteroidetes declined by 50%–80% from young to old chicks (Actinobacteria: 4.1 ± 1.5% to 0.3 ± 0.1%; Bacteroidetes: 5.0 ± 2.2% to 0.1 ± 0.02%; Fig. 5).
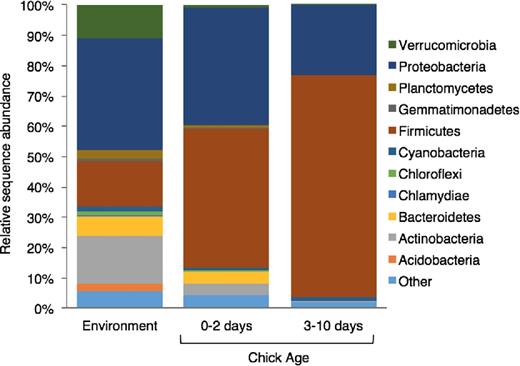
Relative abundance of bacterial phyla in environmental samples and fecal samples of young (0–2 days) and old (3–10 days) chicks of dunlin and red phalaropes at Utqiaġvik, Alaska, 2013. Day 0 represents day of hatching.
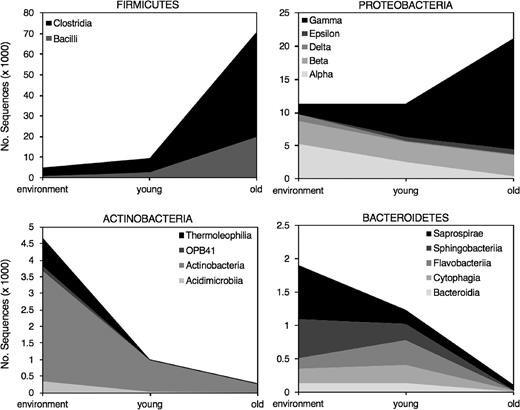
Sequence abundance per bacterial class for the four main phyla found in environmental samples of water and soil, and young (0–2 days) and old (3–10 days) chick fecal samples collected from dunlin and red phalaropes at Utqiaġvik, Alaska, 2013.
Beta diversity of bacterial OTUs was greater among young chicks than among the environmental samples (F2,27 = 3.347, P = 0.03), but did not differ between old chicks and the environment (F2,27 = 3.347, P = 0.26), and only marginally between young and old chicks (F2,27 = 3.347, P = 0.07).
DISCUSSION
Timing of recruitment
Our data largely supported the ‘sterile egg hypothesis’, which posits that embryos hatch from eggs with a sterile gut. Despite detecting low copy number of 16S rRNA genes in embryo samples using conventional and qPCR assays, these were either below detection level (conventional PCR) or indistinguishable from negative controls to which no DNA template had been added. The low copy numbers detected in the negative controls likely represent artifactual sequences, as products appeared late in the amplification process after >30 cycles (Arikawa et al.2008).
Similar to the PCR assays, we documented little evidence of bacterial presence in our embryo samples or the negative controls in the Illumina MiSeq sequencing analyses. The detection of any sequences in our negative controls might be attributed to several potential causes. Carlsen et al. (2012) demonstrated tag switching in 0.7%–1.6% of sequences during pyrosequencing, resulting in erroneous assignment of sequences to samples. In fact, the total number of sequences in our embryos and negative controls comprised only 0.06% of all sequences, a proportion far below error rates estimated by Carlsen et al. (2012). Similarly, barcode switching in Illumina sequencing studies can present a substantial source of error. Sinclair et al. (2015) used both forward and reverse barcoded primers and estimated that barcode switching could represent up to 22% of sequences in the Illumina platform. We used only reverse-barcoded primers and were unable to estimate or detect barcode switching. Sequences in negative controls (or samples without adequate template) may also originate earlier in the library preparation process. Last, DNA extraction kits and PCR reagents may contain some bacterial DNA, leading to false detections in low template samples like our embryos (Salter et al.2014).
The low abundance of bacteria in the guts of shorebird embryos collected under natural conditions contrasts with past reports from domestic chickens. Kizerwetter-Świda and Binek (2008) cultured a variety of enteric bacteria from chicken embryos obtained from a local hatcher. However, if the gut microbiota recruitment and establishment were similar in shorebirds and chickens, we should have detected significantly higher bacterial loads with our culture-independent methods that targeted the entire spectrum of gut bacteria. Differences in environmental conditions between domestic chickens in a commercial-scale production facility and Arctic-breeding birds under field conditions might lead to different routes for bacterial transmission. For example, Salmonella enteritidis can infect chicken embryos via infection of the female chicken's reproductive organs, resulting in incorporation of bacteria into the egg during oogenesis (Gantois et al.2009). The likelihood of a similar infection in wild birds may be low and could possibly be the reason for the limited evidence of bacteria we documented in our wild bird embryos.
Another potential infection route for avian embryos is through the egg shell and membranes, which is referred to as trans-shell infection. We detected minimal gut microbiota in shorebird embryos gut and yolk samples, which also indicated that bacteria do not infect the embryo gastrointestinal tract as a result of trans-shell infection during incubation. Whether infection of the egg after laying reduces hatching success in birds is still under debate (Cook et al.2003; Wang, Firestone and Beissinger 2011), but incubation was shown to reduce and change microbial communities on egg shells (Lee et al.2014). Since we collected our embryos close to hatch and all embryos were alive when the eggs were opened, trans-shell infection likely had either not occurred in our embryos or infection did not lead to reduced viability. Overall, our field data indicate that gut microbiota become established after chicks hatch and are exposed to the environmental microbiome.
Establishment of the microbiota
Based on our qPCR data, the bacterial abundance increased exponentially in chick guts during the first 3 days after hatching. Precocial chicks of Arctic shorebirds leave the nest within a day of hatching and feed independently, and we have observed chicks feeding as early as 1 day of age (personal communication: D.E. Gerik). Since parents do not provision precocial chicks in Arctic-breeding shorebirds, chicks likely acquire gut microbiota from ingestion of prey-associated microbiota. An inoculation route via diet was supported by our ordination results which showed that the microbiota of young chicks were more similar to the environmental microbiota in community composition than old chicks.
A rapid increase in the microbial abundance within the first 3 days after hatching may be enhanced by the supply of yolk to the chick gastrointestinal tract. During the first 2–3 days after hatch, precocial chicks use an invaginated yolk sac for nutrients while they learn to forage (Forsythe 1973; Schekkerman and Boele 2009). The yolk mainly consists of fat, protein and carbohydrates, and also contains vitamins and trace elements (Vieira 2007). Combined with the relatively high body temperature of the chick, yolk provides a rich substrate for bacterial growth, and is commonly used in bacterial pure culture media (Carter 1960; Westblom, Madan and Midkiff 1991; Byrne et al.2008).
We observed higher richness at a phylum, genus and OTU level among gut microbes in young chicks than old chicks. OTU richness was three times higher in young chicks than old chicks for some bacterial phyla, indicating either selective recruitment or host-controlled environmental filtering prior to microbiota stabilization at 3–10 days of age. Community filtering for specific, high abundance gut symbionts was supported by a decrease in evenness, which suggested a rapid increase in abundance of dominant bacterial taxa with age. Depletion of the yolk sac after 2–3 days and the associated decrease in available nutrients to the chick gut microbiota might also explain the decrease in diversity. After nutrients are depleted, only bacteria that use nutrients derived from the chick diet, or those produced by other gastrointestinal bacteria, would be sustained. After 3 days post hatch, we observed establishment of a stable abundance of gut microbiota. Bacterial abundance did not change over the rest of our sampling period, but then differed by two orders of magnitude between 10-day-old chicks and adult birds. We were unable to sample young birds after 10 days of age because chick mortality and enhanced mobility made locating chicks challenging.
As chicks aged from 0 to 10 days old, their gut microbiota became dominated by bacteria within the phyla Firmicutes and Proteobacteria. Increasing abundance of Clostridia was the main driver of the rise in Firmicutes abundance. Clostridia are also early colonizers of the human infant gastrointestinal tract and essential in maintaining gut homeostasis (Lopetuso et al.2013). The prevalence and natural occurrence of Clostridia in the gastrointestinal tract in other wild birds is unknown, but their high abundance in the guts of shorebird chicks suggests a role for commensalism or potentially mutualism.
Within the class Clostridia, C. colinum was the most abundant species with a relative abundance of 70%. Clostridium colinum is an opportunistic avian pathogen and can cause ulcerative enteritis in chickens and quail. Clostridium colinum infects a variety of birds, including domestic poultry, northern bobwhites (Colinus virginianus), American robins (Turdus migratorius), western bluebirds (Sialia mexicana) and lories (Trichoglossus sp. and Eos sp.; Winterfield and Berkhoff 1977; Porter 1998; Bildfell, Eltzroth and Songe 2001; Pizarro et al.2005; Beltran-Alcrudo et al.2008). All previous detections of C. colinum have been associated with disease or death of the host, but the occurrence of C. colinum in shorebird chicks could present evidence of alternative functions in the avian gut. Moreover, our sequences showed 98% similarity to C. colinum, which could indicate that our sequences belong to an alternative, unidentified strain of C. colinum or closely related Clostridium species. Clostridium colinum was not detected in the environmental samples we sequenced, suggesting that chicks may acquire this bacterium from their diet or another source.
Proteobacteria were the second most abundant phylum within the chick gut; their relative abundance decreased from young to old chicks. Similar to Clostridia, human infants experience an early increase in the class Gammaproteobacteria (La Rosa et al.2014). Gammaproteobacteria represent a variety of species, but their functions have not been identified in avian systems. Within the Proteobacteria, we observed a shift from an evenly structured community including all classes of Proteobacteria to a community dominated by Gammaproteobacteria. Rickettsiella was the dominant genus within the Gammaproteobacteria. Rickettsiella spp. are associated with microbiota of terrestrial arthropods (Bouchon, Zimmer and Dittmer 2016; Duron, Cremaschi and McCoy 2016), including several pathogens and endosymbionts (Leclerque and Kleespies 2008; Tsuchida et al.2010). The detection of Rickettsiella in chicks could indicate an important role of diet as a microbial inoculum. Although Rickettsiella spp. are generally classified as arthropod pathogens (Bouchon, Zimmer and Dittmer 2016), the high relative abundance in chicks indicates previously unknown non-pathogenic functions.
The gut microbiota of shorebird chicks of 3+ days old consisted predominantly of Firmicutes (∼73%) and Proteobacteria (∼22%), and to a lesser extent of Bacteroidetes and Actinobacteria. The relative abundance of Firmicutes in chicks was comparable to adult shorebirds sampled in Delaware Bay during spring migratory staging (59%–73%; Ryu et al.2014). However, Firmicutes relative abundance in our study was higher than the majority of wild bird species sampled up to present, including a range of Neotropical bird species, the kakapo (Strigops habroptila), several penguin species and the folivorous hoatzin (Opisthocomus hoazin; Godoy-Vitorino et al.2012; Dewar et al.2014; Waite and Taylor 2014; Hird et al.2015). In addition, gut microbiota of barn swallow chicks consisted of on average 45% Firmicutes (Kreisinger et al.2017). Firmicutes are involved in fermentation of organic molecules in the gut environment, and have been positively linked to weight gain in mammalian and avian models (Turnbaugh et al.2006; Angelakis and Raoult 2010). Shorebird chicks in the Arctic and adults during migratory staging have in common that they both have a need for rapid development and weight gain, which could potentially be facilitated by higher Firmicutes abundances. Gut microbiota of barn swallow (Hirundo rustica) chicks consisted of on average 45% Firmicutes (Kreisinger et al.2017), which could be a result of their slower, altricial developmental strategy.
Based on differences in gut microbiota of chicks and adults, it is likely that chicks continue to acquire more bacteria as they grow. Perhaps the largest expansion of the microbial community occurs when birds migrate for the first time and are exposed to new, diverse environments across a large geographic area. Exposure to new microbial communities in the environment during migration may increase gut microbial abundance, when bacteria occupy previously unused niches, but this question requires further study.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSEC online.
Acknowledgements
We thank Sherry Fleming and Alina Akhunova for help with dissections and molecular analyses. Jenny Cunningham, Patrick Herzog, Franklin Stetler, Leah Rensel, William Kaselow and Vanessa Loverti helped locate shorebird nests and collect eggs at Utqiaġvik, Alaska. We thank Ludek Zurek, Bradley Olson and two anonymous reviewers for their constructive comments on our manuscript. The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the U.S. Fish and Wildlife Service.
FUNDING
This work was supported by the US Fish and Wildlife Service's Avian Health Program [grant number: 2013-02 to KG BKS and RBL]; a Doctoral Dissertation Improvement Grant from the National Science Foundation [grant number: 1501479 to KG]; the Arctic Landscape Conservation Cooperative; the US Fish and Wildlife Service and the Division of Biology at Kansas State University.
Conflicts of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}