





Abstract
Soils are complex ecosystems in which fungi and bacteria co-exist and interact. Fungal highways are a kind of interaction by which bacteria use fungal hyphae to disperse in soils. Despite the fact that fungal highways have been studied in laboratory models, the diversity of fungi and bacteria interacting in this way in soils is still unknown. Fungal highway columns containing two different culture media were used as a selective method to study the identity of fungi and bacteria able to migrate along the hyphae in three forest soils. Regardless of the soil type, fungi of the genus Mortierella (phylum Zygomycota) were selected inside the columns. In contrast, a diverse community of bacteria dominated by Firmicutes and Proteobacteria was observed. The results confirm the importance of bacteria affiliated to Burkholderia as potentially associated migrating bacteria in soils and indicate that other groups such as Bacillus and Clostridium are also highly enriched in the co-colonization of a new habitat (columns) associated to Mortierella. The diversity of potentially associated migrating bacteria brings a novel perspective on the indirect metabolic capabilities that could be favored by r-strategist fungi and supports the fact that these fungi should be considered as crucial actors in soil functioning.
INTRODUCTION
Fungi and bacteria coexist in almost every ecosystem (Frey-Klett et al.2011), yet mycologists and bacteriologists have traditionally conducted their studies separately (Strickland & Rousk 2010). Sharing a common habitat and exploiting common resources has led to numerous and diverse possible interactions between fungi and bacteria (Kobayashi & Crouch 2009). In the specific case of soils, fungi appear to be better adapted than most bacteria to the physical constraints of the habitat (Harms, Schlosser and Wick 2011). In soils, the filamentous growth of fungi allows them to bridge the air-filled voids, thus colonizing water-unsaturated soil patches (Ritz & Young 2004). This is conventionally considered as impossible for bacteria, whose motility is in consequence limited (de Boer et al.2005; Or et al.2007). Another crucial advantage of fungi is their ability to translocate nutrients and carbon inside their hyphae (Schütte 1956; Wells & Boddy 1995; Wells, Boddy and Evans 1995), enabling them to better cross nutrient-poor areas, which are a common feature in soils (Wells & Boddy 1995; Wells, Boddy and Evans 1995; de Boer et al.2005). The specific adaptations of filamentous fungi have led to the presumption of fungi as key metabolic engines in soils, something that can be exploited, for example, in the case of soil bioremediation (Harms, Schlosser and Wick 2011). However, the description of a type of interaction in which fungal hyphae are used by bacteria for dispersal in soils, an interaction termed fungal highways (Kohlmeier et al.2005), offers the possibility for a potential synergistic effect of both microbial groups in soil functioning (Martin et al.2012). The diversity of fungal groups and their associated migrating bacteria (hereafter termed AMB) involved in fungal highways in soils is still poorly documented. Nonetheless, studies on fungal highways made under laboratory conditions have confirmed the positive effect of this type of interaction on microbial activity (Kohlmeier et al.2005; Wick et al.2007; Warmink, Nazir and van Elsas 2009; Furuno et al.2012; Banitz et al.2013; Bravo et al.2013; Knudsen et al.2013; Ellegaard-Jensen et al.2014).
In a previous study, we developed a device, the fungal highway column, that allows for the selective enrichment of fungi and AMB from soils (Simon et al.2015). Microorganisms are attracted towards two culture media within the column: an attracting medium, in direct contact with the soil, and a target medium, separated physically from the soil and colonized by bacteria only through fungal highways. These two media offer a novel habitat that can be colonized by fungi and potential AMB. The validation of the device involved the study of selected fungi and bacteria under laboratory conditions, as well as the identification of easy to cultivate fungi and potential AMB from a soil influenced by the oxalate–carbonate pathway, a biogeochemical pathway for which fungal highways are known to be implicated in microbial activity (Martin et al.2012; Bravo et al.2013). In spite of the known biases of this culturing approach (Rappe & Giovannoni 2003), several fungal–bacterial couples were identified this way. These results demonstrated the potential of the columns as a tool to assess the diversity of fungi and bacteria interacting through fungal highways in soils.
In the present study, fungal highway columns were used to conduct an in situ inventory of the fungal and potential AMB communities in three forest soils from Switzerland. The diversity of fungi and bacteria colonizing the two media compartments and the donor soil was studied by amplification and sequencing of specific molecular markers. Two culture media and three donor soils with different organic carbon and nitrogen contents (and different C:N ratios) were used.
MATERIALS AND METHODS
Sampling and soil characterization
Fungal highway columns have been used for the sampling of fungi and potential AMB. These columns are recently developed devices targeting isolation and separation of soil fungi and bacteria moving along fungal hyphae (Simon et al.2015). The principle of the columns consists in the use of two plugs of agar containing culture medium that would create a novel nutritional habitat within the column. These media are separated from each other and from the soil by a series of physical barriers that limit the colonization to fungi and bacteria. The first medium plug, which is in direct contact with the soil (compartment designated as ‘middle’), is easily accessible to fungi or bacteria. In contrast the second medium (compartment designated as ‘top’) enriches filamentous fungi able to breach the unsaturated space separating the media (filled with glass beads and constricted to avoid the formation of a water film). Because of this, bacteria found in the ‘top’ compartment are presumed to disperse associated to fungal hyphae. This assumption is based on the validation experiments performed in our previous study demonstrating that bacteria required the presence of fungi inside the columns to reach the top medium (Simon et al.2015). As the culture medium placed inside the columns will influence fungal or bacterial diversity, we compared two sets of columns containing a nutrient-rich and a nutrient-poor culture medium, respectively. The nutrient-rich culture medium was a low-malt agar (LMA) medium, composed of 6 g l−1 malt (Mycotec SA, La Chaux-de-Fonds, Switzerland) and 15 g l−1 agar (Biolife Italiana, Milano, Italy). Reasoner's 2 agar (R2A) medium (Reasoner, Blannon and Geldreich 1979) was selected as the nutrient-poor medium. This medium has been used routinely for the enumeration of bacteria in drinking water as it favors the development of slow-growing bacteria that are normally over-competed in richer media (Rice et al.2012). R2A medium was composed of 0.5 g l−1 yeast extract, 0.5 g l−1 Bacto Peptone, 0.5 g l−1 casamino acids, 0.5 g l−1 glucose, 0.5 g l−1 soluble starch, 0.3 g l−1 Na-pyruvate, 0.3 g l−1 K2HPO4, 0.05 g l−1 MgSO4.7H2O and 15 g l−1 purified agar.
The columns were placed in one of the superficial horizons of three soils located in Switzerland: a Folic Histosol (OFnoz3 horizon, 11–22 cm; 46° 55΄ 59.42″ N, 6° 43΄ 36.24″ E; altitude 1170 m), a Dystric Cambisol (A horizon, 0–5 cm; 47° 0΄ 13.31″ N, 6° 56΄ 52.50″ E, altitude 560 m) and a Fluvisol (Jsca1 horizon, 0–9 cm; 46° 32΄ 8.77″ N, 7° 4΄ 3.82΄ E; altitude 735 m; Baize 2009; Jabiol et al.2013; WRB 2014). In each soil, organic carbon content, nitrogen content, pH, water content and the amount of organic matter were measured (Fig. 1). Columns with either medium were placed in triplicate in each soil type. The columns were left in the soil for 7 days, and after this time carried back to the laboratory.

Soils analyzed in this study. (a) The physicochemical properties of the three selected soils, a Fluvisol, a Dystic Cambisol and a Folic Histosol, are indicated alongside an image of the soil profile used for the installation of the columns. C/N ratio (C/N) was calculated using the carbon and nitrogen contents. The soil horizon in which the columns were placed is represented by the colored bands. (b) Geographical location of the three soils in Switzerland. (c) Close view of columns in the Fluvisol.
DNA extraction, sequencing and analysis of taxonomic diversity
In order to identify the microorganisms able to colonize the two media inside the columns, instead of sub-culturing in specific media (as previously performed in Simon et al.2015), total DNA was extracted from three parts: the portion of soil on which the column was placed (termed ‘soil’), the culture medium in direct contact with the soil (‘middle’), and the culture medium at the top (‘top’) of the column. DNA extraction was performed according to the instructions for the FastDNA spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) with an additional bead-beating step of 10 min at 50 beats s−1 (Qiagen, Hilden, Germany). Although DNA from each triplicate column was extracted individually using the entire medium plug, due to the low DNA yield, the extracts obtained from the triplicate columns of each medium (LMA or R2A) were pooled by ethanol precipitation.
In order to identify fungi and bacteria, internal transcribed spacer (ITS) and a fragment of 16S rRNA gene were targeted, respectively. A step of pre-amplification by PCR had to be performed in order to obtain enough DNA material for sequencing in the case of the middle and top samples. Although no control to verify carry-over of contaminating DNA in the pre-amplification step was conducted, community analysis for the two media in the different soils suggest that contamination by foreign DNA at this step was unlikely (see results). For fungi, amplification of a partial fragment of the ITS region was performed with the primers ITS1F (5΄-CTTGGTCATTTAGAGGAAGTAA-3΄) and ITS4 (5΄-TCCTCCGCTTATTGATATGC-3΄; Anderson, Prosser and Campbell 2003). For pre-amplification, the master mix contained (in 25 μl of final volume): 1X buffer (with 1.5 mM MgCl2), 0.5 mM dNTPs mix, 0.5 mM of each primer, 1 mg ml−1 BSA and 0.1 U DNA polymerase (Kapa Biosystems, Inc., Wilmington, MA, USA); 1 μl of DNA template was added (≤2 ng μl−1 of DNA). PCR was carried out in an Arktik thermocycler (Thermo Fisher Scientific, Waltham, MA, USA), with an initial denaturation at 95°C for 2 min, followed by 35 cycles consisting of denaturation at 95°C for 30 s, annealing at 61°C for 30 s and elongation at 72°C for 30 s. Final extension was performed at 72°C for 2 min. Final products contained 19–84 ng μl−1 DNA. For bacteria, PCR amplification was performed on a partial fragment of the 16S rRNA gene using primers EUB 9–27f (5΄-AGAAAGGAGGTGATCCAGCC-3΄) and EUB 1542r (5΄-AGAAAGGAGGTGATCCAGCC-3΄; Liesack, Weyland and Stackebrandt 1991). For pre-amplification, the master mix contained (in 25 μl of final volume): 1X buffer (with 1.5 mM MgCl2), 0.2 mM dNTPs mix, 0.2 mM of each primer, 1 mg ml−1 BSA and 0.5 U DNA polymerase; 1 μl of DNA template was added (≤2 ng μl−1 of DNA). PCR was carried out in an Arktik thermocycler, with an initial denaturation at 95°C for 2 min, followed by 10 cycles consisting of denaturation at 95°C for 30 s, annealing at 60°C (touchdown PCR program decreasing the temperature −0.5°C per cycle) for 20 s, and elongation at 72°C for 1 min, then 25 cycles consisting of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and elongation at 72°C for 1 min. Final extension was performed at 72°C for 1 min. Final products contained 26–250 ng μl−1 DNA. DNA and the pre-amplified PCR products were sent to Eurofins Genomics (Ebersberg, Germany) for tagging using the same primers. Subsequently, 16S rRNA gene and ITS amplicons were sequenced in the forward direction using 454-technology (Roche, Basel, Switzerland).
Fungal and bacterial amplicon sequences were analyzed independently, using the software mothur version 1.36.1 (Schloss et al.2009). Bacterial reads were processed by following a modified standard operating procedure (Schloss, Gevers and Westcott 2011). First, sequencing errors were reduced by implementation of the AmpliconNoise algorithm and low-quality sequences were removed (minimum length of 220 bp, allowing one mismatch to the barcode, two mismatches to the primer and homopolymers no longer than 8 bp). Barcode and primer sequences were removed. Subsequently, sequences were aligned to the SILVA reference database release 119 (Quast et al.2013) and preclustered (pre.cluster, diffs = 1). Chimeras were removed using the chimera.uchime mothur command and singletons were excluded. Finally, sequences were classified using the naïve Bayesian classifier (Wang et al.2007) implemented in mothur with the SILVA reference database release 119 (Quast et al.2013). Operational taxonomic units (OTUs) were generated using the average neighbor algorithm. An OTU was defined at the 97% sequence similarity level. Fungal reads were quality processed with the same parameters as described above. After removing chimeras and singletons, the presence of fungal ITS was checked using ITSx version 1.0.11 (Bengtsson-Palme et al.2013) and non-fungal ITS sequences were discarded. Subsequently, ITS sequences were pairwise aligned to generate a distance matrix using the pairwise.seqs command. Finally, sequences were classified using the naïve Bayesian classifier (Wang et al.2007) implemented in mothur with the UNITE v6_sh_dynamic database (Köljalg et al.2013). OTUs were generated using the average neighbor algorithm, and an OTU was defined at the 97% sequence similarity level. Taxonomic assignment was made using a 80% confidence threshold. The raw sequence data have been deposited in the NCBI Sequence Read Archive under the accession number SRP069959. The matrices of both the fungal and bacterial communities as well as the taxonomic assignment of the representative sequence of each OTU have been submitted to GitHub (https://github.com/vherve/fungal_highway_columns).
Rarefaction curves (calculated from 10 000 iterations) and relative abundances of OTUs were estimated using mothur (Schloss et al.2009). All statistical analyses were computed using R software version 3.2.2 (R Core Team 2013). To evaluate the co-occurrence probability between the bacterial and fungal OTUs, the Veech probabilist model of species co-occurrence (Veech 2013) was applied using the cooccur package (Griffith, Veech and Marsh 2016) in R.
RESULTS
Soil properties
Among the soil horizons studied (Fig. 1), the horizon in the Histosol was the richest in C and N (as well as having the highest C:N ratio) and water and organic matter contents, compared with horizons from the Cambisol and the Fluvisol (Fig. 1). In addition, the pH in the three horizons also varied, with the horizon in the Histosol as the most acidic, followed by horizons from the Cambisol and Fluvisol.
Sequencing and analysis of taxonomic diversity
After leaving the columns for 7 days on site, the fungal and bacterial communities in the soils and those recovered inside the columns were analyzed by performing sequencing and taxonomic assignment on fungal ITS and bacterial 16S rRNA gene amplicons. The analyses were conducted in three compartments of the columns: (i) soil on which the column was placed (soil), (ii) culture medium in direct contact with the soil (middle), and (iii) culture medium at the top of the columns (top). In the case of the culture media, DNA extracts were pooled from the replicates and therefore, considering the lack of proper replication, only the general trends obtained will be indicated hereafter.
After quality filtering, chimera and singleton removals, a total of 167 517 fungal sequences were retained, and clustered into 718 OTUs defined at a similarity level of 97%. One sample of the fungal communities was excluded from the analysis (top of the column with LMA medium placed in the Folic Histosol) because no sequence passed the quality control. For the bacterial sequences, a total of 126 546 reads were retained and clustered into 3229 OTUs. The depth of sequencing was sufficient to describe the fungal communities according to the rarefaction curves obtained for the three soil types and the top compartments, but less for the middle compartment (Supplementary Fig. S1). However, this was not the case for the bacterial communities, with the exception of the middle and top compartments of the columns with R2A medium in the Histosol (Supplementary Fig. S1).
Among the global 718 fungal OTUs, 266 were present in the soil and/or columns from Cambisol, 372 in the Fluvisol and 175 in the Histosol, while in the case of bacteria, of the 3229 bacterial OTUs identified, 1514 were present in the soil/columns from Cambisol, 1458 in the Fluvisol and 795 in the Histosol (Supplementary Fig. S2). Considering that for the attracting and target media we had to perform a pre-amplification step to obtain enough DNA for sequencing, the analysis of the community composition was mainly conducted by adding the number of individual OTUs assigned to the same taxonomic rank (i.e. phylum or genus), rather than by comparing their relative abundance measured in sequence counts. This was considered as an approximation to the richness in OTU composition (number and type of individual entities present) in the samples. We do not use the term OTU diversity, as the relative abundance of each member of the community was not taken into account. There was little overlap in the composition of the fungal and bacterial communities found in soil with those in the middle and top compartments regardless of the soil type (Supplementary Fig. S2). The same is the case for the three soil types (Supplementary Fig. S3).
The analysis of the community composition between the different soils was also performed comparing the two culture media used (Fig. 2). In the case of the fungal communities, Ascomycota and Basiodiomycota were the most represented groups in the three soils. However, a clear shift towards a community dominated by Zygomycota was observed in the two culture media inside the columns (middle and top). This was independent of the media composition and of the soil type. This shift in community composition is the result of the dominance of the genus Mortierella in the columns (both in the middle and top compartments).
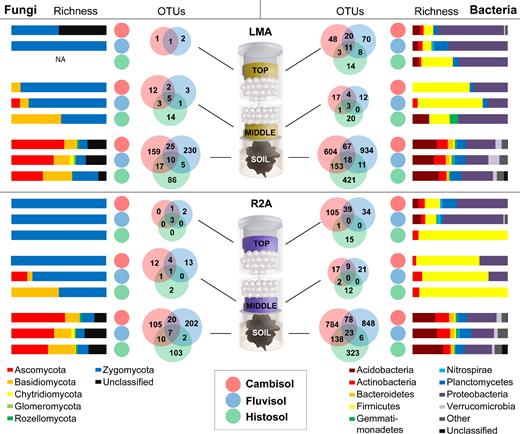
Summary of the fungal (left) and bacterial (right) richness at phylum level for soil, middle and top of the columns containing low malt agar (LMA) or Reasoner's 2 agar (R2A) medium. A color code for the different soil types is shown below. The number of OTUs unique to each soil type and those shared are indicated by Venn diagrams, color coded in the same way as the soil type. The Venn diagrams are presented for each compartment of the columns depicted as an illustration in the middle.
Bacterial community composition was more clearly influenced by the medium used for enrichment in the columns (Fig. 2). While Acidobacteria, followed by different classes of Proteobacteria, dominated the community composition in the three soils, Firmicutes were largely enriched in the middle compartment. Bacterial communities in the medium placed on top of the columns were richer, despite the clear dominance of a single fungal group in the top of the columns.
The distribution of the bacterial OTUs in the soil compared with those in the media inside the columns is presented in Fig. 3. This analysis allowed distinguishing those bacteria able to colonize the culture medium alone (middle), from those corresponding to AMB using fungal highways (top). For all the bacterial groups observed, the overlap between OTUs shared between soil and columns was minimal. This is the case for the OTUs found either in soils and middle (Fig. 3, yellow color) or soils and top (Fig. 3, dark green). This was observed regardless of the culture medium used for the enrichment inside the columns.
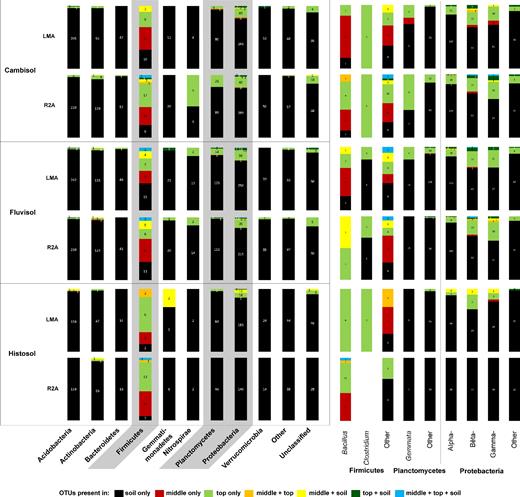
Taxonomical breakdown of the bacterial OTU richness present in the soil only (black), middle only (red), top only (green) and the OTUs shared between compartments (color code shown below the figure) for the two media used for enrichment (see Fig. 2). The comparison is presented as percentage values but the total number of OTUs in each taxonomic group is indicated within the bars. The same analysis for specific genera on the phyla Firmicutes, Planctomycetes and Proteobacteria, with the highest proportion of OTUs in the columns, is shown on the right side of the figure.
A more detailed taxonomic breakdown of the composition of bacterial OTUs assigned to the phyla Firmicutes, Planctomycetes and Proteobacteria, which corresponded to the highest proportion of OTUs present in the top of the columns, is presented in Fig. 3. Within the phylum Firmicutes, the majority of the OTUs affiliated to the genus Bacillus were only found in the middle or the top of the columns, with little to no overlap with the OTUs found in soils. Likewise, OTUs affiliated to Clostridium were either found in the soil only (Fluvisol) or in the top of the columns (all soils except for the R2A-containing column placed in the Histosol). For the phylum Planctomycetes, OTUs affiliated to the genus Gemmata were mainly found in soil (Histosol and column containing R2A medium placed in the Fluvisol), although several OTUs were found also in the top of the columns. In the case of Proteobacteria, most of the OTUs were solely found in soil. However, there were a number of OTUs that were also identified in the middle of the columns. Among the OTUs detected only in the top compartment of the columns, it is worth mentioning the genera Burkholderia (Cambisol, Fluvisol and Histosol LMA medium), Rhizobacter (Cambisol, Fluvisol and Histosol LMA medium), Aquabacterium (Cambisol and Fluvisol), Dechloromonas (Cambisol and Fluvisol), Defluviimonas (Cambisol) and Pseudofulvimonas (Fluvisol).
We analyzed the distribution of the 20 most abundant bacterial OTUs found in soils, middle and top of the columns for the two media used (Fig. 4). The three soil types were clearly separated based on the most abundant bacterial OTUs. Thiobacter, specific OTUs related to Acidobacteria and various genera of Proteobacteria were found for each soil. In spite of the clear differences in the community composition in the Cambisol and Fluvisol, in the middle of the columns, a clustering related to media composition (R2A versus LMA) was observed for the most abundant OTUs. This was the result of the enrichment of different OTUs related to Firmicutes, which are not found in soils. In the top of the columns, a clustering of the samples based once more on soil type was observed. However, clear similarities in the most abundant OTUs found in the Fluvisol and Cambisol were found. This corresponded to the enrichment in OTUs related to various genera of Proteobacteria (for example, Rhizobacter, Burkholderia, Dechloromonas and Aquabacterium), as well as to unclassified OTUs related to Acidobacteria. Various of these OTUs were also enriched in the columns containing LMA medium and placed in the Histosol, but in the case of the R2A medium, this sample is clearly dominated by two OTUs related to Bacillus. Only one OTU was common to the three datasets: Psychrobacillus sp., from the phylum Firmicutes.
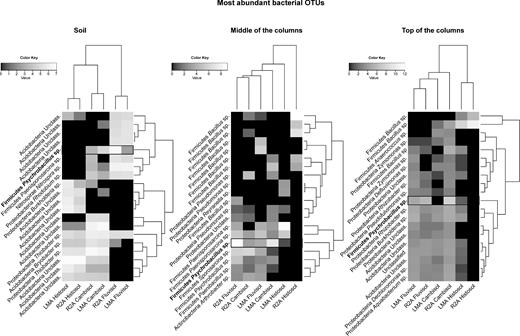
Heat maps representing the relative abundance of the 20 most abundant bacterial OTUs in the soils (left), at the middle (center) and at the top of the columns (right). The only OTU common to all datasets, assigned to the genus Psychrobacillus, is highlighted in the figure. The relative abundances were log(x + 1) transformed.
Finally, we investigated the co-occurrence of fungal and bacterial OTUs in our dataset. The fungal and bacterial OTUs that were preferentially found in association in the soils (P-value ≤0.001) were not found at the top of the columns (Supplementary Table S1).
DISCUSSION
The aim of this study was to establish a first molecular inventory of the diversity of fungi and bacteria potentially engaging in fungal highways in three forest soils. This was performed with the help of an enrichment device that is based on the ability of fungi and bacteria to co-migrate and colonize a new culture medium (Simon et al.2015). This is true, in particular for the second medium in the columns (top), which, according to our previous validation analysis under laboratory conditions (Simon et al.2015), can only be attained after crossing an unsaturated patch containing glass beads. Filamentous fungi are particularly suited for this crossing because of their growth mode (Ritz & Young 2004), as well as for their ability to translocate nutrients (Schütte 1956; Wells, Boddy and Evans 1995). Therefore, bacterial colonization of the top medium is expected to be the result of bacterial dispersal associated to migration along fungal hyphae (fungal highways).
Despite the fact that three different soil types and two kinds of enrichment media were used, all the identified fungal OTUs present in the top of the columns belonged to the genus Mortierella (phylum Zygomycota). Previously, using a culture-dependent method we have observed a limited diversity of culturable fungi able to reach the top medium of the columns, i.e. different strains of Fusarium spp. and one strain of the genus Chaeotomium (Simon et al.2015). However, it was unclear if this extreme selectivity was due to the culture conditions used to recover microorganisms from the columns, or if it reflected a restricted diversity of fungi able to reach the top compartment. Like Fusarium and Chaeotomium, Mortierella spp. are saprotrophic fungi (Cannon & Kirk 2007) that show moderate to fast growth abilities (Yadav et al.2015). Therefore, columns appear to clearly favor fungi with a fast-growing life style (Allison et al.2009; Brabcová et al.2016). This was the case even when a nutrient-poor medium was used and, therefore, it might reflect a bias in the duration of the experiment (7 days in the soil). A way to select for slower growing fungi in soils is still required to assess their implication for bacterial dispersal, and thus microbial community assembly. Nonetheless, our results raise the question of the importance of fast-growing saprophytic fungi in soil ecology. Usually, mycorrhizal and white-rot fungi are the focus of studies on fungal activity in soils (Folman et al.2008; Hoeksema & Classen 2012; Kohl, Lukasiewicz and van der Heijden 2016). However, Mortierellomycotina and Mucoromycotina (phylum Zygomycetes) represented 10.7% of the fungal diversity in a recent global assessment (Tedersoo et al.2014). In addition, a study estimating the global diversity of fungi used as a model the genus Mortierella because of its ubiquity in soils and the fact that it is one of the most widely detected groups in molecular diversity fungal surveys (Nagy et al.2011). Species of Mortierella are widespread in temperate soils, where they are almost cosmopolitan with regard to several ecological factors, occurring in a wide range of habitats. Their physiological characteristics and biochemical properties make them successful competitors among fungi and common inhabitants of soil and organic debris. Alongside Mortierella, the genera Absidia, Fennellomyces, Mucor, Umbelopsis and Zygorhynchus are the most common genera in agricultural and forest soils of the temperate zone, where they constitute almost 100% of Zygomycetes (Richardson 2009). Moreover, various strains of Mortierella have been isolated from pesticide-contaminated soils and showed pesticide degradation potentials (Ellegaard-Jensen et al.2013). In addition, some representatives of Mortierella are known to harbor endosymbiotic bacteria belonging to Burkholderiaceae such as Candidatus Glomeribacter gigasporarum (Sato et al.2010; Kai et al.2012) and Mycoavidus cysteinexigens from Mortierella elongata (Ohshima et al.2016). Although we did not obtain direct proof of this, a hypothesis that could be worth exploring in the future is the potential transport of bacteria not only on the surface of the fungal hyphae but also within the fungal cells. The fact that hyphae of Mortierella spp. are coenocytic (or barely septate) might help this process. This is consistent with a previous study suggesting that endobacteria could be favored in coenocytic hyphae (Desiro et al.2014). In this case, endobacteria will use fungal hyphae as ‘subways’ rather than highways for their dispersal in soils. Here, we refer to subway-like transport as the dispersal of bacteria within the fungal cell. This differs from a previous reference to subway-like dispersal, which was used to explain the passive dispersal of non-motile bacteria (Kohlmeier et al.2005).
The high selectivity exerted by the columns for specific fungal groups able to reach the top medium affected the structure of AMB communities. A strong selection occurred already at the bacterial phylum level between the soils, medium and top of the columns. While the phyla Acidobacteria and Actinobacteria showed a high richness in the soils, they represented a very small fraction of the OTUs in the top of the columns. A higher proportion of Firmicutes, Planctomycetes and specific genus of Proteobacteria was observed at the top of the columns. Some OTUs related to genera such as Clostridium (Firmicutes), Burkholderia, Rhizobacter, Aquabacterium, Defluviimonas and Pseudofulvimonas were only observed inside the columns, and not in the corresponding soils. Among the bacterial groups detected as AMB, some such as Burkholderia are known to establish close associations with fungi in soils, for example through the consumption of fungal-secreted metabolites or by modulating the fungal defense response (Stopnisek et al.2016). Moreover, the migration of Burkholderia terrae BS001 along hyphae of various soil fungi has been shown to provide protection against antifungal agents (Nazir, Tazetdinova and van Elsas 2014). In contrast, the detection of other genera such as Bacillus and Acinetobacter, which were considered as poorly or not interacting with fungi in a previous study investigating the attachment of bacteria to mycorrhizal fungi (Scheublin et al.2010), is more surprising. Our results underpin the fact that fungal highway columns could be used to identify potentially new AMB.
Another aspect analyzed in this study was the effect of organic carbon and nitrogen contents and C:N ratios in soils on the diversity of fungi and AMB. At a phylum level, the relative taxonomic composition was similar between the three soils, for fungi and for bacteria (Fig. 2). For fungi, this observation is consistent with the recently described stochastic assembly processes of fungal communities present in upper soil layers (Powell et al.2015). As indicated previously, despite the differences in the physicochemical conditions in the soil, a single fungal group was attracted to the columns. This is surprising considering that two media with different nutrient levels were used for the enrichment. In contrast, the diversity of bacteria able to colonize the columns and that of AMB was surprisingly large. While in the soil the structure of the bacterial communities clearly reflected the sample origin (Fig. 4), in the middle of the columns the selective medium used for the enrichment was more predominant in the case of the Fluvisol and the Cambisol. In the Histosol, which contained the highest amount of organic carbon and nitrogen and highest C:N ratio, the enriched community was entirely different, and it was less rich in the medium that favors slow growing bacteria (R2A). If the diversity of bacteria dispersed in association to fast-growing fungi is as important as suggested by our results, then the effects of soil properties such as C:N ratio or pH on the composition of bacterial communities might be variable and the physiological status of the fungal host might have a higher influence on the spatial distribution and abundance of AMB in soils. This is in accordance with a recent network analysis showing that modules of co-occurring bacterial and fungal OTUs presented contrasting relationship to various soil properties (de Menezes et al.2015).
CONCLUSION
In this study the analysis of the fungal and bacterial communities interacting through fungal highways in three forest soils revealed the importance of Mortierella in the colonization of novel habitats (culture media inside fungal highway columns), regardless of the soil physicochemical conditions. This study also provides a first insight into the taxonomy of AMB in situ and on the indirect metabolic activities that could be favored by r-strategist fungi as crucial actors in soil functioning. Finally, our results indicate that Mortierella is an ecologically relevant model to study bacterial–fungal interactions.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSEC online.
Acknowledgments
We would like to thank Dylan Tatti and Prof. Jean-Michel Gobat (University of Neuchâtel, Switzerland) for the supply of data concerning soil properties.
FUNDING
This research was supported by the Swiss National Science Foundation through Grants FN CR22I2-137994/1 and FN CR3212-149853/1.
Conflict of interest. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}